
Synergistic Efficacy of CDK4/6 Inhibitor Abemaciclib and HDAC Inhibitor Panobinostat in Pancreatic Cancer Cells
 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemical Reagents
2.2. Cell Culture
2.3. HDAC and Cell Cycle Gene Expression Analysis in Patient Samples
2.4. MTT Cytotoxicity Assay
2.5. Cell Proliferation Assay
2.6. Synergy Study
2.7. Clonogenic Survival Assay
2.8. Cell Cycle Assay
2.9. Scratch Assay
2.10. Invasion Assay
2.11. Apoptosis Assay
2.12. Caspase-3 Enzyme Activity Assay
2.13. Western Blotting
2.14. Spheroid Assay
2.15. Live-Dead Cell Viability of 3D Tumor Spheroids
2.16. Human Dermal Fibroblast Cytotoxicity Assay
2.17. Statistical Analysis
3. Results
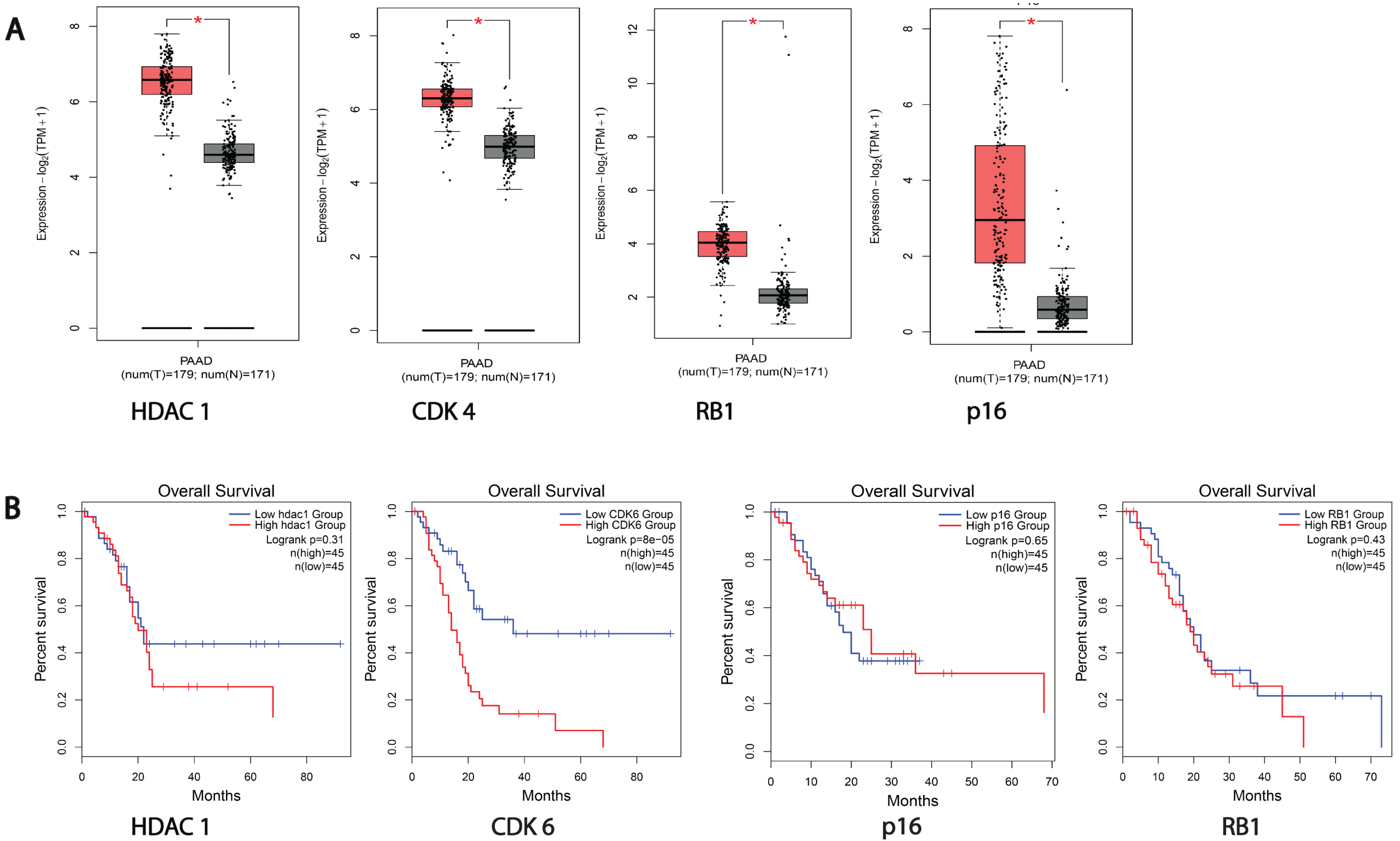
3.1. HDACs and Cell Cycle Proteins Are Overexpressed in Pancreatic Cancer Patients
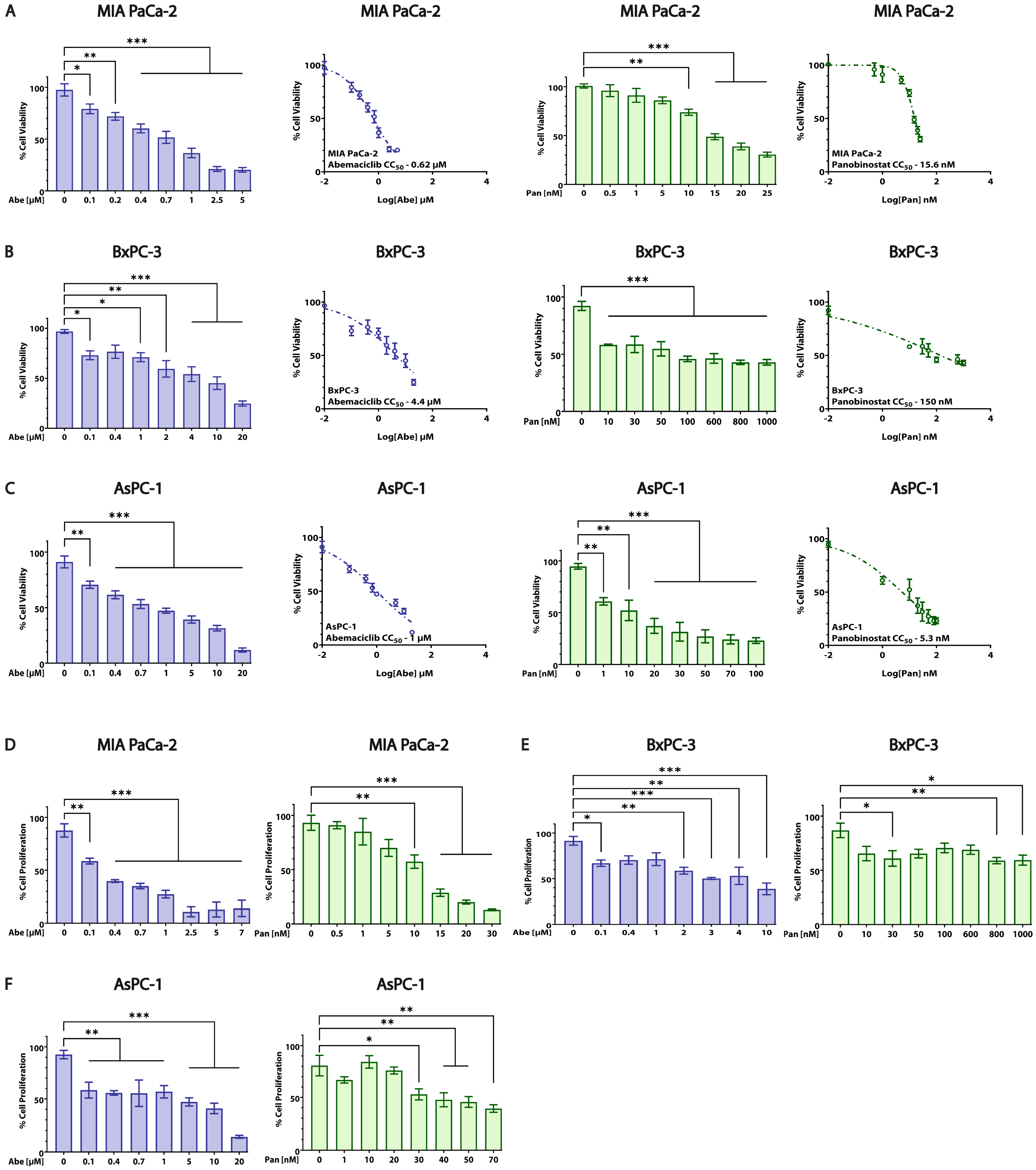
3.2. Abe and Pan Decrease Cell Viability and Affect the Proliferation of PDAC Cells
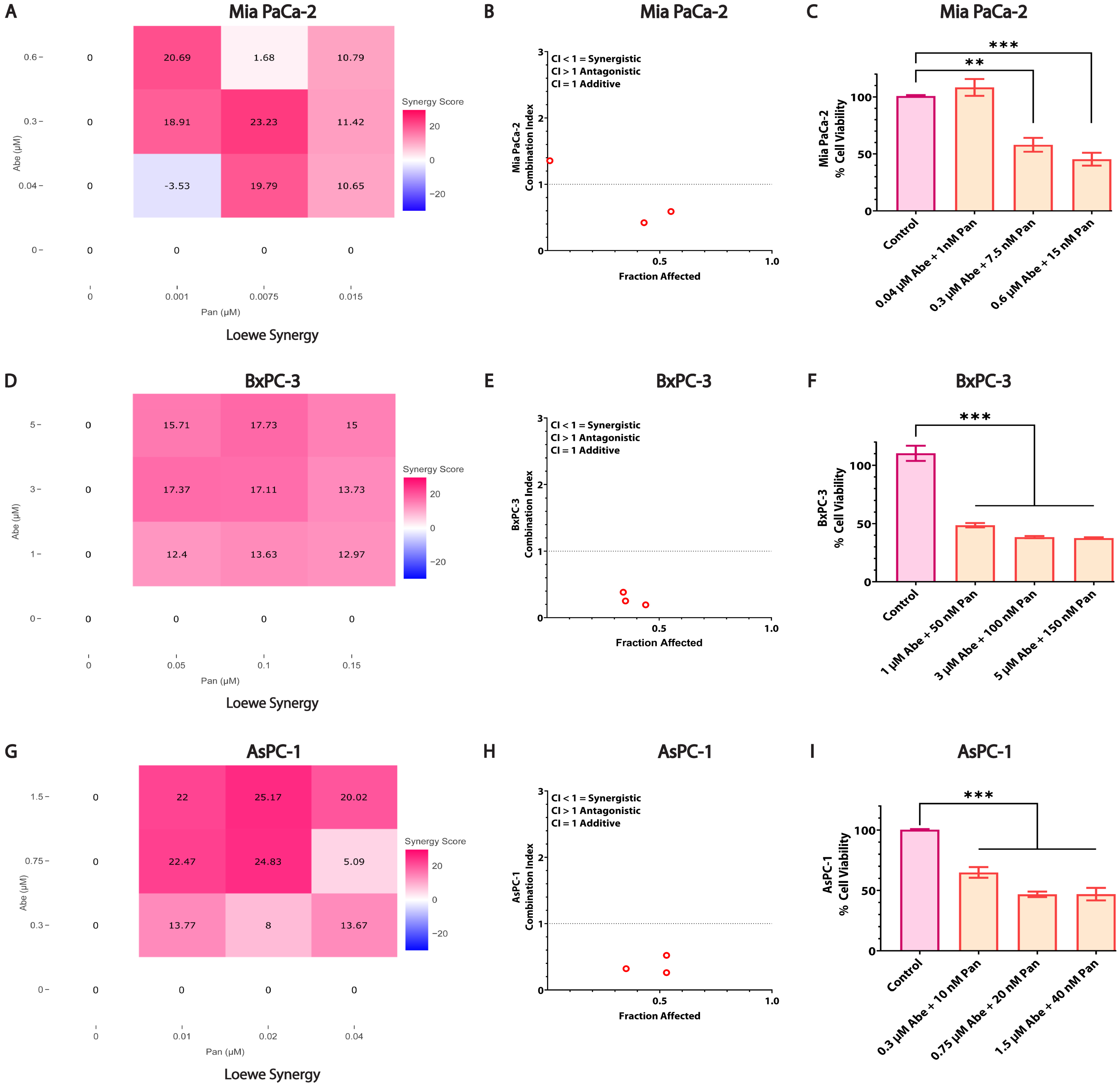
