
Clinical and Histologic Variants of CD8+ Cutaneous T-Cell Lymphomas

 , ,
, ,
Abstract
Simple Summary
Abstract
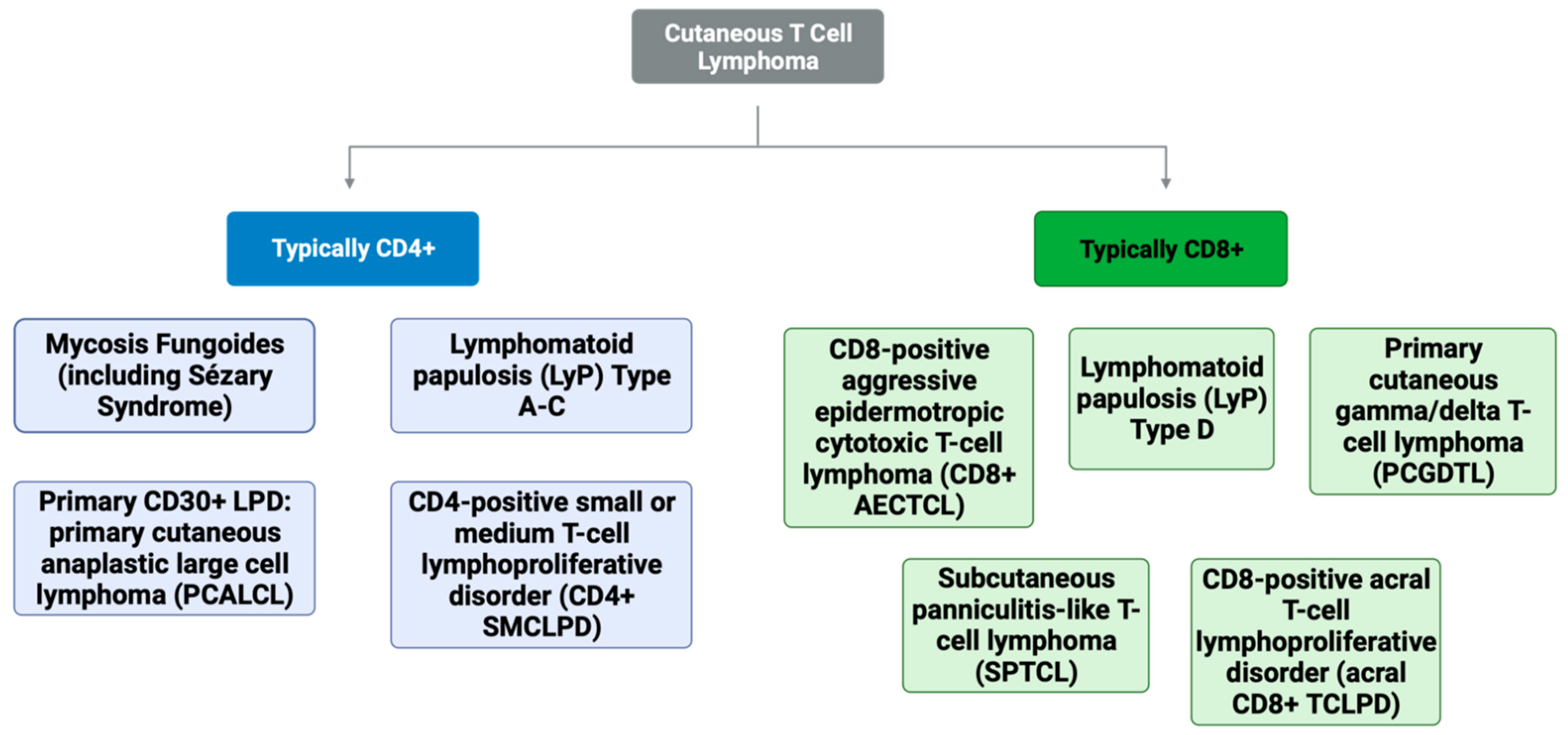
1. Introduction
2. CD8+ Mycosis Fungoides (MF)
3. Lymphomatoid Papulosis (LyP), Type D
4. Subcutaneous Panniculitis-like T-Cell Lymphoma (SPTCL)
5. Primary Cutaneous Gamma/Delta T-Cell Lymphoma (PCGDTL)
6. CD8+ Aggressive Epidermotropic Cutaneous T-Cell Lymphoma (AECTCL)
7. Acral CD8+ T-Cell Lymphoproliferative Disorder (Acral CD8+ TCLPD)
8. Other Diagnostic Considerations: Peripheral T-Cell Lymphomas Not Otherwise Specified (PTCL-NOS) and Natural Killer T-Cell (NK/T) Lymphoma and Lymphoproliferative Disorders
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CD | cluster of differentiation |
| CD4+ SMCLPD | CD4-positive small or medium T-cell lymphoproliferative disorder |
| CD8+ AECTCL | CD8-positive aggressive epidermotropic cytotoxic T-cell lymphoma |
| acral CD8+ TCLPD | CD8-positive acral T-cell lymphoproliferative disorder |
| CTCL | cutaneous T-cell lymphoma |
| FDG | F-18 fluorodeoxyglucose |
| IHC | immunohistochemistry |
| LyP | lymphomatoid papulosis |
| MF | mycosis fungoides |
| PCALCL | primary cutaneous anaplastic large cell lymphoma |
| PCGDTL | primary cutaneous gamma/delta T-cell lymphoma |
| PET-CT | positron emission tomography computed tomography |
| SPTCL | subcutaneous panniculitis-like T-cell lymphoma |
| SS | Sézary syndrome |
| TCL | T-cell lymphoma |
References
- Girardi, M.; Heald, P.W.; Wilson, L.D. The pathogenesis of mycosis fungoides. N. Engl. J. Med. 2004, 350, 1978–1988. [Google Scholar] [CrossRef] [PubMed]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.B.; Goyal, A.; McDivitt Duncan, L. Atlas of Cutaneous Lymphomas; Springer International Publishing: Cham, Switzerland, 2015. [Google Scholar]
- Kempf, W.; Petrella, T.; Willemze, R.; Jansen, P.; Berti, E.; Santucci, M.; Geissinger, E.; Cerroni, L.; Maubec, E.; Battistella, M.; et al. Clinical, histopathological and prognostic features of primary cutaneous acral CD8+ T-cell lymphoma and other dermal CD8+ cutaneous lymphoproliferations: Results of an EORTC Cutaneous Lymphoma Group workshop. Br. J. Dermatol. 