
Optimizing ctDNA: An Updated Review of a Promising Clinical Tool for the Management of Uveal Melanoma
 , ,
, ,
Abstract
Simple Summary
Abstract

1. Introduction
2. Sources of Liquid Biopsies
2.1. Blood-Based Liquid Biopsy in Uveal Melanoma
2.2. Vitreous Liquid Biopsy in Uveal Melanoma
2.3. Aqueous Humor Liquid Biopsy in Uveal Melanoma
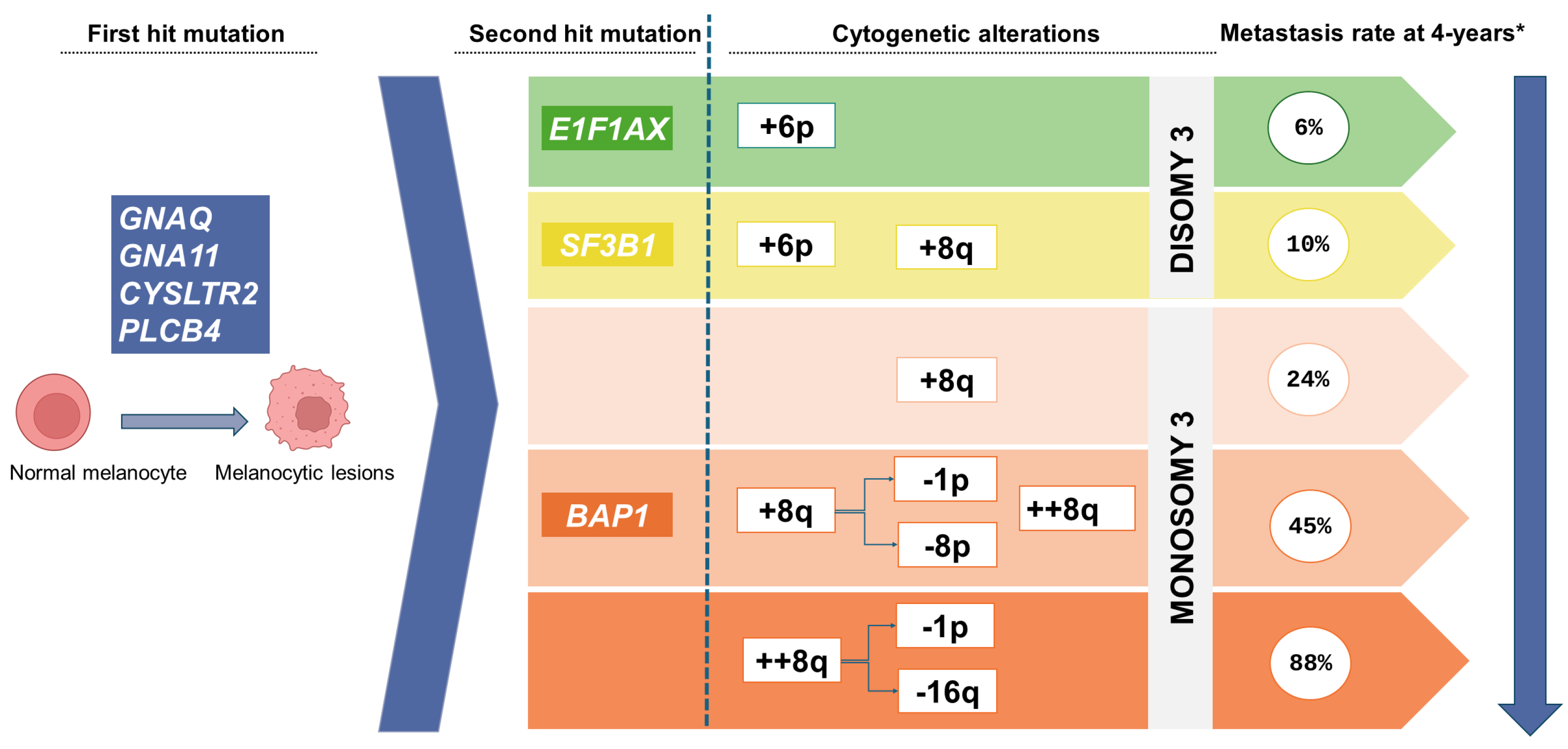
3. The Molecular Landscape of Uveal Melanoma
4. Isolation Techniques for ctDNA
5. Molecular Techniques for ctDNA Detection
5.1. PCR-Based Detection Techniques
5.1.1. Droplet Digital PCR (ddPCR) and Digital PCR (dPCR)
5.1.2. COLD-PCR
5.1.3. BEAMing
5.1.4. ARMS-PCR
5.1.5. CRISPR/Cas12a Technology
5.2. NGS-Based Detection Techniques
5.2.1. Whole-Genome Sequencing (WGS)
5.2.2. WES
5.2.3. Targeted Deep Sequencing
5.2.4. MAPS
5.2.5. CAPP-Seq
5.2.6. Tam-Seq
5.2.7. WGBS-Seq
6. Clinical Use of ctDNA in UM
7. Future Directions and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- De Mattos-Arruda, L.; Siravegna, G. How to Use Liquid Biopsies to Treat Patients with Cancer. ESMO Open 2021, 6, 100060. [Google Scholar] [CrossRef] [PubMed]
- Batool, S.M.; Yekula, A.; Khanna, P.; Hsia, T.; Gamblin, A.S.; Ekanayake, E.; Escobedo, A.K.; You, D.G.; Castro, C.M.; Im, H.; et al. The Liquid Biopsy Consortium: Challenges and Opportunities for Early Cancer Detection and Monitoring. Cell Rep. Med. 2023, 4, 101198. [Google Scholar] [CrossRef] [PubMed]
- Martel, A.; Baillif, S.; Nahon-Esteve, S.; Gastaud, L.; Bertolotto, C.; Roméo, B.; Mograbi, B.; Lassalle, S.; Hofman, P. Liquid Biopsy for Solid Ophthalmic Malignancies: An Updated Review and Perspectives. Cancers 2020, 12, 3284. [Google Scholar] [CrossRef] [PubMed]
- Schefler, A.C.; Gologorsky, D.; Marr, B.P.; Shields, C.L.; Zeolite, I.; Abramson, D.H. Extraocular Extension of Uveal Melanoma After Fine-Needle Aspiration, Vitrectomy, and Open Biopsy. JAMA Ophthalmol. 2013, 131, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Nikanjam, M.; Kato, S.; Kurzrock, R. Liquid Biopsy: Current Technology and Clinical Applications. J. Hematol. Oncol. 2022, 15, 131. [Google Scholar] [CrossRef]
- Gilson, P.; Merlin, J.L.; Harlé, A. Deciphering Tumour Heterogeneity: From Tissue to Liquid Biopsy. Cancers 2022, 14, 1384. [Google Scholar] [CrossRef]
- Imperial, R.; Nazer, M.; Ahmed, Z.; Kam, A.E.; Pluard, T.J.; Bahaj, W.; Levy, M.; Kuzel, T.M.; Hayden, D.M.; Pappas, S.G.; et al. Matched Whole-Genome Sequencing (WGS) and Whole-Exome Sequencing (WES) of Tumor Tissue with Circulating Tumor DNA (CtDNA) Analysis: Complementary Modalities in Clinical Practice. Cancers 2019, 11, 1399. [Google Scholar] [CrossRef]
- Vellanki, P.J.; Ghosh, S.; Pathak, A.; Fusco, M.J.; Bloomquist, E.W.; Tang, S.; Singh, H.; Philip, R.; Pazdur, R.; Beaver, J.A. Regulatory Implications of CtDNA in Immuno-Oncology for Solid Tumors. J. Immunother. Cancer 2023, 11, e005344. [Google Scholar] [CrossRef]
- Sato, Y. Clinical Utility of Liquid Biopsy-Based Companion Diagnostics in the Non-Small-Cell Lung Cancer Treatment. Explor. Target. Antitumor Ther. 2022, 3, 630–642. [Google Scholar] [CrossRef]
- Spagnolo, F.; Caltabiano, G.; Queirolo, P. Uveal Melanoma. Cancer Treat. Rev. 2012, 38, 549–553. [Google Scholar] [CrossRef]
- Wolf, J.; Franco, J.A.; Yip, R.; Dabaja, M.Z.; Velez, G.; Liu, F.; Bassuk, A.G.; Mruthyunjaya, P.; Dufour, A.; Mahajan, V.B. Liquid Biopsy Proteomics in Ophthalmology. J. Proteome Res. 2024, 23, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Beasley, A.B.; Chen, F.K.; Isaacs, T.W.; Gray, E.S. Future Perspectives of Uveal Melanoma Blood Based Biomarkers. Br. J. Cancer 2022, 126. [Google Scholar] [CrossRef]
- Kim, V.; Guberina, M.; Bechrakis, N.E.; Lohmann, D.R.; Zeschnigk, M.; Le Guin, C.H.D. Release of Cell-Free Tumor DNA in the Plasma of Uveal Melanoma Patients Under Radiotherapy. Investig. Ophthalmol. Vis. Sci. 2023, 64, 35. [Google Scholar] [CrossRef]
- De Bruyn, D.P.; van Poppelen, N.M.; Brands, T.; van den Boom, S.C.; Eikenboom, E.; Wagner, A.; van Veghel-Plandsoen, M.M.; Geeven, G.; Beverloo, B.; van Rij, C.M.; et al. Evaluation of Circulating Tumor DNA as a Liquid Biomarker in Uveal Melanoma. Investig. Ophthalmol. Vis. Sci. 2024, 65, 11. [Google Scholar] [CrossRef]
- Mishra, K.; Velez, G.; Chemudupati, T.; Tang, P.H.; Mruthyunjaya, P.; Sanislo, S.R.; Mahajan, V.B. Intraoperative Complications with Vitreous Biopsy for Molecular Proteomics. Ophthalmic Surg. Lasers Imaging Retin. 2023, 54, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Velez, G.; Nguyen, H.V.; Chemudupati, T.; Ludwig, C.A.; Toral, M.; Reddy, S.; Mruthyunjaya, P.; Mahajan, V.B. Liquid Biopsy Proteomics of Uveal Melanoma Reveals Biomarkers Associated with Metastatic Risk. Mol. Cancer 2021, 20, 39. [Google Scholar] [CrossRef]
