
Inhibition of Insulin-like Growth Factor 1 Receptor/Insulin Receptor Signaling by Small-Molecule Inhibitor BMS-754807 Leads to Improved Survival in Experimental Esophageal Adenocarcinoma

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice, Cell Lines, and Reagents
2.2. Cell Viability Assay
2.3. Western Blot Analysis
2.4. Scratch Wound Healing Assessment
2.5. Animal Studies
2.6. Immunohistochemistry (IHC)
2.7. Statistical Test
3. Results
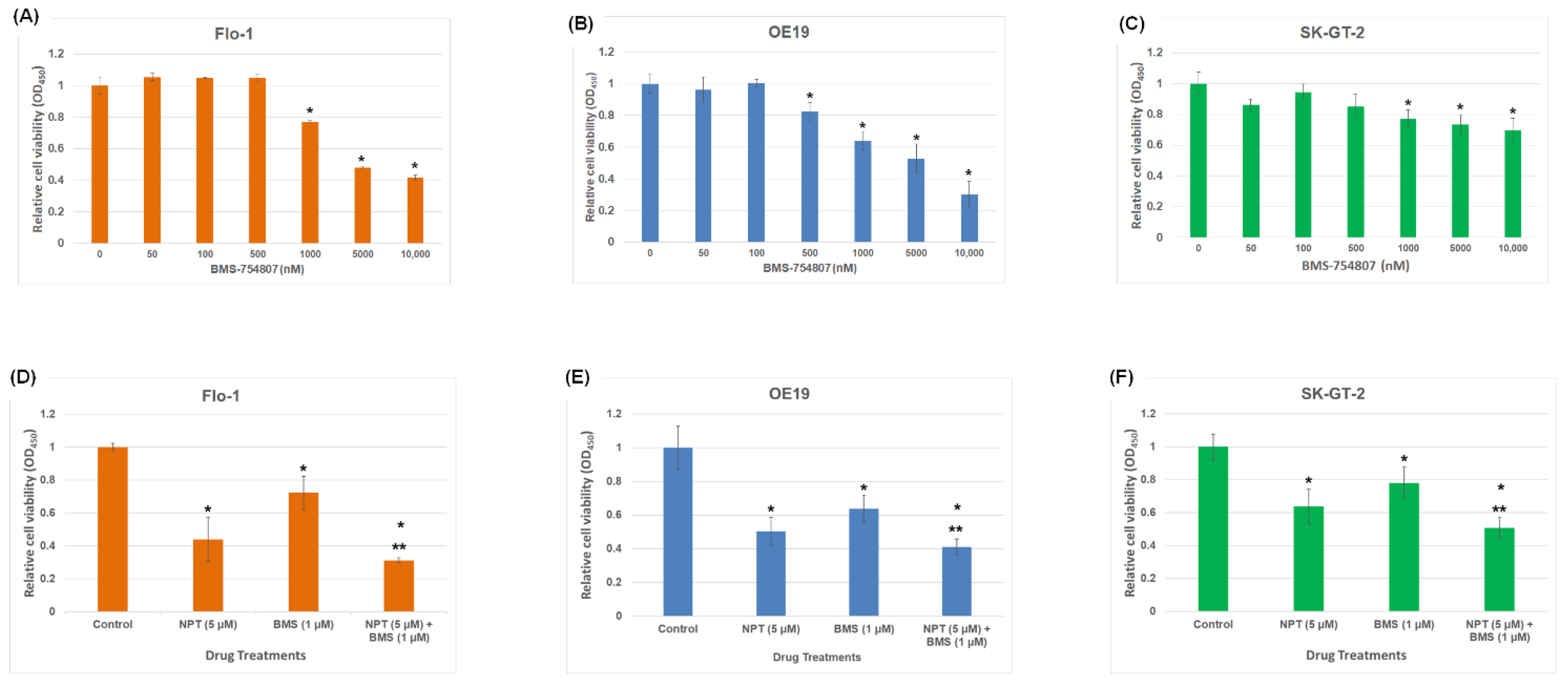
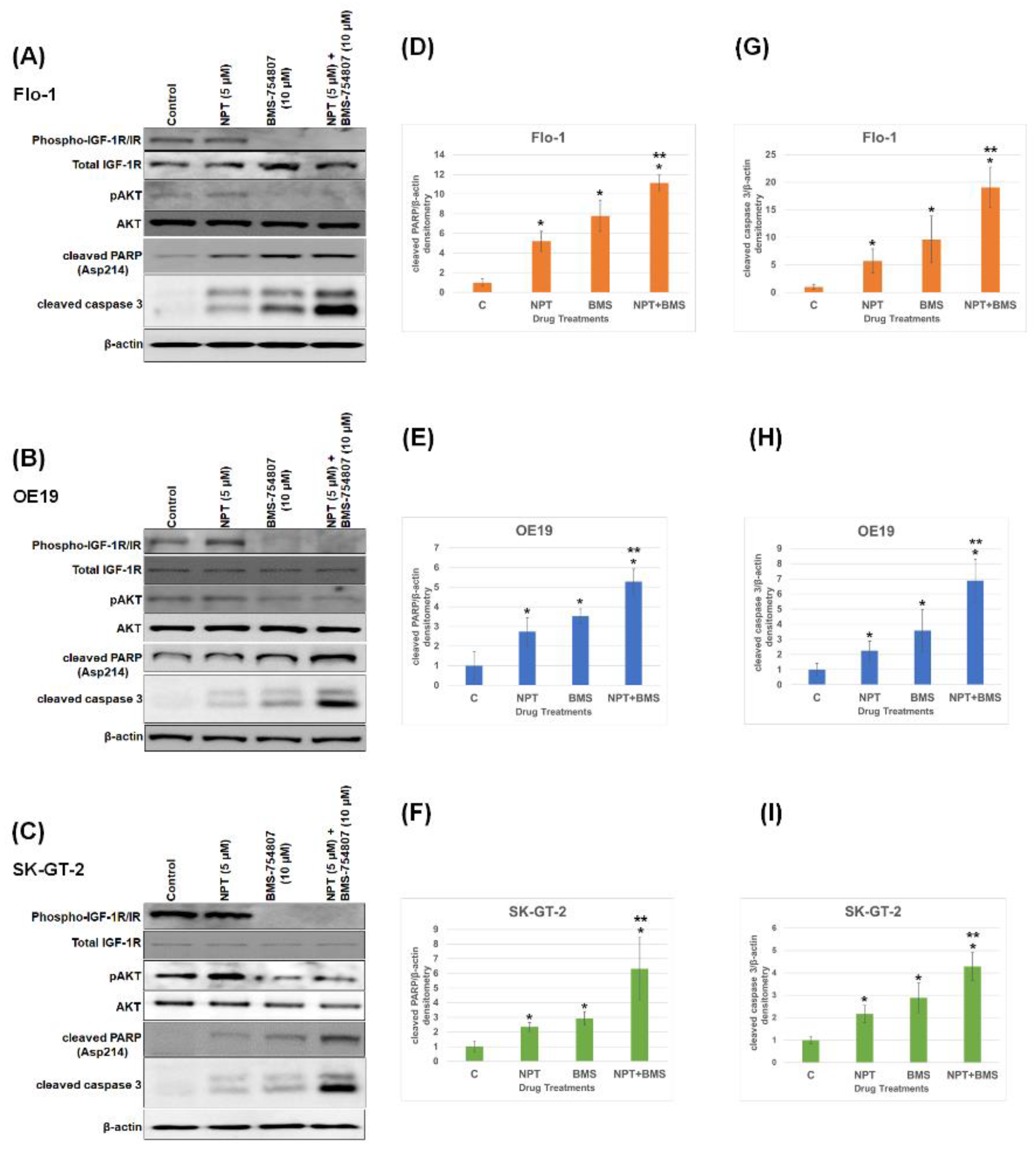
3.1. Effects of BMS-754807 Alone and in Combination with Nab-Paclitaxel on EAC Cell Proliferation and Apoptosis
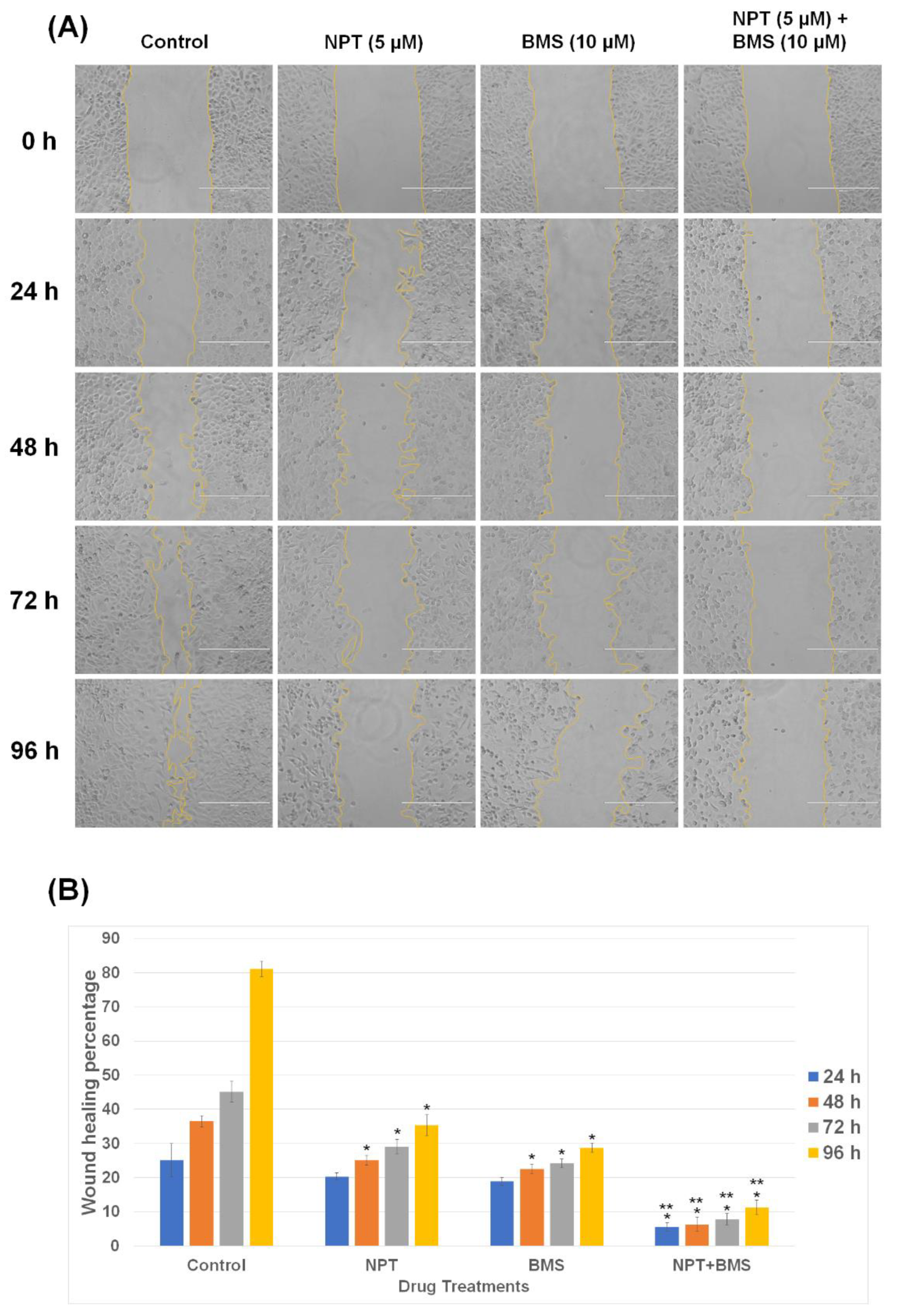
3.2. Effects of BMS-754807 Alone and in Combination with Nab-Paclitaxel on EAC Cell Scratch Wound Closure
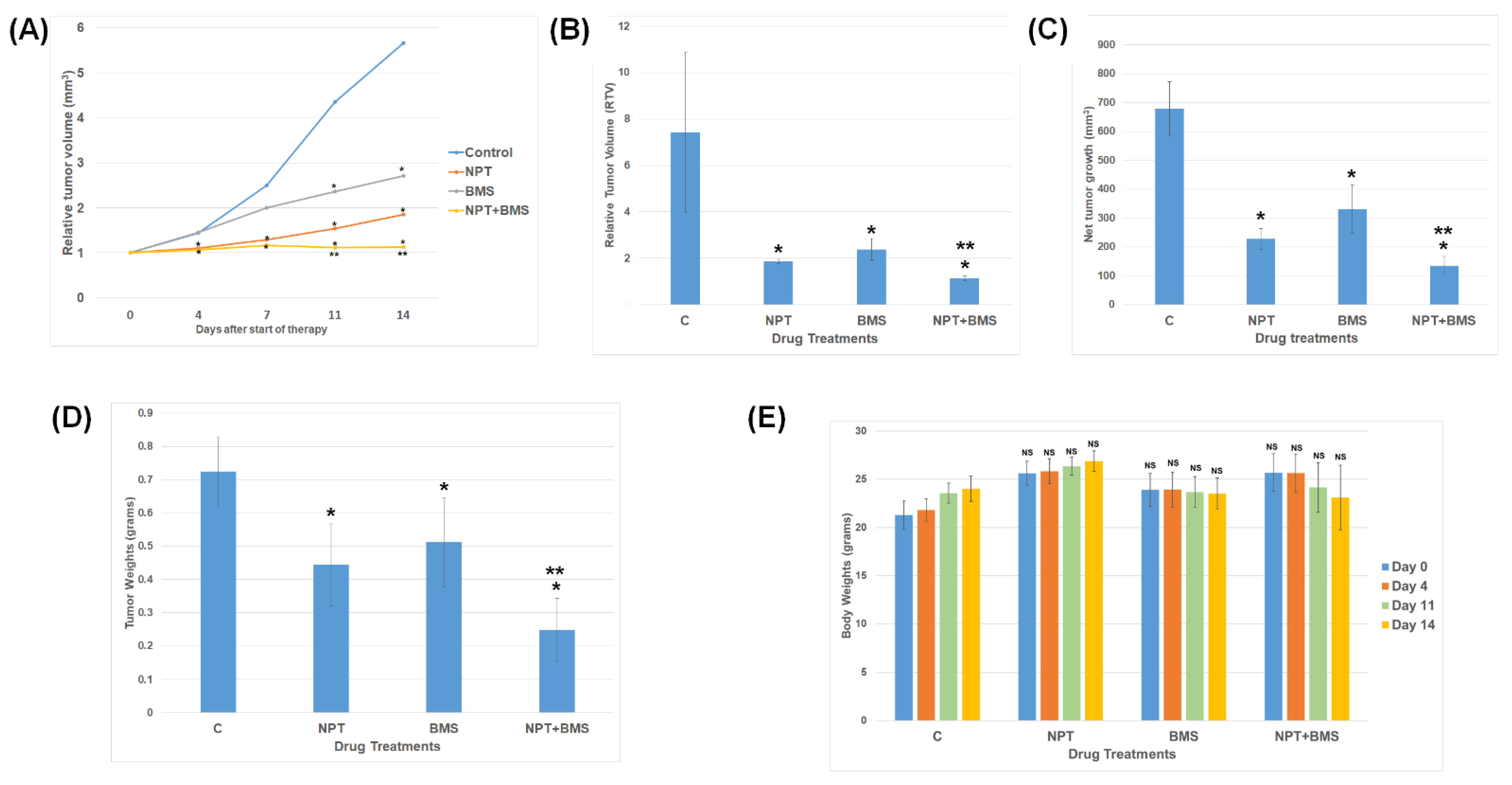
3.3. Effects of BMS-754807 Alone and in Combination with Nab-Paclitaxel on EAC Xenograft Tumor Size
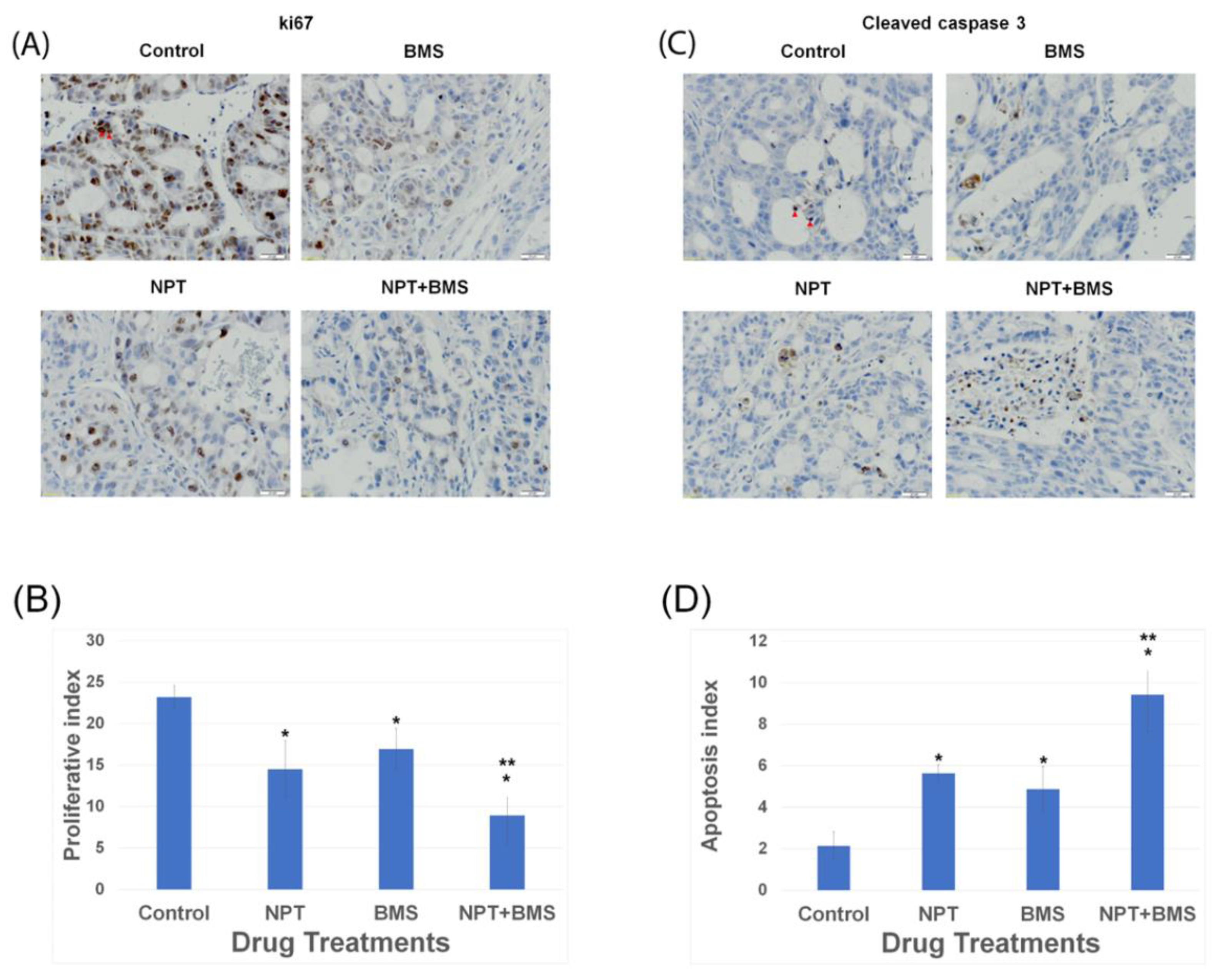
3.4. Effects of BMS-754807 Alone and in Combination with Nab-Paclitaxel on Proliferation and Apoptosis within the Tumor In Vivo
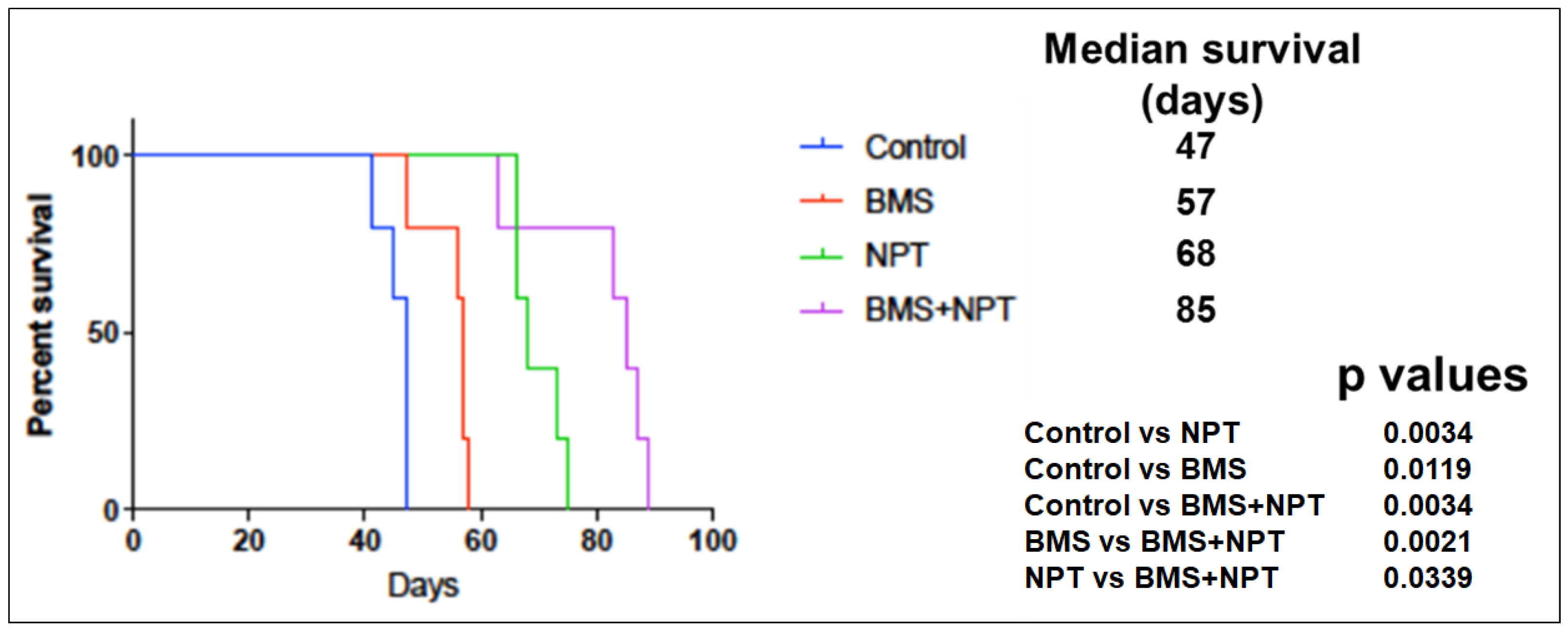
3.5. Effects of BMS-754807 Alone and in Combination with Nab-Paclitaxe on EAC Animal Survival
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pandilla, R.; Kotapalli, V.; Gowrishankar, S.; Chigurupati, M.; Patnaik, S.; Uppin, S.; Rao, S.; Kalidindi, N.; Regulagadda, S.; Sundaram, C.; et al. Distinct genetic aberrations in oesophageal adeno and squamous carcinoma. Eur. J. Clin. Investig. 2013, 43, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Jamal, M.M. Esophageal malignancy: A growing concern. World J. Gastroenterol. 2012, 18, 6521–6526. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.H.; Shaheen, N.J. Epidemiology, Diagnosis, and Management of Esophageal Adenocarcinoma. Gastroenterology 2015, 149, 302–317.e1. [Google Scholar] [CrossRef] [PubMed]