3.3. Combined Treatment of Abe and Pan Synergistically Affects the Viability of Pancreatic Cancer Cells
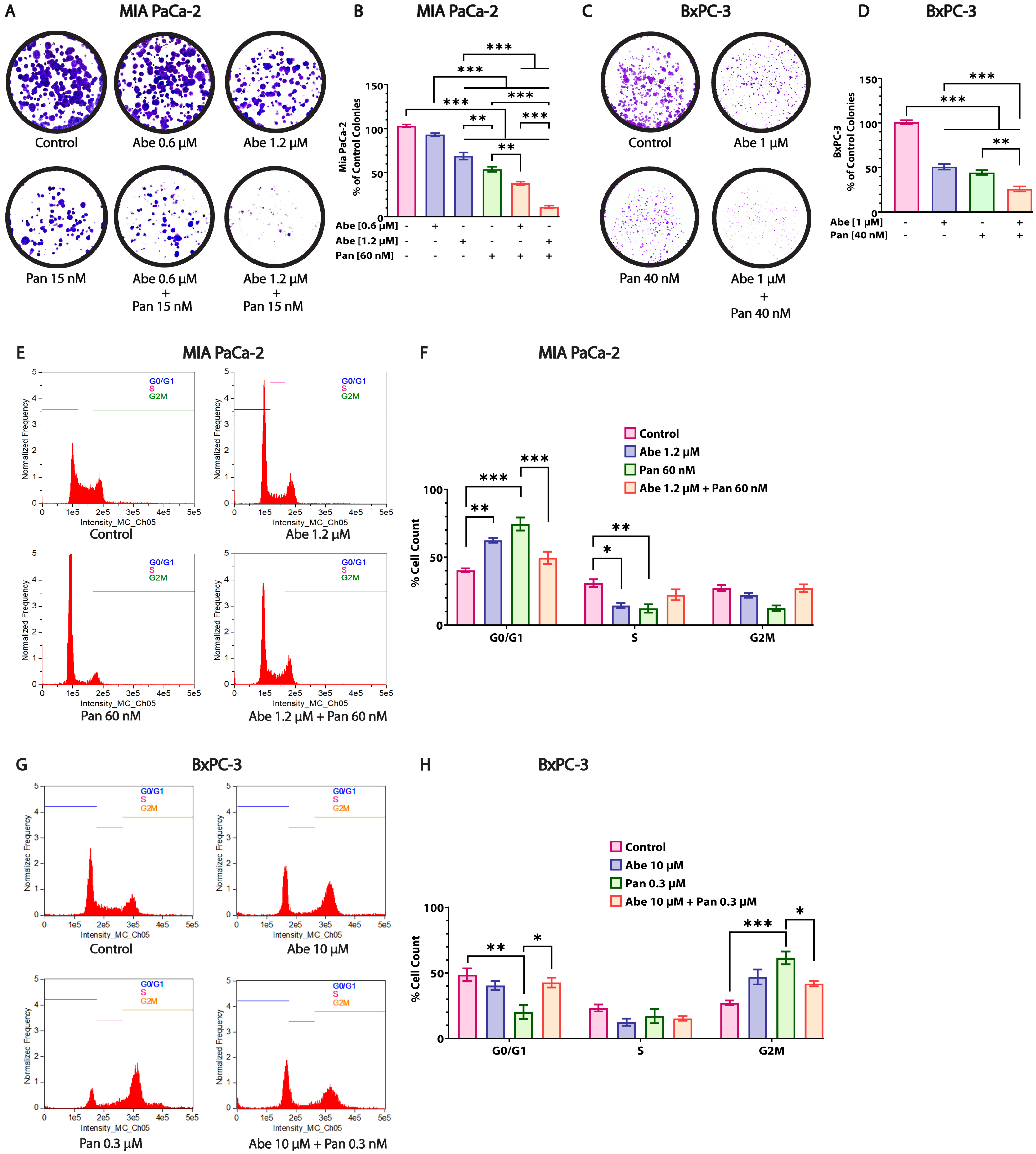
3.4. Abe and Pan Treatment Drastically Affects the Colony Formation Ability of Pancreatic Cancer Cells and Induces Cell Cycle Arrest
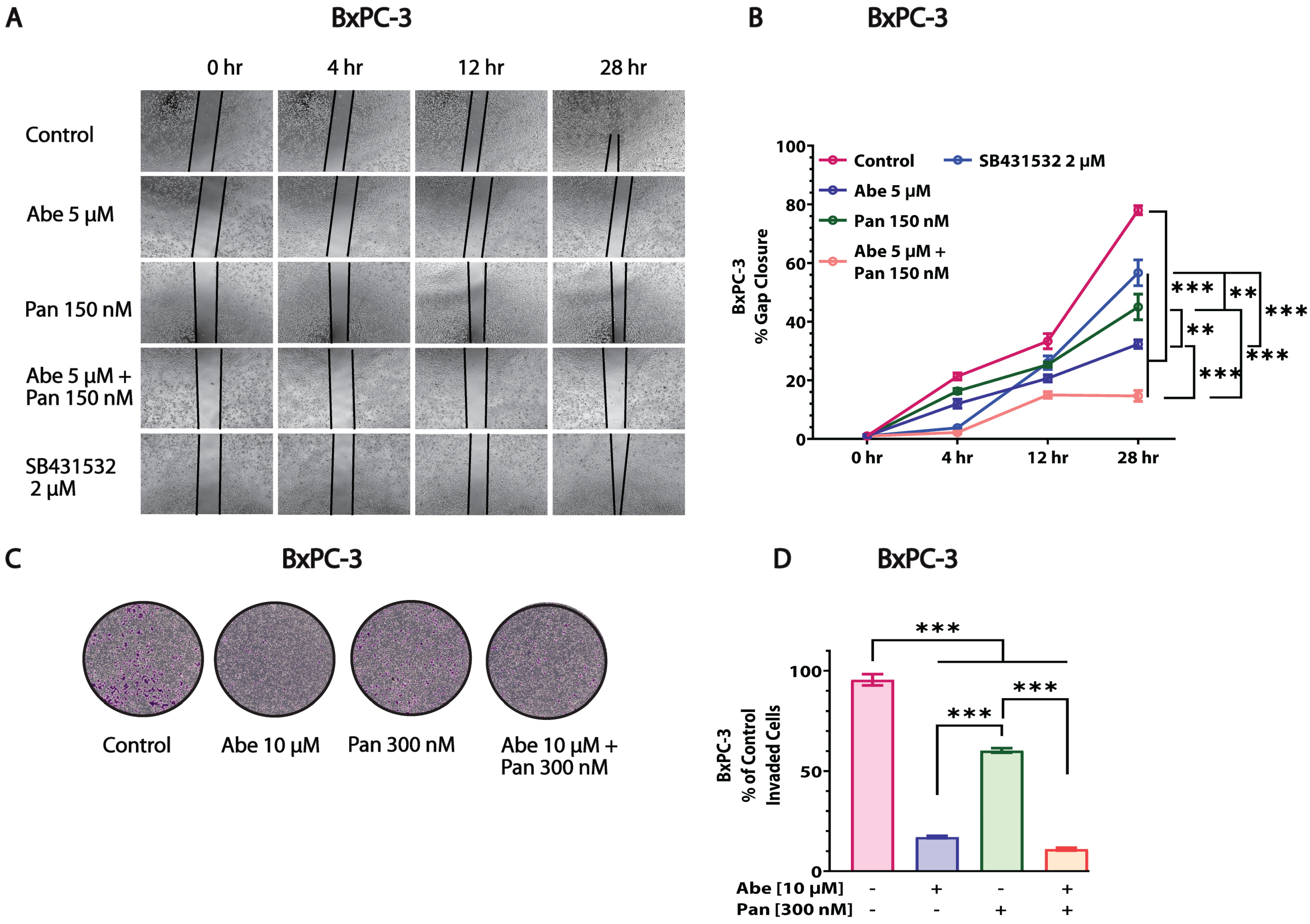
3.5. Abe and Pan Combination Treatment Inhibited the In Vitro Invasion and Migration of Pancreatic Cancer Cells
3.6. Abe and Pan Treatment Alters the Target Protein Levels in MIA PaCa-2 Cells
3.7. Pan and Abe Combination Treatment Leads to Robust Apoptosis in Pancreatic Cancer Cells
3.8. Abe and Pan Treatment Severely Reduces the 3D Spheroid Growth of PDAC Cells and Moderately Affects Human Dermal Fibroblast (HDF) Cell Viability
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic Adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, A.; Andersson, R.; Ansari, D. The Actual 5-Year Survivors of Pancreatic Ductal Adenocarcinoma Based on Real-World Data. Sci. Rep. 2020, 10, 16425. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Underwood, P.W.; Korc, M.; Trevino, J.G.; Munshi, H.G.; Rana, A. The Current Treatment Paradigm for Pancreatic Ductal Adenocarcinoma and Barriers to Therapeutic Efficacy. Front. Oncol. 2021, 11, 688377. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with Nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Ito, A.; Kawaguchi, Y.; Lai, C.-H.; Kovacs, J.J.; Higashimoto, Y.; Appella, E.; Yao, T.-P. MDM2-HDAC1-Mediated Deacetylation of P53 Is Required for Its Degradation. EMBO J. 2002, 21, 6236–6245. [Google Scholar] [CrossRef]
- Wilson, A.J.; Byun, D.-S.; Nasser, S.; Murray, L.B.; Ayyanar, K.; Arango, D.; Figueroa, M.; Melnick, A.; Kao, G.D.; Augenlicht, L.H.; et al. HDAC4 Promotes Growth of Colon Cancer Cells via Repression of P21. Mol. Biol. Cell 2008, 19, 4062–4075. [Google Scholar] [CrossRef]
- Rodríguez, S.; Bermúdez, L.G.; González, D.; Bernal, C.; Cañas, A.; Morales-Ruíz, T.; Henríquez, B.; Rojas, A. Transcriptional Regulation of CDKN2A/P16 by Sirtuin 7 in Senescence. Mol. Med. Rep. 2022, 26, 345. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Niu, X.; Zhang, W.; Caldwell, J.T.; Edwards, H.; Chen, W.; Taub, J.W.; Zhao, L.; Ge, Y. Synergistic Antitumor Interactions between MK-1775 and Panobinostat in Preclinical Models of Pancreatic Cancer. Cancer Lett. 2015, 356, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Mehdi, O.; Françoise, S.; Sofia, C.L.; Urs, G.; Kevin, Z.; Bernard, S.; Igor, S.; Anabela, C.-S.; Dominique, L.; Eric, M.; et al. HDAC Gene Expression in Pancreatic Tumor Cell Lines Following Treatment with the HDAC Inhibitors Panobinostat (LBH589) and Trichostatine (TSA). Pancreatology 2012, 12, 146–155. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves Panobinostat for Some Patients with Multiple Myeloma—NCI. Available online: https://www.cancer.gov/news-events/cancer-currents-blog/2015/fda-approves-panobinostat (accessed on 6 May 2023).