- Wang, X.; Su, W.; Gao, Y.; Feng, Y.; Wang, X.; Chen, X.; Hu, Y.; Ma, Y.; Ou, Q.; Liang, D.; et al. A Pilot Study of the Use of Dynamic Analysis of Cell-Free DNA from Aqueous Humor and Vitreous Fluid for the Diagnosis and Treatment Monitoring of Vitreoretinal Lymphomas. Haematologica 2022, 107, 2154–2162. [Google Scholar] [CrossRef] [PubMed]
- Cani, A.K.; Hovelson, D.H.; Demirci, H.; Johnson, M.W.; Tomlins, S.A.; Rao, R.C. Next Generation Sequencing of Vitreoretinal Lymphomas from Small-Volume Intraocular Liquid Biopsies: New Routes to Targeted Therapies. Oncotarget 2017, 8, 7989–7998. [Google Scholar] [CrossRef]
- Gu, J.; Jiang, T.; Liu, S.; Ping, B.; Li, R.; Chen, W.; Wang, L.; Huang, X.; Xu, G.; Chang, Q. Cell-Free DNA Sequencing of Intraocular Fluid as Liquid Biopsy in the Diagnosis of Vitreoretinal Lymphoma. Front. Oncol. 2022, 12, 1. [Google Scholar] [CrossRef]
- Nell, R.J.; Versluis, M.; Menger, N.V.; Gelmi, M.C.; Vu, T.H.K.; Verdijk, R.M.; Luyten, G.P.M.; Jager, M.J.; van der Velden, P.A. Vitreous Fluid-Isolated DNA for the Genetic Analysis of Primary Uveal Melanoma: A Proof-of-Concept Study. medRxiv 2024. [Google Scholar] [CrossRef]
- Pike, S.B.; Reid, M.W.; Peng, C.C.; Chang, C.; Xu, B.Y.; Gombos, D.S.; Patel, S.; Xu, L.; Berry, J.L. Multicentre Analysis of Nucleic Acid Quantification Using Aqueous Humour Liquid Biopsy in Uveal Melanoma: Implications for Clinical Testing. Can. J. Ophthalmol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Im, D.H.; Peng, C.C.; Xu, L.; Kim, M.E.; Ostrow, D.; Yellapantula, V.; Bootwalla, M.; Biegel, J.A.; Gai, X.; Prabakar, R.K.; et al. Potential of Aqueous Humor as a Liquid Biopsy for Uveal Melanoma. Int. J. Mol. Sci. 2022, 23, 6226. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.C.; Sirivolu, S.; Pike, S.; Kim, M.E.; Reiser, B.; Li, H.T.; Liang, G.; Xu, L.; Berry, J.L. Diagnostic Aqueous Humor Proteome Predicts Metastatic Potential in Uveal Melanoma. Int. J. Mol. Sci. 2023, 24, 6825. [Google Scholar] [CrossRef] [PubMed]
- Wierenga, A.P.A.; Cao, J.; Mouthaan, H.; van Weeghel, C.; Verdijk, R.M.; van Duinen, S.G.; Kroes, W.G.M.; Dogrusöz, M.; Marinkovic, M.; van der Burg, S.S.H.; et al. Aqueous Humor Biomarkers Identify Three Prognostic Groups in Uveal Melanoma. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4740–4747. [Google Scholar] [CrossRef] [PubMed]
- Bechrakis, N.E.; Foerster, M.H.; Bornfeld, N. Biopsy in Indeterminate Intraocular Tumors. Ophthalmology 2002, 109, 235–242. [Google Scholar] [CrossRef]
- Frizziero, L.; Midena, E.; Trainiti, S.; Londei, D.; Bonaldi, L.; Bini, S.; Parrozzani, R. Uveal Melanoma Biopsy: A Review. Cancers 2019, 11, 1075. [Google Scholar] [CrossRef]
- Bakhoum, M.F.; Esmaeli, B. Molecular Characteristics of Uveal Melanoma: Insights from the Cancer Genome Atlas (TCGA) Project. Cancers 2019, 11, 1061. [Google Scholar] [CrossRef]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2017, 32, 204. [Google Scholar] [CrossRef]
- Rostami, A.; Lambie, M.; Yu, C.W.; Stambolic, V.; Waldron, J.N.; Bratman, S.V. Senescence, Necrosis, and Apoptosis Govern Circulating Cell-Free DNA Release Kinetics. Cell Rep. 2020, 31, 107830. [Google Scholar] [CrossRef]
- Bustamante, P.; Tsering, T.; Coblentz, J.; Mastromonaco, C.; Abdouh, M.; Fonseca, C.; Proença, R.P.; Blanchard, N.; Dugé, C.L.; Andujar, R.A.S.; et al. Circulating Tumor DNA Tracking through Driver Mutations as a Liquid Biopsy-Based Biomarker for Uveal Melanoma. J. Exp. Clin. Cancer Res. 2021, 40, 196. [Google Scholar] [CrossRef]
- Decatur, C.L.; Ong, E.; Garg, N.; Anbunathan, H.; Bowcock, A.M.; Field, M.G.; Harbour, J.W. Driver Mutations in Uveal Melanoma: Associations with Gene Expression Profile and Patient Outcomes. JAMA Ophthalmol. 2016, 134, 728–733. [Google Scholar] [CrossRef]
- Yavuzyigitoglu, S.; Koopmans, A.E.; Verdijk, R.M.; Vaarwater, J.; Eussen, B.; Van Bodegom, A.; Paridaens, D.; Kiliç, E.; De Klein, A. Uveal Melanomas with SF3B1 Mutations A Distinct Subclass Associated with Late-Onset Metastases. Ophthalmology 2016, 123, 1118–1128. [Google Scholar] [CrossRef]