- Rustgi, A.K.; El-Serag, H.B. Esophageal carcinoma. N. Engl. J. Med. 2014, 371, 2499–2509. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Hang, T.-V.P.; Spiritos, Z.; Gamboa, A.M.; Chen, Z.; Force, S.; Patel, V.; Chawla, S.; Keilin, S.; Saba, N.F.; El-Rayes, B.; et al. Epidemiology of Early Esophageal Adenocarcinoma. Clin. Endosc. 2022, 55, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Corley, D.A. The Troublesome Epidemiology of Barrett’s Esophagus and Esophageal Adenocarcinoma. Gastrointest. Endosc. Clin. N. Am. 2017, 27, 353–364. [Google Scholar] [CrossRef]
- Brown, L.M.; Devesa, S.S.; Chow, W.-H. Incidence of adenocarcinoma of the esophagus among white americans by sex, stage, and age. J. Natl. Cancer Inst. 2008, 100, 1184–1187. [Google Scholar] [CrossRef]
- Domper Arnal, M.J.; Ferrández Arenas, Á.; Lanas Arbeloa, Á. Esophageal cancer: Risk factors, screening and endoscopic treatment in Western and Eastern countries. World J. Gastroenterol. 2015, 21, 7933–7943. [Google Scholar] [CrossRef]
- Glasgow, R.; Mawhinney, M. Current treatment options for the management of esophageal cancer. Cancer Manag. Res. 2012, 4, 367–377. [Google Scholar] [CrossRef]
- Bollschweiler, E.; Hölscher, A.H.; Schmidt, M.; Warnecke-Eberz, U. Neoadjuvant treatment for advanced esophageal cancer: Response assessment before surgery and how to predict response to chemoradiation before starting treatment. Chin. J. Cancer Res. 2015, 27, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Sawas, T.; Katzka, D.A. Esophageal adenocarcinoma phenotypes and risk factors. Curr. Opin. Gastroenterol. 2022, 38, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wang, Z.; Li, J.; Wu, X.; Fan, N.; Li, D.; Liu, F.; Plum, P.S.; Hoppe, S.; Hillmer, A.M.; et al. Aldo-Keto Reductase 1C3 Mediates Chemotherapy Resistance in Esophageal Adenocarcinoma via ROS Detoxification. Cancers 2021, 13, 2403. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.A.; Reynolds, J.V. Visceral Obesity, Metabolic Syndrome, and Esophageal Adenocarcinoma. Front. Oncol. 2021, 11, 627270. [Google Scholar] [CrossRef]