2022, 186, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Guitart, J.; Martinez-Escala, M.E.; Subtil, A.; Duvic, M.; Pulitzer, M.P.; Olsen, E.A.; Kim, E.; Rook, A.H.; Samimi, S.S.; Wood, G.S.; et al. Primary cutaneous aggressive epidermotropic cytotoxic T-cell lymphomas: Reappraisal of a provisional entity in the 2016 WHO classification of cutaneous lymphomas. Mod. Pathol. 2017, 30, 761–772. [Google Scholar] [CrossRef]
- Kalay Yildizhan, I.; Sanli, H.; Akay, B.N.; Surgun, E.; Heper, A. CD8+ cytotoxic mycosis fungoides: A retrospective analysis of clinical features and follow-up results of 29 patients. Int. J. Dermatol. 2020, 59, 127–133. [Google Scholar] [CrossRef]
- Ahn, H.J.; Shin, E.J.; Gwak, M.J.; Jeong, K.H.; Shin, M.K. Sudden aggravated CD8+ mycosis fungoides accompanied by hidden adenocarcinoma of the colon. JAAD Case Rep. 2017, 3, 83–86. [Google Scholar] [CrossRef]
- Martinez-Escala, M.E.; Kantor, R.W.; Cices, A.; Zhou, X.A.; Kaplan, J.B.; Pro, B.; Choi, J.; Guitart, J. CD8+ mycosis fungoides: A low-grade lymphoproliferative disorder. J. Am. Acad. Dermatol. 2017, 77, 489–496. [Google Scholar] [CrossRef]
- Song, S.X.; Willemze, R.; Swerdlow, S.H.; Kinney, M.C.; Said, J.W. Mycosis fungoides: Report of the 2011 Society for Hematopathology/European Association for Haematopathology workshop. Am. J. Clin. Pathol. 2013, 139, 466–490. [Google Scholar] [CrossRef]
- Cao, S.; Kruglov, O.; Akilov, O.E. CD8+ T Lymphocytes in Hypopigmented Mycosis Fungoides: Malignant Cells or Reactive Clone? J. Investig. Dermatol. 2023, 143, 521–524.e3. [Google Scholar] [CrossRef]
- Mangold, A.R.; Thompson, A.K.; Davis, M.D.; Saulite, I.; Cozzio, A.; Guenova, E.; Hodak, E.; Amitay-Laish, I.; Pujol, R.M.; Pittelkow, M.R.; et al. Early clinical manifestations of Sezary syndrome: A multicenter retrospective cohort study. J. Am. Acad. Dermatol. 2017, 77, 719–727. [Google Scholar] [CrossRef]
- Mehrany, K.; El-Azhary, R.A.; Bouwhuis, S.A.; Pittelkow, M.R. Cutaneous T-cell lymphoma and atopy: Is there an association? Br. J. Dermatol. 2003, 149, 1013–1017. [Google Scholar] [CrossRef]