- Martin, M.; Maßhöfer, L.; Temming, P.; Rahmann, S.; Metz, C.; Bornfeld, N.; Van De Nes, J.; Hitpass, L.K.; Hinnebusch, A.G.; Horsthemke, B.; et al. Exome Sequencing Identifies Recurrent Somatic Mutations in EIF1AX and SF3B1 in Uveal Melanoma with Disomy 3. Nat. Genet. 2013, 45, 933–936. [Google Scholar] [CrossRef]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.O.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent Mutation of BAP1 in Metastasizing Uveal Melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef] [PubMed]
- Ewens, K.G.; Lalonde, E.; Richards-Yutz, J.; Shields, C.L.; Ganguly, A. Comparison of Germline versus Somatic BAP1 Mutations for Risk of Metastasis in Uveal Melanoma. BMC Cancer 2018, 18, 1172. [Google Scholar] [CrossRef]
- Prescher, G.; Bornfeld, N.; Hirche, H.; Horsthemke, B.; Jöckel, K.H.; Becher, R. Prognostic Implications of Monosomy 3 in Uveal Melanoma. Lancet 1996, 347, 1222–1225. [Google Scholar] [CrossRef] [PubMed]
- Lalonde, E.; Ewens, K.; Richards-Yutz, J.; Ebrahimzedeh, J.; Terai, M.; Gonsalves, C.F.; Sato, T.; Shields, C.L.; Ganguly, A. Improved Uveal Melanoma Copy Number Subtypes Including an Ultra–High-Risk Group. Ophthalmol. Sci. 2022, 2, 100121. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Chen, H.; Long, Y.; Li, P.; Gu, Y. The Main Sources of Circulating Cell-Free DNA: Apoptosis, Necrosis and Active Secretion. Crit. Rev. Oncol. Hematol. 2021, 157, 103166. [Google Scholar] [CrossRef]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Marass, F.; Heider, K.; et al. Enhanced Detection of Circulating Tumor DNA by Fragment Size Analysis. Sci. Transl. Med. 2018, 10, eaat4921. [Google Scholar] [CrossRef] [PubMed]
- Greytak, S.R.; Engel, K.B.; Parpart-Li, S.; Murtaza, M.; Bronkhorst, A.J.; Pertile, M.D.; Moore, H.M. Harmonizing Cell-Free DNA Collection and Processing Practices through Evidence-Based Guidance. Clin. Cancer Res. 2020, 26, 3104–3109. [Google Scholar] [CrossRef]
- Danesi, R.; Lo, Y.M.D.; Oellerich, M.; Beck, J.; Galbiati, S.; Del Re, M.; Lianidou, E.; Neumaier, M.; van Schaik, R.H.N. What Do We Need to Obtain High Quality Circulating Tumor DNA (CtDNA) for Routine Diagnostic Test in Oncology?—Considerations on Pre-Analytical Aspects by the IFCC Workgroup CfDNA. Clin. Chim. Acta 2021, 520, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Lampignano, R.; Neumann, M.H.D.; Weber, S.; Kloten, V.; Herdean, A.; Voss, T.; Groelz, D.; Babayan, A.; Tibbesma, M.; Schlumpberger, M.; et al. Multicenter Evaluation of Circulating Cell-Free DNA Extraction and Downstream Analyses for the Development of Standardized (Pre)Analytical Work Flows. Clin. Chem. 2020, 66, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Barrios, C.; Nieto-Alcolado, I.; Torrente, M.; Jiménez-Sánchez, C.; Calvo, V.; Gutierrez-Sanz, L.; Palka, M.; Donoso-Navarro, E.; Provencio, M.; Romero, A. Comparison of Methods for Circulating Cell-Free DNA Isolation Using Blood from Cancer Patients: Impact on Biomarker Testing. Transl. Lung Cancer Res. 2016, 5, 665–672. [Google Scholar] [CrossRef]
- Polatoglou, E.; Mayer, Z.; Ungerer, V.; Bronkhorst, A.J.; Holdenrieder, S. Isolation and Quantification of Plasma Cell-Free DNA Using Different Manual and Automated Methods. Diagnostics 2022, 12, 2550. [Google Scholar] [CrossRef]
- Terp, S.K.; Pedersen, I.S.; Stoico, M.P. Extraction of Cell-Free DNA. Evaluation of Efficiency, Quantity, and Quality. J. Mol. Diagn. 2024, 26, 310–319. [Google Scholar] [CrossRef]
- Lehle, S.; Emons, J.; Hack, C.C.; Heindl, F.; Hein, A.; Preuß, C.; Seitz, K.; Zahn, A.L.; Beckmann, M.W.; Fasching, P.A.; et al. Evaluation of Automated Techniques for Extraction of Circulating Cell-Free DNA for Implementation in Standardized High-Throughput Workflows. Sci. Rep. 2023, 13, 373. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, R.J.; Lee, J.H.; Kefford, R.F.; Rizos, H. Evaluation of Commercial Kits for Purification of Circulating Free DNA. Cancer Genet. 2018, 228–229, 21–27. [Google Scholar] [CrossRef]
- Wang, M.; Huang, X.; Li, X.; Guo, Q.; Xu, W.; Zhao, M.; Wang, X.; Wang, L.; Lou, J. Performance Comparison of Commercial Kits for Isolating and Detecting Circulating Tumor DNA. Scand. J. Clin. Lab. Investig. 2021, 81, 276–281. [Google Scholar] [CrossRef]
- Van Der Leest, P.; Ketelaar, E.M.; Van Noesel, C.J.M.; Van Den Broek, D.; Van Boerdonk, R.A.A.; Deiman, B.; Rifaela, N.; Van Der Geize, R.; Huijsmans, C.J.J.; Speel, E.J.M.; et al. Dutch National Round Robin Trial on Plasma-Derived Circulating Cell-Free DNA Extraction Methods Routinely Used in Clinical Pathology for Molecular Tumor Profiling. Clin. Chem. 2022, 68, 963–972. [Google Scholar] [CrossRef]