- Arcidiacono, D.; Zaramella, A.; Fabris, F.; Sánchez-Rodríguez, R.; Nucci, D.; Fassan, M.; Nardi, M.; Benna, C.; Cristofori, C.; Morbin, T.; et al. Insulin/IGF-1 Signaling Is Downregulated in Barrett’s Esophagus Patients Undergoing a Moderate Calorie and Protein Restriction Program: A Randomized 2-Year Trial. Nutrients 2021, 13, 3638. [Google Scholar] [CrossRef]
- Doyle, S.L.; Donohoe, C.L.; Finn, S.P.; Howard, J.M.; E Lithander, F.; Reynolds, J.V.; Pidgeon, G.P.; Lysaght, J. IGF-1 and its receptor in esophageal cancer: Association with adenocarcinoma and visceral obesity. Am. J. Gastroenterol. 2012, 107, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Greer, K.B.; Kresak, A.; Bednarchik, B.; Dawson, D.; Li, L.; Chak, A.; Willis, J. Insulin/Insulin-Like Growth Factor-1 Pathway in Barrett’s Carcinogenesis. Clin. Transl. Gastroenterol. 2013, 4, e31. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, C.L.; Doyle, S.L.; McGarrigle, S.; Cathcart, M.C.; Daly, E.E.; O’Grady, A.A.; Lysaght, J.; Pidgeon, G.P.; Reynolds, J.V. Role of the insulin-like growth factor 1 axis and visceral adiposity in oesophageal adenocarcinoma. Br. J. Surg. 2012, 99, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Larsson, O.; Girnita, A.; Girnita, L. Role of insulin-like growth factor 1 receptor signalling in cancer. Br. J. Cancer 2005, 92, 2097–2101. [Google Scholar] [CrossRef] [PubMed]
- Samani, A.A.; Yakar, S.; LeRoith, D.; Brodt, P. The role of the igf system in cancer growth and metastasis: Overview and recent insights. Endocr. Rev. 2007, 28, 20–47. [Google Scholar] [CrossRef]
- Vigneri, R.; Goldfine, I.D.; Frittitta, L. Insulin, insulin receptors, and cancer. J. Endocrinol. Investig. 2016, 39, 1365–1376. [Google Scholar] [CrossRef]
- Denduluri, S.K.; Idowu, O.; Wang, Z.; Liao, Z.; Yan, Z.; Mohammed, M.K.; Ye, J.; Wei, Q.; Wang, J.; Zhao, L.; et al. Insulin-like growth factor (IGF) signaling in tumorigenesis and the development of cancer drug resistance. Genes Dis. 2015, 2, 13–25. [Google Scholar] [CrossRef]