- Cordel, N.; Tressieres, B.; D’Incan, M.; Machet, L.; Grange, F.; Esteve, E.; Dalac, S.; Ingen-Housz-Oro, S.; Bagot, M.; Beylot-Barry, M.; et al. Frequency and Risk Factors for Associated Lymphomas in Patients With Lymphomatoid Papulosis. Oncologist 2016, 21, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Wieser, I.; Oh, C.W.; Talpur, R.; Duvic, M. Lymphomatoid papulosis: Treatment response and associated lymphomas in a study of 180 patients. J. Am. Acad. Dermatol. 2016, 74, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Wakumoto, K.; Sugita, K.; Yamamoto, O. Subcutaneous Panniculitis-like T-Cell Lymphoma Without Erythema and Subcutaneous Tumors. Yonago Acta Med. 2021, 64, 126–128. [Google Scholar] [CrossRef] [PubMed]
- Ukishima, S.; Miyagami, T.; Arikawa, M.; Kushiro, S.; Takaku, T.; Naito, T. Subcutaneous panniculitis-like T-cell lymphoma post-mRNA-1273 COVID-19 vaccination. Clin. Case Rep. 2023, 11, e7143. [Google Scholar] [CrossRef]
- Kreher, M.A.; Ahn, J.; Werbel, T.; Motaparthi, K. Subcutaneous panniculitis-like T-cell lymphoma after COVID-19 vaccination. JAAD Case Rep. 2022, 28, 18–20. [Google Scholar] [CrossRef]
- Ou, W.; Zhao, Y.; Wei, A.; Ma, H.; Zhang, L.; Lian, H.; Zhang, Q.; Wang, D.; Li, Z.; Wang, T.; et al. Subcutaneous panniculitis-like T-cell lymphoma associated with hemophagocytic lymphohistiocytosis: A systematic review of 63 patients reported in the literature. Clin. Exp. Med. 2023, 23, 4575–4583. [Google Scholar] [CrossRef]
- Willemze, R.; Jansen, P.M.; Cerroni, L.; Berti, E.; Santucci, M.; Assaf, C.; Canninga-van Dijk, M.R.; Carlotti, A.; Geerts, M.L.; Hahtola, S.; et al. Subcutaneous panniculitis-like T-cell lymphoma: Definition, classification, and prognostic factors: An EORTC Cutaneous Lymphoma Group Study of 83 cases. Blood 2008, 111, 838–845. [Google Scholar] [CrossRef]
- Guenova, E.; Schanz, S.; Hoetzenecker, W.; DeSimone, J.A.; Mehra, T.; Voykov, B.; Urosevic-Maiwald, M.; Berneburg, M.; Dummer, R.; French, L.E.; et al. Systemic corticosteroids for subcutaneous panniculitis-like T-cell lymphoma. Br. J. Dermatol. 2014, 171, 891–894. [Google Scholar] [CrossRef]
- Mehta, N.; Wayne, A.S.; Kim, Y.H.; Hale, G.A.; Alvarado, C.S.; Myskowski, P.; Jaffe, E.S.; Busam, K.J.; Pulitzer, M.; Zwerner, J.; et al. Bexarotene is active against subcutaneous panniculitis-like T-cell lymphoma in adult and pediatric populations. Clin. Lymphoma Myeloma Leuk. 2012, 12, 20–25. [Google Scholar] [CrossRef]