- Mendes, M.S.M.; Rosa, M.E.; Ramalho, F.; Freire, M.G.; e Silva, F.A. Aqueous Two-Phase Systems as Multipurpose Tools to Improve Biomarker Analysis. Sep. Purif. Technol. 2023, 317, 123875. [Google Scholar] [CrossRef]
- Janku, F.; Huang, H.J.; Pereira, D.Y.; Kobayashi, M.; Chiu, C.H.; Call, S.G.; Woodbury, K.T.; Chao, F.; Marshak, D.R.; Chiu, R.Y.T. A Novel Method for Liquid-Phase Extraction of Cell-Free DNA for Detection of Circulating Tumor DNA. Sci. Rep. 2021, 11, 19653. [Google Scholar] [CrossRef] [PubMed]
- Van Der Leest, P.; Schuuring, E. Critical Factors in the Analytical Work Flow of Circulating Tumor DNA-Based Molecular Profiling. Clin. Chem. 2024, 70, 220–233. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, Y.; Fan, Y.; Liu, Y.; Yang, M. Recent Advances in Droplet-Based Microfluidics in Liquid Biopsy for Cancer Diagnosis. Droplet 2024, 3, e92. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Masunaga, N.; Kagara, N.; Abe, K.; Yoshinami, T.; Tsukabe, M.; Sota, Y.; Miyake, T.; Tanei, T.; Shimoda, M.; et al. Detection of Ultra-Rare ESR1 Mutations in Primary Breast Cancer Using LNA-Clamp DdPCR. Cancers 2023, 15, 2632. [Google Scholar] [CrossRef] [PubMed]
- Crucitta, S.; Ruglioni, M.; Novi, C.; Manganiello, M.; Arici, R.; Petrini, I.; Pardini, E.; Cucchiara, F.; Marmorino, F.; Cremolini, C.; et al. Comparison of Digital PCR Systems for the Analysis of Liquid Biopsy Samples of Patients Affected by Lung and Colorectal Cancer. Clin. Chim. Acta 2023, 541, 117239. [Google Scholar] [CrossRef]
- How-Kit, A.; Lebbé, C.; Bousard, A.; Daunay, A.; Mazaleyrat, N.; Daviaud, C.; Mourah, S.; Tost, J. Ultrasensitive Detection and Identification of BRAF V600 Mutations in Fresh Frozen, FFPE, and Plasma Samples of Melanoma Patients by E-Ice-COLD-PCR. Anal. Bioanal. Chem. 2014, 406, 5513–5520. [Google Scholar] [CrossRef]
- Freidin, M.B.; Freydina, D.V.; Leung, M.; Fernandez, A.M.; Nicholson, A.G.; Lim, E. Circulating Tumor DNA Outperforms Circulating Tumor Cells for KRAS Mutation Detection in Thoracic Malignancies. Clin. Chem. 2015, 61, 1299–1304. [Google Scholar] [CrossRef]
- Lanman, R.B.; Mortimer, S.A.; Zill, O.A.; Sebisanovic, D.; Lopez, R.; Blau, S.; Collisson, E.A.; Divers, S.G.; Hoon, D.S.B.; Scott Kopetz, E.; et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PLoS ONE 2015, 10, e0140712. [Google Scholar] [CrossRef] [PubMed]
- Khagi, Y.; Goodman, A.M.; Daniels, G.A.; Patel, S.P.; Sacco, A.G.; Randall, J.M.; Bazhenova, L.A.; Kurzrock, R. Hypermutated Circulating Tumor DNA: Correlation with Response to Checkpoint Inhibitor-Based Immunotherapy. Clin. Cancer Res. 2017, 23, 5729–5736. [Google Scholar] [CrossRef]
- Zhang, X.; Chang, N.; Yang, G.; Zhang, Y.; Ye, M.; Cao, J.; Xiong, J.; Han, Z.; Wu, S.; Shang, L.; et al. A Comparison of ARMS-Plus and Droplet Digital PCR for Detecting EGFR Activating Mutations in Plasma. Oncotarget 2017, 8, 112014–112023. [Google Scholar] [CrossRef]
- Escalona-Noguero, C.; Alarcón-Iniesta, H.; López-Valls, M.; del Carpio, L.P.; Piulats, J.M.; Somoza, Á.; Sot, B. Detection of the Uveal Melanoma-Associated Mutation GNAQ Q209P from Liquid Biopsy Using CRISPR/Cas12a Technology. Anal. Chem. 2023, 95, 16692–16700. [Google Scholar] [CrossRef] [PubMed]
- Rickles-Young, M.; Tinoco, G.; Tsuji, J.; Pollock, S.; Haynam, M.; Lefebvre, H.; Glover, K.; Owen, D.H.; Collier, K.A.; Ha, G.; et al. Assay Validation of Cell-Free DNA Shallow Whole-Genome Sequencing to Determine Tumor Fraction in Advanced Cancers. J. Mol. Diagn. 2024, 26, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Bos, M.K.; Angus, L.; Nasserinejad, K.; Jager, A.; Jansen, M.P.H.M.; Martens, J.W.M.; Sleijfer, S. Whole Exome Sequencing of Cell-Free DNA—A Systematic Review and Bayesian Individual Patient Data Meta-Analysis. Cancer Treat. Rev. 2020, 83, 101951. [Google Scholar] [CrossRef]
- Zhao, J.; Reuther, J.; Scozzaro, K.; Hawley, M.; Metzger, E.; Emery, M.; Chen, I.; Barbosa, M.; Johnson, L.; O’Connor, A.; et al. Personalized Cancer Monitoring Assay for the Detection of CtDNA in Patients with Solid Tumors. Mol. Diagn. Ther. 2023, 27, 753–768. [Google Scholar] [CrossRef]
- Garcia, J.; Kamps-Hughes, N.; Geiguer, F.; Couraud, S.; Sarver, B.; Payen, L.; Ionescu-Zanetti, C. Sensitivity, Specificity, and Accuracy of a Liquid Biopsy Approach Utilizing Molecular Amplification Pools. Sci. Rep. 2021, 11, 10761. [Google Scholar] [CrossRef]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An Ultrasensitive Method for Quantitating Circulating Tumor DNA with Broad Patient Coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Plagnol, V.; Woodhouse, S.; Howarth, K.; Lensing, S.; Smith, M.; Epstein, M.; Madi, M.; Smalley, S.; Leroy, C.; Hinton, J.; et al. Analytical Validation of a next Generation Sequencing Liquid Biopsy Assay for High Sensitivity Broad Molecular Profiling. PLoS ONE 2018, 13, e0193802. [Google Scholar] [CrossRef]