- Singh, P.; Alex, J.M.; Bast, F. Insulin receptor (IR) and insulin-like growth factor receptor 1 (IGF-1R) signaling systems: Novel treatment strategies for cancer. Med. Oncol. 2014, 31, 805. [Google Scholar] [CrossRef] [PubMed]
- Buck, E.; Gokhale, P.C.; Koujak, S.; Brown, E.; Eyzaguirre, A.; Tao, N.; Miglarese, M.R. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): Rationale for cotargeting IGF-1R and IR in cancer. Mol. Cancer Ther. 2010, 9, 2652–2664. [Google Scholar] [CrossRef]
- LeRoith, D.; Holly, J.M.; Forbes, B.E. Insulin-like growth factors: Ligands, binding proteins, and receptors. Mol. Metab. 2021, 52, 101245. [Google Scholar] [CrossRef] [PubMed]
- Zha, J.; Lackner, M.R. Targeting the insulin-like growth factor receptor-1R pathway for cancer therapy. Clin. Cancer Res. 2010, 16, 2512–2517. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Ohashi, H.; Imsumran, A.; Yamamoto, H.; Matsunaga, Y.; Taniguchi, H.; Nosho, K.; Suzuki, H.; Sasaki, Y.; Arimura, Y.; et al. The effect of IGF-I receptor blockade for human esophageal squamous cell carcinoma and adenocarcinoma. Tumor Biol. 2014, 35, 973–985. [Google Scholar] [CrossRef]
- Jiang, D.M.; Sim, H.-W.; Espin-Garcia, O.; Chan, B.A.; Natori, A.; Lim, C.H.; Moignard, S.; Chen, E.X.; Liu, G.; Darling, G.; et al. Chemoradiotherapy Using Carboplatin plus Paclitaxel versus Cisplatin plus Fluorouracil for Esophageal or Gastroesophageal Junction Cancer. Oncology 2021, 99, 49–56. [Google Scholar] [CrossRef]
- Keresztes, R.; Port, J.; Pasmantier, M.; Korst, R.; Altorki, N. Preoperative chemotherapy for esophageal cancer with paclitaxel and carboplatin: Results of a phase II trial. J. Thorac. Cardiovasc. Surg. 2003, 126, 1603–1608. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.J. Albumin-bound paclitaxel: A next-generation taxane. Expert Opin. Pharmacother. 2006, 7, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.; Trieu, V.; Yao, Z.; Louie, L.; Ci, S.; Yang, A.; Tao, C.; De, T.; Beals, B.; Dykes, D.; et al. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin. Cancer Res. 2006, 12, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.S.; Awasthi, N.; Li, J.; Williams, F.; Schwarz, M.A.; Schwarz, R.E.; von Holzen, U. Superior Therapeutic Efficacy of Nanoparticle Albumin Bound Paclitaxel Over Cremophor-Bound Paclitaxel in Experimental Esophageal Adenocarcinoma. Transl. Oncol. 2018, 11, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Wittman, M.D.; Carboni, J.M.; Yang, Z.; Lee, F.Y.; Antman, M.; Attar, R.; Balimane, P.; Chang, C.; Chen, C.; Discenza, L.; et al. Discovery of a 2,4-Disubstituted Pyrrolo[1,2-f][1,2,4]triazine inhibitor (BMS-754807) of insulin-like growth factor receptor (IGF-1R) kinase in clinical development. J. Med. Chem. 2009, 52, 7360–7363. [Google Scholar] [CrossRef] [PubMed]
- King, E.R.; Wong, K.-K. Insulin-like growth factor: Current concepts and new developments in cancer therapy. Recent Patents Anti-Cancer Drug Discov. 2012, 7, 14–30. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, N.; Zhang, C.; Ruan, W.; Schwarz, M.A.; Schwarz, R.E. BMS-754807, a small-molecule inhibitor of insulin-like growth factor-1 receptor/insulin receptor, enhances gemcitabine response in pancreatic cancer. Mol. Cancer Ther. 2012, 11, 2644–2653. [Google Scholar] [CrossRef] [PubMed]
- Carboni, J.M.; Wittman, M.; Yang, Z.; Lee, F.; Greer, A.; Hurlburt, W.; Gottardis, M.M. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol. Cancer Ther. 2009, 8, 3341–3349. [Google Scholar] [CrossRef]
- Awasthi, N.; Scire, E.; Monahan, S.; Grojean, M.; Zhang, E.; Schwarz, M.A.; Schwarz, R.E. Augmentation of response to nab-paclitaxel by inhibition of insulin-like growth factor (IGF) signaling in preclinical pancreatic cancer models. Oncotarget 2016, 7, 46988–47001. [Google Scholar] [CrossRef]
- Huang, F.; Hurlburt, W.; Greer, A.; Reeves, K.A.; Hillerman, S.; Chang, H.; Carboni, J.M. Differential mechanisms of acquired resistance to insulin-like growth factor-i receptor antibody therapy or to a small-molecule inhibitor, BMS-754807, in a human rhabdomyosarcoma model. Cancer Res. 2010, 70, 7221–7231. [Google Scholar] [CrossRef]