- Inc, S.B. Secura Bio Announces U.S. Withdrawal of FARYDAK® (Panobinostat) NDA. Available online: https://www.prnewswire.com/news-releases/secura-bio-announces-us-withdrawal-of-farydak--panobinostat-nda-301434428.html (accessed on 6 May 2023).
- Jones, S.F.; Bendell, J.C.; Infante, J.R.; Spigel, D.R.; Thompson, D.S.; Yardley, D.A.; Greco, F.A.; Murphy, P.B.; Burris, H.A. A Phase I Study of Panobinostat in Combination with Gemcitabine in the Treatment of Solid Tumors. Clin. Adv. Hematol. Oncol. HO 2011, 9, 225–230. [Google Scholar]
- Tarhini, A.A.; Zahoor, H.; Mclaughlin, B.; Gooding, W.E.; Schmitz, J.C.; Siegfried, J.M.; Socinski, M.A.; Argiris, A. Phase I Trial of Carboplatin and Etoposide in Combination with Panobinostat in Patients with Lung Cancer. Anti-Cancer Res. 2013, 33, 4475–4481. [Google Scholar]
- Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A.; Cicenas, J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers 2017, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Lemire, M.; Zhang, A.; Chan-Seng-Yue, M.; Wilson, G.; Grant, R.C.; Merico, D.; Lungu, I.; et al. Integration of Genomic and Transcriptional Features in Pancreatic Cancer Reveals Increased Cell Cycle Progression in Metastases. Cancer Cell 2019, 35, 267–282.e7. [Google Scholar] [CrossRef] [PubMed]
- Dhir, T.; Schultz, C.W.; Jain, A.; Brown, S.Z.; Haber, A.; Goetz, A.; Xi, C.; Su, G.H.; Xu, L.; Posey, J.; et al. Abemaciclib Is Effective against Pancreatic Cancer Cells and Synergizes with HuR and YAP1 Inhibition. Mol. Cancer Res. MCR 2019, 17, 2029–2041. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, S.; Sun, N.; Chen, J. Differential Expression of STAT1 and P21 Proteins Predicts Pancreatic Cancer Progression and Prognosis. Pancreas 2014, 43, 619–623. [Google Scholar] [CrossRef]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone Deacetylase Inhibitor Selectively Induces p21WAF1 Expression and Gene-Associated Histone Acetylation. Proc. Natl. Acad. Sci. USA 2000, 97, 10014–10019. [Google Scholar] [CrossRef]
- Venkannagari, S.; Fiskus, W.; Peth, K.; Atadja, P.; Hidalgo, M.; Maitra, A.; Bhalla, K.N. Superior Efficacy of Co-Treatment with Dual PI3K/mTOR Inhibitor NVP-BEZ235 and Pan-Histone Deacetylase Inhibitor against Human Pancreatic Cancer. Oncotarget 2012, 3, 1416–1427. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Choy, M.L.; Marks, P.A. Chapter Two—Mechanisms of Resistance to Histone Deacetylase Inhibitors. In Advances in Cancer Research; Grant, S., Ed.; Histone Deacetylase Inhibitors as Cancer Therapeutics; Academic Press: Cambridge, MA, USA, 2012; Volume 116, pp. 39–86. [Google Scholar]
- Lee, J.; Lim, B.; Pearson, T.; Tripathy, D.; Ordentlich, P.; Ueno, N. Abstract P5-21-15: The Synergistic Antitumor Activity of Entinostat (MS-275) in Combination with Palbociclib (PD 0332991) in Estrogen Receptor-Positive and Triple-Negative Breast Cancer. Cancer Res. 2018, 78 (Suppl. S4), P5-21-15. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A Web Server for Cancer and Normal Gene Expression Profiling and Interactive Analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 3.0: An Interactive Analysis and Consensus Interpretation of Multi-Drug Synergies across Multiple Samples. Nucleic Acids Res. 2022, 50, W739–W743. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C.; Talalay, P. Quantitative Analysis of Dose-Effect Relationships: The Combined Effects of Multiple Drugs or Enzyme Inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Alnakhli, A.; Vemana, H.P.; Bhutkar, S.; Muth, A.; Dukhande, V.V. Repurposing an Antiepileptic Drug for the Treatment of Glioblastoma. Pharm. Res. 2022, 39, 2871–2883. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Chen, Y.; Lin, Z.; Chen, C.; Dong, Y. Overexpressing PRMT1 Inhibits Proliferation and Invasion in Pancreatic Cancer by Inverse Correlation of ZEB1. IUBMB Life 2018, 70, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Barot, S.; Stephenson, O.J.; Priya Vemana, H.; Yadav, A.; Bhutkar, S.; Trombetta, L.D.; Dukhande, V.V. Metabolic Alterations and Mitochondrial Dysfunction Underlie Hepatocellular Carcinoma Cell Death Induced by a Glycogen Metabolic Inhibitor. Biochem. Pharmacol. 2022, 203, 115201. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Yamato, T.; Furukawa, T.; Ohnishi, Y.; Kijima, H.; Horii, A. Characterization of the Mutations of the K-Ras, P53, P16, and SMAD4 Genes in 15 Human Pancreatic Cancer Cell Lines. Oncol. Rep. 2001, 8, 89–92. [Google Scholar] [CrossRef]