- Gale, D.; Lawson, A.R.J.; Howarth, K.; Madi, M.; Durham, B.; Smalley, S.; Calaway, J.; Blais, S.; Jones, G.; Clark, J.; et al. Development of a Highly Sensitive Liquid Biopsy Platform to Detect Clinically-Relevant Cancer Mutations at Low Allele Fractions in Cell-Free DNA. PLoS ONE 2018, 13, e0194630. [Google Scholar] [CrossRef]
- Taylor, S.C.; Laperriere, G.; Germain, H. Droplet Digital PCR versus QPCR for Gene Expression Analysis with Low Abundant Targets: From Variable Nonsense to Publication Quality Data. Sci. Rep. 2017, 7, 2409. [Google Scholar] [CrossRef]
- Zhang, B.; Xu, C.W.; Shao, Y.; Wang, H.T.; Wu, Y.F.; Song, Y.Y.; Li, X.B.; Zhang, Z.; Wang, W.J.; Li, L.Q.; et al. Comparison of Droplet Digital PCR and Conventional Quantitative PCR for Measuring EGFR Gene Mutation. Exp. Ther. Med. 2015, 9, 1383. [Google Scholar] [CrossRef]
- Li, H.; Jing, C.; Wu, J.; Ni, J.I.E.; Sha, H.; Xu, X.; Du, Y.; Lou, R.U.I.; Dong, S.; Feng, J. Circulating Tumor DNA Detection: A Potential Tool for Colorectal Cancer Management. Oncol. Lett. 2019, 17, 1409. [Google Scholar] [CrossRef] [PubMed]
- Beasley, A.B.; de Bruyn, D.P.; Calapre, L.; Al-Ogaili, Z.; Isaacs, T.W.; Bentel, J.; Reid, A.L.; Dwarkasing, R.S.; Pereira, M.R.; Khattak, M.A.; et al. Detection of Metastases Using Circulating Tumour DNA in Uveal Melanoma. J. Cancer Res. Clin. Oncol. 2023, 149, 14953–14963. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.; Jabbar, K.J. COLD-PCR: Applications and Advantages. Methods Mol. Biol. 2016, 1392, 17–25. [Google Scholar] [CrossRef]
- Rodríguez, J.; Avila, J.; Rolfo, C.; Ruíz-Patiño, A.; Russo, A.; Ricaurte, L.; Ordóñez-Reyes, C.; Arrieta, O.; Zatarain-Barrón, Z.L.; Recondo, G.; et al. When Tissue Is an Issue the Liquid Biopsy Is Nonissue: A Review. Oncol. Ther. 2021, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhao, C.; Zhao, J.; Wang, Q.; Chu, X.; Li, J.; Zhou, F.; Ren, S.; Li, X.; Su, C.; et al. Re-Biopsy and Liquid Biopsy for Patients with Non-Small Cell Lung Cancer after EGFR-Tyrosine Kinase Inhibitor Failure. Thorac. Cancer 2019, 10, 957–965. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Stephens, D.; Luo, L.; White, J.R.; Stewart, C.M.; Rousseau, B.; Tsui, D.W.Y.; Diaz, L.A. Genome-Wide Mutational Signatures in Low-Coverage Whole Genome Sequencing of Cell-Free DNA. Nat. Commun. 2022, 13, 4953. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, E.; Yellapantula, V.; O’Halloran, K.; Xu, L.; Berry, J.L.; Cotter, J.A.; Zdanowicz, A.; Mascarenhas, L.; Amatruda, J.F.; Ostrow, D.; et al. Combined Low-Pass Whole Genome and Targeted Sequencing in Liquid Biopsies for Pediatric Solid Tumors. Npj Precis. Oncol. 2023, 7, 21. [Google Scholar] [CrossRef]
- Johansson, P.A.; Brooks, K.; Newell, F.; Palmer, J.M.; Wilmott, J.S.; Pritchard, A.L.; Broit, N.; Wood, S.; Carlino, M.S.; Leonard, C.; et al. Whole Genome Landscapes of Uveal Melanoma Show an Ultraviolet Radiation Signature in Iris Tumours. Nat. Commun. 2020, 11, 2408. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, J.; Nilsson, L.M.; Mitra, S.; Alsén, S.; Shelke, G.V.; Sah, V.R.; Forsberg, E.M.V.; Stierner, U.; All-Eriksson, C.; Einarsdottir, B.; et al. Molecular Profiling of Driver Events in Metastatic Uveal Melanoma. Nat. Commun. 2020, 11, 1894. [Google Scholar] [CrossRef]
- Koeppel, F.; Blanchard, S.; Jovelet, C.; Genin, B.; Marcaillou, C.; Martin, E.; Rouleau, E.; Solary, E.; Soria, J.C.; André, F.; et al. Whole Exome Sequencing for Determination of Tumor Mutation Load in Liquid Biopsy from Advanced Cancer Patients. PLoS ONE 2017, 12, e0188174. [Google Scholar] [CrossRef]
- Leenanitikul, J.; Chanchaem, P.; Mankhong, S.; Denariyakoon, S.; Fongchaiya, V.; Arayataweegool, A.; Angspatt, P.; Wongchanapai, P.; Prapanpoj, V.; Chatamra, K.; et al. Concordance between Whole Exome Sequencing of Circulating Tumor DNA and Tumor Tissue. PLoS ONE 2023, 18, e0292879. [Google Scholar] [CrossRef]