- Kolb, E.A.; Gorlick, R.; Lock, R.; Carol, H.; Morton, C.L.; Keir, S.T.; Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Billups, C.; et al. Initial testing (stage 1) of the IGF-1 receptor inhibitor BMS-754807 by the pediatric preclinical testing program. Pediatr. Blood Cancer 2011, 56, 595–603. [Google Scholar] [CrossRef]
- Hassan, M.S.; Awasthi, N.; Li, J.; Schwarz, M.A.; Schwarz, R.E.; Holzen, U.V. A novel intraperitoneal metastatic xenograft mouse model for survival outcome assessment of esophageal adenocarcinoma. PLoS ONE 2017, 12, e0171824. [Google Scholar] [CrossRef]
- Hassan, M.S.; Cwidak, N.; Johnson, C.; Däster, S.; Eppenberger-Castori, S.; Awasthi, N.; von Holzen, U. Therapeutic Potential of the Cyclin-Dependent Kinase Inhibitor Flavopiridol on c-Myc Overexpressing Esophageal Cancer. Front. Pharmacol. 2021, 12, 746385. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.S.; Williams, F.; Awasthi, N.; Schwarz, M.A.; Schwarz, R.E.; Li, J.; von Holzen, U. Combination effect of lapatinib with foretinib in HER2 and MET co-activated experimental esophageal adenocarcinoma. Sci. Rep. 2019, 9, 17608. [Google Scholar] [CrossRef] [PubMed]
- Grada, A.; Otero-Viñas, M.; Prieto-Castrillo, F.P.; Obagi, Z.; Falanga, V. Research Techniques Made Simple: Analysis of Collective Cell Migration Using the Wound Healing Assay. J. Investig. Dermatol. 2017, 137, e11–e16. [Google Scholar] [CrossRef]
- Wang, P.; Gao, X.-Y.; Yang, S.-Q.; Sun, Z.-X.; Dian, L.-L.; Qasim, M.; Phyo, A.T.; Liang, Z.-S.; Sun, Y.-F. Jatrorrhizine inhibits colorectal carcinoma proliferation and metastasis through Wnt/β-catenin signaling pathway and epithelial–mesenchymal transition. Drug Des. Dev. Ther. 2019, 13, 2235–2247. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Metzinger, M.N.; Lewellen, K.A.; Cripps, S.N.; Carey, K.D.; Harper, E.I.; Stack, M.S. Obesity Contributes to Ovarian Cancer Metastatic Success through Increased Lipogenesis, Enhanced Vascularity, and Decreased Infiltration of M1 Macrophages. Cancer Res. 2015, 75, 5046–5057. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. Epidemiology of esophageal cancer. World J. Gastroenterol. 2013, 19, 5598–5606. [Google Scholar] [CrossRef]
- Prithviraj, G.K.; Baksh, K.; Fulp, W.; Meredith, K.; Hoffe, S.; Shridhar, R.; Almhanna, K. Carboplatin and paclitaxel as first-line treatment of unresectable or metastatic esophageal or gastric cancer. Dis. Esophagus 2015, 28, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef]
- Tan, C.; Qian, X.; Guan, Z.; Yang, B.; Ge, Y.; Wang, F.; Cai, J. Potential biomarkers for esophageal cancer. SpringerPlus 2016, 5, 467. [Google Scholar] [CrossRef]
- Fatehi Hassanabad, A.; Chehade, R.; Breadner, D.; Raphael, J. Esophageal carcinoma: Towards targeted therapies. Cell Oncol. 2020, 43, 195–209. [Google Scholar] [CrossRef]
- Hassan, M.S.; Makuru, V.; von Holzen, U. Targeted therapy in esophageal cancer. Dig. Med. Res. 2021, 4, 29. [Google Scholar] [CrossRef]
- Nagaraja, V.; Shaw, N.; Morey, A.L.; Cox, M.R.; Eslick, G.D. HER2 expression in oesophageal carcinoma and Barrett’s oesophagus associated adenocarcinoma: An Australian study. Eur. J. Surg. Oncol. 2016, 42, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.-S.; Jiang, L.-P.; He, C.-Y.; Tong, Y.-N.; Liu, Y.-Y. Up-Regulation of MicroRNA-133a Inhibits the MEK/ERK Signaling Pathway to Promote Cell Apoptosis and Enhance Radio-Sensitivity by Targeting EGFR in Esophageal Cancer In Vivo and In Vitro. J. Cell. Biochem. 2017, 118, 2625–2634. [Google Scholar] [CrossRef] [PubMed]
- Ladeira, K.; Macedo, F.; Longatto-Filho, A.; Martins, S.F. Angiogenic factors: Role in esophageal cancer, a brief review. Esophagus 2018, 15, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.D.; Grabsch, H.I.; Mauer, M.; Marreaud, S.; Caballero, C.; Thuss-Patience, P.; Mueller, L.; Elme, A.; Moehler, M.H.; Martens, U.; et