- Jothishankar, B.; Espinosa, M.L.; Zain, J.; Parekh, V.; Di Raimondo, C.; Abdulla, F. Complete response to romidepsin as monotherapy in treatment-resistant subcutaneous panniculitis-like T-cell lymphoma. JAAD Case Rep. 2020, 6, 1245–1247. [Google Scholar] [CrossRef]
- Torres-Cabala, C.A.; Huen, A.; Iyer, S.P.; Miranda, R.N. Gamma/Delta Phenotype in Primary Cutaneous T-cell Lymphomas and Lymphoid Proliferations: Challenges for Diagnosis and Classification. Surg. Pathol. Clin. 2021, 14, 177–194. [Google Scholar] [CrossRef]
- Onkarappa Mangala, Y.; Onukogu, I.D.; Breen, C.M.; Colvin, G.A. Primary Cutaneous Gamma-Delta T-cell Lymphoma: A Case Report and Review of Literature. Cureus 2023, 15, e35442. [Google Scholar] [CrossRef] [PubMed]
- Guitart, J.; Weisenburger, D.D.; Subtil, A.; Kim, E.; Wood, G.; Duvic, M.; Olsen, E.; Junkins-Hopkins, J.; Rosen, S.; Sundram, U.; et al. Cutaneous γδ T-cell lymphomas: A spectrum of presentations with overlap with other cytotoxic lymphomas. Am. J. Surg. Pathol. 2012, 36, 1656–1665. [Google Scholar] [CrossRef] [PubMed]
- Alberti-Violetti, S.; Maronese, C.A.; Venegoni, L.; Merlo, V.; Berti, E. Primary Cutaneous Gamma-Delta T Cell Lymphomas: A Case Series and Overview of the Literature. Dermatopathology 2021, 8, 515–524. [Google Scholar] [CrossRef]
- Oh, Y.; Stoll, J.R.; Moskowitz, A.; Pulitzer, M.; Horwitz, S.; Myskowski, P.; Noor, S.J. Primary cutaneous T-cell lymphomas other than mycosis fungoides and Sezary syndrome. Part II: Prognosis and management. J. Am. Acad. Dermatol. 2021, 85, 1093–1106. [Google Scholar] [CrossRef] [PubMed]
- Nofal, A.; Abdel-Mawla, M.Y.; Assaf, M.; Salah, E. Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma: Proposed diagnostic criteria and therapeutic evaluation. J. Am. Acad. Dermatol. 2012, 67, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Ormerod, E.; Murigu, T.; Pawade, J.; Beasley, M.; Dunnill, G. Primary cutaneous acral CD8+ T-cell lymphoma of the ear: A case report. J. Cutan. Pathol. 2019, 46, 790–793. [Google Scholar] [CrossRef]
- Oluwasanjo, A.; Kartan, S.; Johnson, W.; Alpdogan, O.; Gru, A.; Mishra, A.; Haverkos, B.M.; Gong, J.; Porcu, P. Peripheral T-Cell Lymphoma, not Otherwise Specified (PTCL-NOS). Cancer Treat. Res. 2019, 176, 83–98. [Google Scholar] [CrossRef]