- Grinshpun, A.; Tsuji, J.; Li, T.; Russo, D.; Anderson, L.; Rees, R.; Cibulskis, C.; Leshchiner, I.; Stewart, C.; Tung, N.M.; et al. Longitudinal Circulating Tumor DNA (CtDNA) Whole-Exome Sequencing (WES) in the Phase Ib/II Trial of Palbociclib and Bazedoxifene Reveals Genomic Dynamics and Clonal Evolution with the Acquisition of Treatment Resistance in Hormone Receptor-Positive, HER2-Negative (HR+ HER2-), Advanced Breast Cancer (ABC). J. Clin. Oncol. 2022, 40, 1058. [Google Scholar] [CrossRef]
- Van der Linden, M.; Van Gaever, B.; Raman, L.; Vermaelen, K.; Demedts, I.; Surmont, V.; Himpe, U.; Lievens, Y.; Ferdinande, L.; Dedeurwaerdere, F.; et al. Application of an Ultrasensitive NGS-Based Blood Test for the Diagnosis of Early-Stage Lung Cancer: Sensitivity, a Hurdle Still Difficult to Overcome. Cancers 2022, 14, 2031. [Google Scholar] [CrossRef]
- Alsina, K.M.; Sholl, L.M.; Covington, K.R.; Arnal, S.M.; Durante, M.A.; Decatur, C.L.; Stone, J.F.; Oelschlager, K.M.; Harbour, J.W.; Monzon, F.A.; et al. Analytical Validation and Performance of a 7-Gene Next-Generation Sequencing Panel in Uveal Melanoma. Ocul. Oncol. Pathol. 2021, 7, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Park, J.J.; Diefenbach, R.J.; Byrne, N.; Long, G.V.; Scolyer, R.A.; Gray, E.S.; Carlino, M.S.; Rizos, H. Circulating Tumor Dna Reflects Uveal Melanoma Responses to Protein Kinase c Inhibition. Cancers 2021, 13, 1740. [Google Scholar] [CrossRef]
- Le Guin, C.H.D.; Bornfeld, N.; Bechrakis, N.E.; Jabbarli, L.; Richly, H.; Lohmann, D.R.; Zeschnigk, M. Early Detection of Metastatic Uveal Melanoma by the Analysis of Tumor-Specific Mutations in Cell-Free Plasma DNA. Cancer Med. 2021, 10, 5974–5982. [Google Scholar] [CrossRef] [PubMed]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.Y.; Kaper, F.; Dawson, S.J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive Identification and Monitoring of Cancer Mutations by Targeted Deep Sequencing of Plasma DNA. Sci. Transl. Med. 2012, 4, 136ra68. [Google Scholar] [CrossRef] [PubMed]
- Hon, G.C.; Hawkins, R.D.; Caballero, O.L.; Lo, C.; Lister, R.; Pelizzola, M.; Valsesia, A.; Ye, Z.; Kuan, S.; Edsall, L.E.; et al. Global DNA Hypomethylation Coupled to Repressive Chromatin Domain Formation and Gene Silencing in Breast Cancer. Genome Res. 2012, 22, 246–258. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, H.; An, K.; Liu, Z.; Hai, L.; Li, R.; Zhou, Y.; Zhao, W.; Jia, Y.; Wu, N.; et al. Whole-genome Bisulfite Sequencing Analysis of Circulating Tumour DNA for the Detection and Molecular Classification of Cancer. Clin. Transl. Med. 2022, 12, e1014. [Google Scholar] [CrossRef]
- Bustamante, P.; Coblentz, J.; Mastromonaco, C.; Youhnovska, E.; Ito, H.; Proença, R.P.; Fonseca, C.; Dickinson, K.; Marcotte, E.; MacDonald, M.; et al. Comprehensive Clinical Imaging, Histopathological Analysis and Liquid Biopsy-Based Surveillance of Human Uveal Melanoma in a Prolonged Rabbit Xenograft Model. Melanoma Res. 2024, 34, 285–295. [Google Scholar] [CrossRef]
- Grisanti, S.; Schindler, F.; Merz, H.; Kakkassery, V.; Sonntag, S.R.; Tura, A. Detection of Circulating Tumor Cells in Patients with Small Choroidal Melanocytic Lesions. Ophthalmology 2023, 130, 1290–1303. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Butler, M.O.; Shoushtari, A.N.; Hassel, J.C.; Ikeguchi, A.; Hernandez-Aya, L.; Nathan, P.; Hamid, O.; Piulats, J.M.; Rioth, M.; et al. Clinical and Molecular Response to Tebentafusp in Previously Treated Patients with Metastatic Uveal Melanoma: A Phase 2 Trial. Nat. Med. 2022, 28, 2364–2373. [Google Scholar] [CrossRef]
- Ny, L.; Jespersen, H.; Karlsson, J.; Alsén, S.; Filges, S.; All-Eriksson, C.; Andersson, B.; Carneiro, A.; Helgadottir, H.; Levin, M.; et al. The PEMDAC Phase 2 Study of Pembrolizumab and Entinostat in Patients with Metastatic Uveal Melanoma. Nat. Commun. 2021, 12, 5155. [Google Scholar] [CrossRef]
- Gerard, C.; Shum, B.; Nathan, P.; Turajlic, S. Immuno-Oncology Approaches in Uveal Melanoma: Tebentafusp and Beyond. Immuno-Oncol. Technol. 2023, 19, 100386. [Google Scholar] [CrossRef] [PubMed]
- Sacco, J.J.; Carvajal, R.D.; Butler, M.O.; Shoushtari, A.N.; Hassel, J.C.; Ikeguchi, A.; Hernandez-Aya, L.; Nathan, P.; Hamid, O.; Piulats, J.M.; et al. Long-Term Survival Follow-up for Tebentafusp in Previously Treated Metastatic Uveal Melanoma. J. Immunother. Cancer 2024, 12, e009028. [Google Scholar] [CrossRef]
- Kim, M.E.; Xu, L.; Prabakar, R.K.; Shen, L.; Peng, C.C.; Kuhn, P.; Gai, X.; Hicks, J.; Berry, J.L. Aqueous Humor as a Liquid Biopsy for Retinoblastoma: Clear Corneal Paracentesis and Genomic Analysis. J. Vis. Exp. 2021, 2021, e62939. [Google Scholar] [CrossRef]