- Chien, W.; Lee, D.H.; Zheng, Y.; Wuensche, P.; Alvarez, R.; Wen, D.L.; Aribi, A.M.; Thean, S.M.; Doan, N.B.; Said, J.W.; et al. Growth Inhibition of Pancreatic Cancer Cells by Histone Deacetylase Inhibitor Belinostat Through Suppression of Multiple Pathways, Including HIF, NFkB, and mTOR Signaling In Vitro and In Vivo. Mol. Carcinog. 2014, 53, 722–735. [Google Scholar] [CrossRef]
- Hu, H.; Ye, Z.; Qin, Y.; Xu, X.; Yu, X.; Zhuo, Q.; Ji, S. Mutations in Key Driver Genes of Pancreatic Cancer: Molecularly Targeted Therapies and Other Clinical Implications. Acta Pharmacol. Sin. 2021, 42, 1725–1741. [Google Scholar] [CrossRef] [PubMed]
- Mancarella, D.; Plass, C. Epigenetic Signatures in Cancer: Proper Controls, Current Challenges and the Potential for Clinical Translation. Genome Med. 2021, 13, 23. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-C.; Baylin, S.B. Cancer Epigenetics: Linking Basic Biology to Clinical Medicine. Cell Res. 2011, 21, 502–517. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.; Kaur, P.; Singh, P.; Singh, S.; Munshi, A. Differential Molecular Mechanistic Behavior of HDACs in Cancer Progression. Med. Oncol. 2022, 39, 171. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.-H.; Xu, X.-G.; Yan, S.-L.; Sun, Z.; Ying, Y.; Wang, B.-K.; Tu, Y.-X. Depletion of HDAC1, 7 and 8 by Histone Deacetylase Inhibition Confers Elimination of Pancreatic Cancer Stem Cells in Combination with Gemcitabine. Sci. Rep. 2018, 8, 1621. [Google Scholar] [CrossRef] [PubMed]
- Rana, Z.; Diermeier, S.; Hanif, M.; Rosengren, R.J. Understanding Failure and Improving Treatment Using HDAC Inhibitors for Prostate Cancer. Biomedicines 2020, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Miyake, K.; Yoshizumi, T.; Imura, S.; Sugimoto, K.; Batmunkh, E.; Kanemura, H.; Morine, Y.; Shimada, M. Expression of Hypoxia-Inducible Factor-1alpha, Histone Deacetylase 1, and Metastasis-Associated Protein 1 in Pancreatic Carcinoma: Correlation with Poor Prognosis with Possible Regulation. Pancreas 2008, 36, e1–e9. [Google Scholar] [CrossRef] [PubMed]
- Salvador-Barbero, B.; Álvarez-Fernández, M.; Zapatero-Solana, E.; El Bakkali, A.; Menéndez, M.D.C.; López-Casas, P.P.; Di Domenico, T.; Xie, T.; VanArsdale, T.; Shields, D.J.; et al. CDK4/6 Inhibitors Impair Recovery from Cytotoxic Chemotherapy in Pancreatic Adenocarcinoma. Cancer Cell 2020, 37, 340–353.e6. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.; Balaji, U.; Freinkman, E.; Witkiewicz, A.K.; Knudsen, E.S. Metabolic Re-Programming of Pancreatic Cancer Mediated by CDK4/6 Inhibition Elicits Unique Vulnerabilities. Cell Rep. 2016, 14, 979–990. [Google Scholar] [CrossRef]
- Li, Z.; Zou, W.; Zhang, J.; Zhang, Y.; Xu, Q.; Li, S.; Chen, C. Mechanisms of CDK4/6 Inhibitor Resistance in Luminal Breast Cancer. Front. Pharmacol. 2020, 11, 580251. [Google Scholar] [CrossRef]
- Fantin, V.R.; Richon, V.M. Mechanisms of Resistance to Histone Deacetylase Inhibitors and Their Therapeutic Implications. Clin. Cancer Res. 2007, 13, 7237–7242. [Google Scholar] [CrossRef]
- Burgess, A.J.; Pavey, S.; Warrener, R.; Hunter, L.-J.K.; Piva, T.J.; Musgrove, E.A.; Saunders, N.; Parsons, P.G.; Gabrielli, B.G. Up-Regulation of P21WAF1/CIP1 by Histone Deacetylase Inhibitors Reduces Their Cytotoxicity. Mol. Pharmacol. 2001, 60, 828. [Google Scholar]
- Derjuga, A.; Richard, C.; Crosato, M.; Wright, P.S.; Chalifour, L.; Valdez, J.; Barraso, A.; Crissman, H.A.; Nishioka, W.; Bradbury, E.M.; et al. Expression of p21Waf1/Cip1 and Cyclin D1 Is Increased in Butyrate-Resistant HeLa Cells. J. Biol. Chem. 2001, 276, 37815–37820. [Google Scholar] [CrossRef]
- Quiñonero, F.; Mesas, C.; Doello, K.; Cabeza, L.; Perazzoli, G.; Jimenez-Luna, C.; Rama, A.R.; Melguizo, C.; Prados, J. The Challenge of Drug Resistance in Pancreatic Ductal Adenocarcinoma: A Current Overview. Cancer Biol. Med. 2019, 16, 688–699. [Google Scholar] [CrossRef]
- Biermann, M.; Quintero, C.; Ferguson, P.; Rajbhandari, N.; Park, D.E.; Patel, H.; Reya, T. Repurposing HDAC and mTOR Inhibitors for Pancreatic Cancer. J. Clin. Oncol. 2022, 40, e16234. [Google Scholar] [CrossRef]
- Goodwin, C.M.; Waters, A.M.; Klomp, J.E.; Javaid, S.; Bryant, K.L.; Stalnecker, C.A.; Drizyte-Miller, K.; Papke, B.; Yang, R.; Amparo, A.M.; et al. Combination Therapies with CDK4/6 Inhibitors to Treat KRAS-Mutant Pancreatic Cancer. Cancer Res. 2023, 83, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Zhang, B.; Cai, D.; Zou, X. Therapeutic Potential of Histone Deacetylase Inhibitors in Pancreatic Cancer. Cancer Lett. 2014, 347, 183–190. [Google Scholar] [CrossRef]