- Cardwell, L.A.; Majerowski, J.; Chiu, Y.E.; Harrington, A.M.; Sokumbi, O. Post-transplant primary cutaneous peripheral T-cell lymphoma not otherwise specified in a pediatric patient. J. Cutan. Pathol. 2021, 48, 706–712. [Google Scholar] [CrossRef]
- de Matos, P.R.; Amoedo, P.; Nogueira, A.; Lisboa, C.; Carvalhais, I.; Fonseca, E.; Azevedo, F. Primary cutaneous peripheral T-cell lymphoma not otherwise specified (PTCL-NOS) simulating periorbital cellulitis. Int. J. Dermatol. 2023, 62, e125–e128. [Google Scholar] [CrossRef]
- García-Herrera, A.; Calonje, E. Cutaneous Lymphomas with Cytotoxic Phenotype. Surg. Pathol. Clin. 2017, 10, 409–427. [Google Scholar] [CrossRef]
- Geller, S.; Myskowski, P.L.; Pulitzer, M. NK/T-cell lymphoma, nasal type, γδ T-cell lymphoma, and CD8-positive epidermotropic T-cell lymphoma-clinical and histopathologic features, differential diagnosis, and treatment. Semin. Cutan. Med. Surg. 2018, 37, 30–38. [Google Scholar] [CrossRef]
- Schukow, C.; Ahmed, A. Dermatopathology, Cutaneous Lymphomas. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]

{kind=link}
{kind=link}
| Entity Name | Frequency (% of T-Cell Lymphomas) | 5-Year Prognosis (%) | Immunohistochemistry Findings | |||||
|---|---|---|---|---|---|---|---|---|
| CD3 | CD4 | CD8 | CD30 | CD56 | Other | |||
| MF | 65% | 75–98%, can vary by subtype | + | + | - (rarely +) | - | - | CD25- |
| SS | 4% | 10–33% | + | + | - | - | - | CD26-, CD27+ |
| LyP: Type A,C | 16% | 100% | + | + | - | + | - | ALK-, CD15- |
| LyP: Type B | + | + | - | - | - | ALK- | ||
| LyP: Type D | + | - | + | + | - | ALK-, βF1+ | ||
| PCALCL | 10% | 90% | + | + | - | + | - | ALK- |
| SPTCL | 1% | 85–90% | + | - | + | ± | - | βF1+ |
| PCGDTL | <1% | N/A, median survival is 15 months | + | - | ± | ± | + | Beta F1-, TCRγ+ |
| CD8+ AECTCL | <1% | 18% | + | - | + (rarely -) | - | - | CD45RA- or partially +, βF1+ |
| Acral CD8+ TCLPD | <1% | 75–100% | + | - | + | - | - | |
| CD4+ SMCLPD | <1% | >90% | + | + | - | - | - | |
| CD8+ Subtype | Clinical Features | IHC | Treatment |
|---|---|---|---|
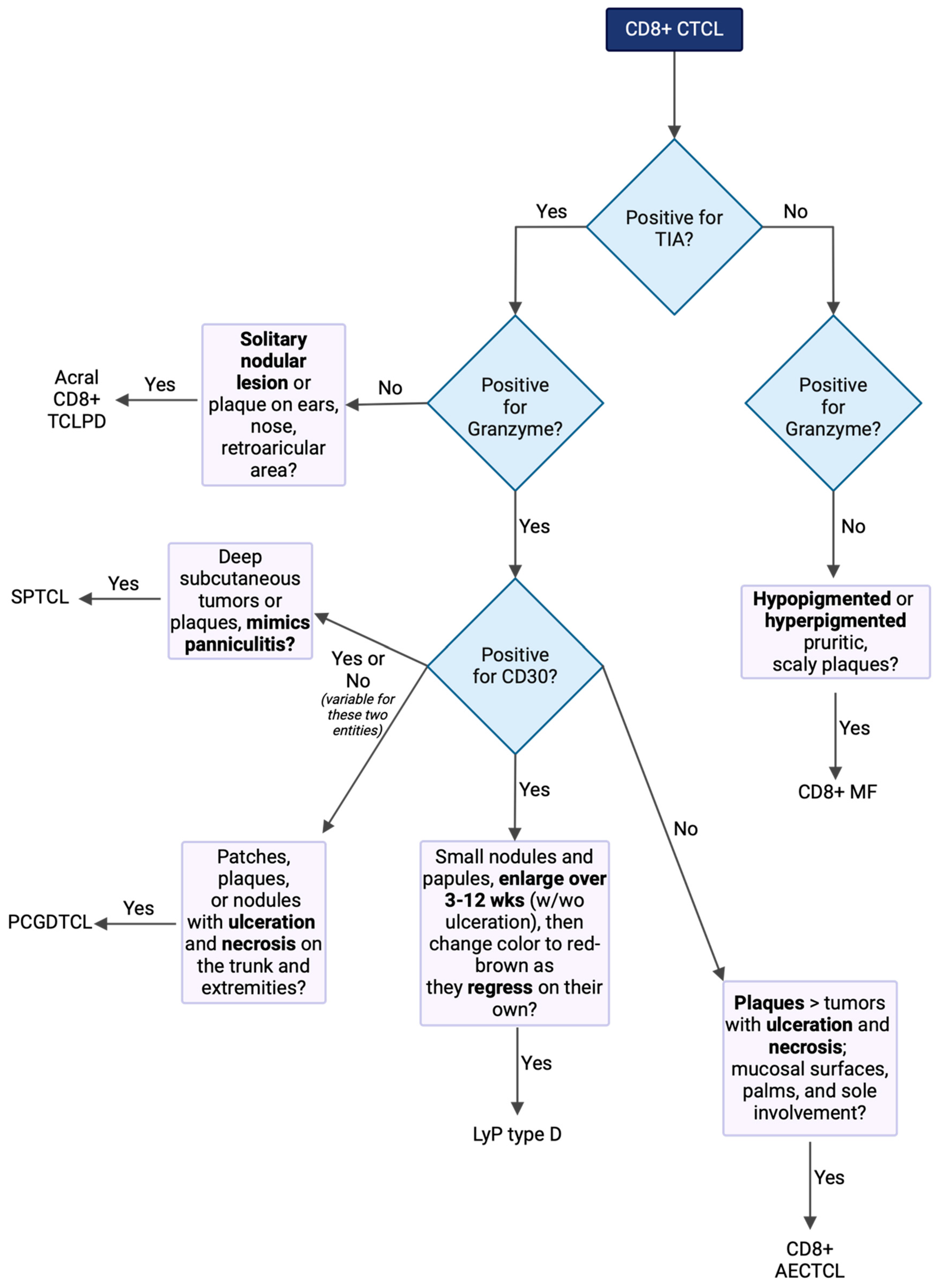