- Ghose, N.; Kaliki, S. Liquid Biopsy in Retinoblastoma: A Review. Semin. Ophthalmol. 2022, 37, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Montazeri, K.; Gragoudas, E.S.; Lane, A.M.; Aronow, M.B.; Cohen, J.V.; Boland, G.M.; Banks, E.; Kachulis, C.; Fleharty, M.; et al. Detection of Copy-Number Variation in Circulating Cell-Free DNA in Patients with Uveal Melanoma. JCO Precis. Oncol. 2024, 8, e2300368. [Google Scholar] [CrossRef]
- Jin, E.; Burnier, J.V. Liquid Biopsy in Uveal Melanoma: Are We There Yet? Ocul. Oncol. Pathol. 2021, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Cabel, L.; Riva, F.; Servois, V.; Livartowski, A.; Daniel, C.; Rampanou, A.; Lantz, O.; Romano, E.; Milder, M.; Buecher, B.; et al. Circulating Tumor DNA Changes for Early Monitoring of Anti-PD1 Immunotherapy: A Proof-of-Concept Study. Ann. Oncol. 2017, 28, 1996–2001. [Google Scholar] [CrossRef]
- Iwama, E.; Sakai, K.; Azuma, K.; Harada, T.; Harada, D.; Nosaki, K.; Hotta, K.; Ohyanagi, F.; Kurata, T.; Fukuhara, T.; et al. Monitoring of Somatic Mutations in Circulating Cell-Free DNA by Digital PCR and next-Generation Sequencing during Afatinib Treatment in Patients with Lung Adenocarcinoma Positive for EGFR Activating Mutations. Ann. Oncol. 2017, 28, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Cai, P.; Xie, J.; Wei, Y. The Diagnostic Accuracy of Digital PCR, ARMS and NGS for Detecting KRAS Mutation in Cell-Free DNA of Patients with Colorectal Cancer: A Systematic Review and Meta-Analysis. PLoS ONE 2021, 16, e0248775. [Google Scholar] [CrossRef] [PubMed]
- Connors, D.; Allen, J.; Alvarez, J.D.; Boyle, J.; Cristofanilli, M.; Hiller, C.; Keating, S.; Kelloff, G.; Leiman, L.; McCormack, R.; et al. International Liquid Biopsy Standardization Alliance White Paper. Crit. Rev. Oncol. Hematol. 2020, 156, 103112. [Google Scholar] [CrossRef]
- Dunavoelgyi, R.; Milman, T.; Shields, C.L.; Schmidt-Erfurth, U.; Pulido, J.S. Minimal Residual Disease—A Novel Concept in Uveal Melanoma. Eye 2020, 35, 702–704. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Technique | Sensitivity | Specificity | Detection Limit, % ctDNA | Reference | |
|---|---|---|---|---|---|
| PCR-based | ddPCR | 73.3–100% | 93.3% | 0.10–0.01 | Shi et al. [53] |
| ddPCR + LNA-Clamp ddPCR | NA | NA | 0.003 | Hashimoto et al. [54] | |
| IC3D ddPCR | NA | NA | 0.005–0.001 | Shi et al. [53] | |
| dPCR | 100% | 100% | 0.01 | Crucitta et al. [55] | |
| COLD-PCR/E-ice-COLD-PCR | 96–97% | 96% | 0.025–0.01 | How-Kit et al., Freidin et al. [56,57] | |
| BEAMing | 85% | 99.99% | 0.01 | Lanman et al., Khagi et al. [58,59] | |
| ARMS-PCR | 77.27% | 97.22% | 0.015 | Khagi et al., Zhang et al. [59,60] | |
| CRISPR/Cas12a | NA | NA | 3 | Escalona-Noguero et al. [61] | |
| NGS-based | WGS | 97.20% | 100% | 3 | Rickles-Young et al. [62] |
| WES | 99.8% | 99.9% | 0.1 | Bos et al. [63] | |
| Targeted NGS | 95.7% | 99.9% | 0.008 | Zhao et al. [64] | |
| MAPS | 98.5% | 98.9% | 0.1 | Garcia et al. [65] | |
| CAPP-Seq | 50–100% | 96% | 0.02 | Newman et al. [66] | |
| Tam-Seq/eTam-Seq | 97–99.48% | 97–99.99% | 2–0.02 | Plagnol et al., Gale et al. [67,68] |
| Advantages | Limitations |
|---|---|
| Non-invasive genetic uveal melanoma profile; | Demanding technology; |
| Early detection of malignancy in indeterminate small choroidal melanocytic lesions; | The method has not been fully standardized, and reproducibility is challenging; |
| High sensitivity to detect early metastatic disease; | Difficult to detect ctDNA in early, low-tumor burden disease; |
| Useful for prognosis assessment and therapeutic response. | Not clinically validated (although validation studies are underway). |
| Authors | Fluid Type | Study Population | Number of Patients | System Detection | Detection Rate | Main Findings |
|---|---|---|---|---|---|---|
| Pike et al. [21] | AH | Primary | n = 66; samples collected pre- and post-brachytherapy. | Quantification of nucleic acids | Analytes were quantifiable in >70% of diagnostic samples with tumors < 2 mm tall. | AH is a rich repository of analytes. Tumors with poorer prognostic features have increased concentrations of analytes compared with tumors with lower risk. |
| Im et al. [22] | AH | Primary | n = 20; samples taken before or after radiation. | WGS and targeted NGS | SCNAs were found in 75% (6/8) of post-radiation CB samples. | UM, SCNAs and mutations can be identified from the AH, especially in CB tumors. |
| Bustamante et al. [30] | PB | Primary | n = 14. | ddPCR | 100% efficiency of UM mutant ctDNA detection. | Potential of ctDNA as a biomarker of early diagnosis and disease progression. |