- Mathew, O.P.; Ranganna, K.; Yatsu, F.M. Butyrate, an HDAC Inhibitor, Stimulates Interplay between Different Post-translationaltranslational Modifications of Histone H3 and Differently Alters G1-Specific Cell Cycle Proteins in Vascular Smooth Muscle Cells. Biomed. Pharmacother. 2010, 64, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, C.S.; Poulaki, V.; McMullan, C.; Negri, J.; Fanourakis, G.; Goudopoulou, A.; Richon, V.M.; Marks, P.A.; Mitsiades, N. Novel Histone Deacetylase Inhibitors in the Treatment of Thyroid Cancer. Clin. Cancer Res. 2005, 11, 3958–3965. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, L.; Zhang, B.; Feng, J.; Wang, Y.; Zhang, B.; Jin, H.; Ding, L.; Wang, N.; He, S. Discovery of Novel Polysubstituted N-Alkyl Acridone Analogues as Histone Deacetylase Isoform-Selective Inhibitors for Cancer Therapy. J. Enzyme Inhib. Med. Chem. 2023, 38, 2206581. [Google Scholar] [CrossRef]
- Zhou, W.; Feng, X.; Han, H.; Guo, S.; Wang, G. Synergistic Effects of Combined Treatment with Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid and TRAIL on Human Breast Cancer Cells. Sci. Rep. 2016, 6, 28004. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.G.; Pugliese, M.; Gargantini, E.; Grange, C.; Bussolati, B.; Asioli, S.; Bosco, O.; Poli, R.; Compagnone, A.; Bandino, A.; et al. Cytotoxic Activity of the Histone Deacetylase Inhibitor Panobinostat (LBH589) in Anaplastic Thyroid Cancer in Vitro and in Vivo. Int. J. Cancer 2012, 130, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Valentini, A.; Gravina, P.; Federici, G.; Bernardini, S. Valproic Acid Induces Apoptosis, p16INK4A Upregulation and Sensitization to Chemotherapy in Human Melanoma Cells. Cancer Biol. Ther. 2007, 6, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Milton, S.G.; Mathew, O.P.; Yatsu, F.M.; Ranganna, K. Differential Cellular and Molecular Effects of Butyrate and Trichostatin A on Vascular Smooth Muscle Cells. Pharmaceuticals 2012, 5, 925–943. [Google Scholar] [CrossRef] [PubMed]
- Mathew, O.P.; Ranganna, K.; Mathew, J.; Zhu, M.; Yousefipour, Z.; Selvam, C.; Milton, S.G. Cellular Effects of Butyrate on Vascular Smooth Muscle Cells Are Mediated through Disparate Actions on Dual Targets, Histone Deacetylase (HDAC) Activity and PI3K/Akt Signaling Network. Int. J. Mol. Sci. 2019, 20, 2902. [Google Scholar] [CrossRef] [PubMed]
- Patra, N.; De, U.; Kim, T.H.; Lee, Y.J.; Ahn, M.Y.; Kim, N.D.; Yoon, J.H.; Choi, W.S.; Moon, H.R.; Lee, B.M.; et al. A Novel Histone Deacetylase (HDAC) Inhibitor MHY219 Induces Apoptosis via up-Regulation of Androgen Receptor Expression in Human Prostate Cancer Cells. Biomed. Pharmacother. 2013, 67, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Kerl, K.; Ries, D.; Unland, R.; Borchert, C.; Moreno, N.; Hasselblatt, M.; Jürgens, H.; Kool, M.; Görlich, D.; Eveslage, M.; et al. The Histone Deacetylase Inhibitor SAHA Acts in Synergism with Fenretinide and Doxorubicin to Control the Growth of Rhabdoid Tumor Cells. BMC Cancer 2013, 13, 286. [Google Scholar] [CrossRef] [PubMed]
- Juengel, E.; Dauselt, A.; Makarević, J.; Wiesner, C.; Tsaur, I.; Bartsch, G.; Haferkamp, A.; Blaheta, R.A. Acetylation of Histone H3 Prevents Resistance Development Caused by Chronic mTOR Inhibition in Renal Cell Carcinoma Cells. Cancer Lett. 2012, 324, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Shirsath, N.; Rathos, M.; Chaudhari, U.; Sivaramakrishnan, H.; Joshi, K. Potentiation of anticancer effect of valproic acid, an antiepileptic agent with histone deacetylase inhibitory activity, by the cyclin-dependent kinase inhibitor P276-00 in human non-small-cell lung cancer cell lines. Lung Cancer Amst. Neth. 2013, 82, 214–221. [Google Scholar] [CrossRef]
- Herrera-Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia-Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J.; et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 2016, 76, 2301–2313. [Google Scholar] [CrossRef]
- Teh, J.L.F.; Aplin, A.E. Arrested Developments: CDK4/6 Inhibitor Resistance and Alterations in the Tumor Immune Microenvironment. Clin. Cancer Res. 2019, 25, 921–927. [Google Scholar] [CrossRef]
- Cai, Z.; Wang, J.; Li, Y.; Shi, Q.; Jin, L.; Li, S.; Zhu, M.; Wang, Q.; Wong, L.L.; Yang, W.; et al. Overexpressed Cyclin D1 and CDK4 Proteins Are Responsible for the Resistance to CDK4/6 Inhibitor in Breast Cancer That Can Be Reversed by PI3K/mTOR Inhibitors. Sci. China Life Sci. 2023, 66, 94–109. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Kumarasamy, V.; Ruiz, A.; Sivinski, J.; Chung, S.; Grant, A.; Vail, P.; Chauhan, S.S.; Jie, T.; Riall, T.S.; et al. Cell Cycle Plasticity Driven by MTOR Signaling: Integral Resistance to CDK4/6 Inhibition in Patient-Derived Models of Pancreatic Cancer. Oncogene 2019, 38, 3355–3370. [Google Scholar] [CrossRef] [PubMed]
- Wiedemeyer, W.R.; Dunn, I.F.; Quayle, S.N.; Zhang, J.; Chheda, M.G.; Dunn, G.P.; Zhuang, L.; Rosenbluh, J.; Chen, S.; Xiao, Y.; et al. Pattern of Retinoblastoma Pathway Inactivation Dictates Response to CDK4/6 Inhibition in GBM. Proc. Natl. Acad. Sci. USA 2010, 107, 11501–11506. [Google Scholar] [CrossRef]