| CD8+ MF | Hypopigmented or hyperpigmented pruritic, scaly plaques | CD3+, CD7− CD8+, CD30+/− CD56−, CCR4+ Cytotoxic granules- (TIA-GZM-) | Early: topical steroids and/or retinoids, phototherapy, RT Advanced: bexarotene, IFNα, ECP, chemotherapy, stem cell transplant |
| LyP type D | Small nodules and papules, enlarge over 3–12 wks (w/wo ulceration), then change color to red-brown as they regress on their own | CD3+, CD4− CD8+, CD25+ CD30+, CD45RO+ CD56+/− Cytotoxic granules+ (TIA+GZM+) | Topical steroids, phototherapy, low-dose MTX Recurrence is common |
| SPTCL | Deep subcutaneous tumors or plaques, mimics panniculitis | CD4−, CD5−, CD8+, CD30+/−, and CD56− Cytotoxic granules+ (TIA+GZM+) | Prednisone, MTX, bexarotene, romidepsin, chemotherapy |
| PCGDTL | Erythematous to violaceous patches, plaques, or nodules with ulceration and necrosis on trunk and extremities | CD2+, CD3+, CD4−, CD5−, CD8 +/−, CD30+/−, CD56+, Cytotoxic granules+ (TIA+GZM+) | Chemotherapy, though almost always fatal |
| CD8+ AECTCL | Plaques > tumors with ulceration and necrosis; mucosal surfaces, palms, and sole involvement | CD3+, CD4− CD5−, CD8+ CD30− Cytotoxic granules+ (TIA+GZM+) | Chemotherapy, localized RT IFNα may worsen disease |
| Acral CD8+ TCLPD | Solitary nodular lesion or plaque on ears, nose, or retroarticular area | CD3+, CD4− CD8+, CD30− CD56− Cytotoxic granules+/− (TIA+, GZM−) | RT or surgical excision |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swallow, M.A.; Micevic, G.; Zhou, A.; Carlson, K.R.; Foss, F.M.; Girardi, M. Clinical and Histologic Variants of CD8+ Cutaneous T-Cell Lymphomas. Cancers 2024, 16, 3087. https://doi.org/10.3390/cancers16173087
Swallow MA, Micevic G, Zhou A, Carlson KR, Foss FM, Girardi M. Clinical and Histologic Variants of CD8+ Cutaneous T-Cell Lymphomas. Cancers. 2024; 16(17):3087. https://doi.org/10.3390/cancers16173087
Chicago/Turabian StyleSwallow, Madisen A., Goran Micevic, Amanda Zhou, Kacie R. Carlson, Francine M. Foss, and Michael Girardi. 2024. "Clinical and Histologic Variants of CD8+ Cutaneous T-Cell Lymphomas" Cancers 16, no. 17: 3087. https://doi.org/10.3390/cancers16173087
APA StyleSwallow, M. A., Micevic, G., Zhou, A., Carlson, K. R., Foss, F. M., & Girardi, M. (2024). Clinical and Histologic Variants of CD8+ Cutaneous T-Cell Lymphomas. Cancers, 16(17), 3087. https://doi.org/10.3390/cancers16173087