| Beasley et al. [72] | PB | Primary and metastatic | Three cohorts: a retrospective cohort of 30 primary tumor patients; a prospective cohort of 37 primary tumors in patients with known mutations, and six patients with metastatic UM. | ddPCR | In a retrospective cohort, ctDNA was detectable in 8/30 cases (26%). In the prospective cohort, ctDNA was detectable in 17/25 (68%) patients that developed metastases. In the metastatic cohort, ctDNA was detectable in 6/6 (100%). | ctDNA levels in primary UM are not associated with survival, but this was the strongest predictor of OS in MetUM. Decreases in ctDNA levels are an indicator of response to immunotherapy. |
| de Bruyn et al. [14] | PB | Primary and metastatic | n = 34; for ctDNA detection (n = 18) and/or SCNA analysis (n = 26) at various time points. | ddPCR and sWGS | ctDNA was detectable in 38% (5/13) of patients at diagnosis, in 77% (10/13) upon detection of metastatic disease, and in 50% (3/6) during fSRT. Loss of Chr 3 was detected in 70% (7/10) of patients with MetUM. | No SCNA profiles and ctDNA levels were low or undetectable in localized disease. ctDNA levels in metastatic patients could be a biomarker of disease progression. |
| Nell et al. [20] | Vitreous fluid | Primary | n = 65. | ddPCR | 39/65 (60%) patients had Gαq signaling mutations; 13/15 (87%) had a BSE mutation; Chr 3p losses were detected in 13/15 (87%) samples; Chr 8q gains were identified in 15/17 samples (88%). | cfDNA was associated with larger tumors of BSE mutation. and CNA results could be inferred from vitreous fluid liquid biopsy. |
| Park et al. [85] | PB | Metastatic | n = 17; samples were collected at baseline, EDT and on-treatment. | ddPCR and NGS | At baseline, ctDNA was detected by ddPCR in 94% (16/17) of patients, by NGS in 88% (15/17), and for those on treatment it was identfied by NGS in 94% (16/17). | Absolute level of EDT ctDNA is indicative of treatment response. Increasing UM ctDNA preceded radiological progression. |
| Carvajal et al. [92] | PB | Metastatic | n = 127 | Multiplex PCR and NGS | 80% (94/118) had mutations detected in one or more genes (Gαq and SF3B1). | ctDNA as an early indicator of clinical benefit from tebentafusp; Post-treatment reduction in ctDNA levels correlated with survival benefit. |
| Kim et al. [13] | PB | Primary | n = 26; samples were collected at various time points. | NGS | In 31% (8/26) of patients, ctDNA was detected during or after brachytherapy. No ctDNA was detected in any of the samples collected before brachytherapy. | Brachytherapy increases the presence of ctDNA. The allele fraction detected correlates with the largest basal diameter and tumor thickness. |
| Ny et al. [93] | PB | Metastatic | n = 29; samples were collected at various time points. | NGS | 75% (12/16) of patients with PD and 37% (3/8) with stable disease had detectable ctDNA levels. | Low baseline ctDNA levels predicted long OS but not PFS. The ctDNA levels were lower (not significantly) in patients with PR vs. those with PD. |
| Sato et al. [98] | PB | Primary and metastatic | 14 MetUM and two non-metastatic patients; samples collected at various time points | ULP-WGS | 78% (11/14) of patients with MetUM had detectable ctDNA. 8q gain was detected in all; Loss of Chr 3 was detectable in 59% (10/17). | ctDNA in metastatic patients can be detected by ULP-WGS, and ctDNA levels correlate with disease status. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varela, M.; Villatoro, S.; Lorenzo, D.; Piulats, J.M.; Caminal, J.M. Optimizing ctDNA: An Updated Review of a Promising Clinical Tool for the Management of Uveal Melanoma. Cancers 2024, 16, 3053. https://doi.org/10.3390/cancers16173053
Varela M, Villatoro S, Lorenzo D, Piulats JM, Caminal JM. Optimizing ctDNA: An Updated Review of a Promising Clinical Tool for the Management of Uveal Melanoma. Cancers. 2024; 16(17):3053. https://doi.org/10.3390/cancers16173053
Chicago/Turabian StyleVarela, Mar, Sergi Villatoro, Daniel Lorenzo, Josep Maria Piulats, and Josep Maria Caminal. 2024. "Optimizing ctDNA: An Updated Review of a Promising Clinical Tool for the Management of Uveal Melanoma" Cancers 16, no. 17: 3053. https://doi.org/10.3390/cancers16173053
APA StyleVarela, M., Villatoro, S., Lorenzo, D., Piulats, J. M., & Caminal, J. M. (2024). Optimizing ctDNA: An Updated Review of a Promising Clinical Tool for the Management of Uveal Melanoma. Cancers, 16(17), 3053. https://doi.org/10.3390/cancers16173053