- Lee, H.-A.; Chu, K.-B.; Moon, E.-K.; Quan, F.-S. Histone Deacetylase Inhibitor-Induced CDKN2B and CDKN2D Contribute to G2/M Cell Cycle Arrest Incurred by Oxidative Stress in Hepatocellular Carcinoma Cells via Forkhead Box M1 Suppression. J. Cancer 2021, 12, 5086–5098. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Wang, D.; Wei, X.; Liu, C.; Xiao, Z.; Qian, W.; Song, Y.; Hou, X. MS275 as Class I HDAC Inhibitor Displayed Therapeutic Potential on Malignant Ascites by iTRAQ-Based Quantitative Proteomic Analysis. BMC Gastroenterol. 2022, 22, 29. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, S.; Ku, S.; Ciamporcero, E.; Miles, K.M.; Attwood, K.; Chintala, S.; Shen, L.; Ellis, L.; Sotomayor, P.; Swetzig, W.; et al. HDAC 1 and 6 Modulate Cell Invasion and Migration in Clear Cell Renal Cell Carcinoma. BMC Cancer 2016, 16, 617. [Google Scholar] [CrossRef]
- Rhodes, L.V.; Tate, C.R.; Segar, H.C.; Burks, H.E.; Phamduy, T.B.; Hoang, V.; Elliott, S.; Gilliam, D.; Pounder, F.N.; Anbalagan, M.; et al. Suppression of Triple-Negative Breast Cancer Metastasis by Pan-DAC Inhibitor Panobinostat via Inhibition of ZEB Family of EMT Master Regulators. Breast Cancer Res. Treat. 2014, 145, 593–604. [Google Scholar] [CrossRef]
- Song, X.; Wang, J.; Zheng, T.; Song, R.; Liang, Y.; Bhatta, N.; Yin, D.; Pan, S.; Liu, J.; Jiang, H.; et al. LBH589 Inhibits Proliferation and Metastasis of Hepatocellular Carcinoma via Inhibition of Gankyrin/Stat3/Akt Pathway. Mol. Cancer 2013, 12, 114. [Google Scholar] [CrossRef]
- Scheidemann, E.R.; Demas, D.M.; Hou, C.; Ma, J.; He, W.; Sharma, G.; Schultz, E.; Weilbaecher, K.N.; Shajahan-Haq, A.N. Resistance to Abemaciclib Is Associated with Increased Metastatic Potential and Lysosomal Protein Deregulation in Breast Cancer Cells. Mol. Carcinog. 2024, 63, 209–223. [Google Scholar] [CrossRef]
- Liu, F.; Korc, M. Cdk4/6 Inhibition Induces Epithelial-Mesenchymal Transition and Enhances Invasiveness in Pancreatic Cancer Cells. Mol. Cancer Ther. 2012, 11, 2138–2148. [Google Scholar] [CrossRef] [PubMed]
- Abutorabi, E.S.; Poursheikhani, A.; Kashani, B.; Shamsaiegahkani, S.; Haghpanah, V.; Bashash, D.; Mousavi, S.A.; Momeny, M.; Ghaffari, S.H. The Effects of Abemaciclib on Cell Cycle and Apoptosis Regulation in Anaplastic Thyroid Cancer Cells. Mol. Biol. Rep. 2023, 50, 4073–4082. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, M.S. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Gilardini Montani, M.S.; Granato, M.; Santoni, C.; Del Porto, P.; Merendino, N.; D’Orazi, G.; Faggioni, A.; Cirone, M. Histone Deacetylase Inhibitors VPA and TSA Induce Apoptosis and Autophagy in Pancreatic Cancer Cells. Cell. Oncol. 2017, 40, 167–180. [Google Scholar] [CrossRef]
- Ala, M. Target C-Myc to Treat Pancreatic Cancer. Cancer Biol. Ther. 2022, 23, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, S.; Chen, X.; Zheng, X.; Yao, Y.; Lu, G.; Zhou, J. Palbociclib, a Selective CDK4/6 Inhibitor, Enhances the Effect of Selumetinib in RAS-Driven Non-Small Cell Lung Cancer. Cancer Lett. 2017, 408, 130–137. [Google Scholar] [CrossRef]
- Rencuzogulları, O.; Yerlikaya, P.O.; Gürkan, A.Ç.; Arısan, E.D.; Telci, D. Palbociclib, a Selective CDK4/6 Inhibitor, Restricts Cell Survival and Epithelial-Mesenchymal Transition in Panc-1 and MiaPaCa-2 Pancreatic Cancer Cells. J. Cell. Biochem. 2020, 121, 508–523. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Abemaciclib CC50 ± SEM (µM) | Panobinostat CC50 ± SEM (nM) |
|---|---|---|
| MIA PaCa-2 (KRAS-mut, p16-mut, p53-mut) | 0.62 ± 0.059 | 15.6 ± 0.785 |
| BxPC-3 (KRAS-wt, p16-mut, p53-mut) | 4.4 ± 2.168 | 150 ± 0.204 |
| AsPC-1 (KRAS-mut, p16-mut, p53-mut) | 1 ± 0.251 | 5.3 ± 0.783 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhutkar, S.; Yadav, A.; Patel, H.; Barot, S.; Patel, K.; Dukhande, V.V. Synergistic Efficacy of CDK4/6 Inhibitor Abemaciclib and HDAC Inhibitor Panobinostat in Pancreatic Cancer Cells. Cancers 2024, 16, 2713. https://doi.org/10.3390/cancers16152713
Bhutkar S, Yadav A, Patel H, Barot S, Patel K, Dukhande VV. Synergistic Efficacy of CDK4/6 Inhibitor Abemaciclib and HDAC Inhibitor Panobinostat in Pancreatic Cancer Cells. Cancers. 2024; 16(15):2713. https://doi.org/10.3390/cancers16152713
Chicago/Turabian StyleBhutkar, Shraddha, Anjali Yadav, Himaxi Patel, Shrikant Barot, Ketan Patel, and Vikas V. Dukhande. 2024. "Synergistic Efficacy of CDK4/6 Inhibitor Abemaciclib and HDAC Inhibitor Panobinostat in Pancreatic Cancer Cells" Cancers 16, no. 15: 2713. https://doi.org/10.3390/cancers16152713