al. EORTC-1203-GITCG—The “INNOVATION”-trial: Effect of chemotherapy alone versus chemotherapy plus trastuzumab, versus chemotherapy plus trastuzumab plus pertuzumab, in the perioperative treatment of HER2 positive, gastric and gastroesophageal junction adenocarcinoma on pathologic response rate: A randomized phase II-intergroup trial of the EORTC-Gastrointestinal Tract Cancer Group, Korean Cancer Study Group and Dutch Upper GI-Cancer group. BMC Cancer 2019, 19, 494. [Google Scholar] [CrossRef]
- Gerson, J.N.; Skariah, S.; Denlinger, C.S.; Astsaturov, I. Perspectives of HER2-targeting in gastric and esophageal cancer. Expert Opin. Investig. Drugs 2017, 26, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.; Adachi, Y.; Yamamoto, H.; Imsumran, A.; Arimura, Y.; Endo, T.; Hinoda, Y.; Lee, C.-T.; Nadaf, S.; Carbone, D.P.; et al. Insulin-like growth factor I receptor blockade enhances chemotherapy and radiation responses and inhibits tumour growth in human gastric cancer xenografts. Gut 2005, 54, 591–600. [Google Scholar] [CrossRef]
- Imsumran, A.; Adachi, Y.; Yamamoto, H.; Li, R.; Wang, Y.; Min, Y.; Piao, W.; Nosho, K.; Arimura, Y.; Shinomura, Y.; et al. Insulin-like growth factor-I receptor as a marker for prognosis and a therapeutic target in human esophageal squamous cell carcinoma. Carcinogenesis 2007, 28, 947–956. [Google Scholar] [CrossRef]
- Arcidiacono, D.; Dedja, A.; Giacometti, C.; Fassan, M.; Nucci, D.; Francia, S.; Fabris, F.; Zaramella, A.; Gallagher, E.J.; Cassaro, M.; et al. Hyperinsulinemia Promotes Esophageal Cancer Development in a Surgically-Induced Duodeno-Esophageal Reflux Murine Model. Int. J. Mol. Sci. 2018, 19, 1198. [Google Scholar] [CrossRef]
- Simpson, A.; Petnga, W.; Macaulay, V.M.; Weyer-Czernilofsky, U.; Bogenrieder, T. Insulin-Like Growth Factor (IGF) Pathway Targeting in Cancer: Role of the IGF Axis and Opportunities for Future Combination Studies. Target. Oncol. 2017, 12, 571–597. [Google Scholar] [CrossRef]
- LeRoith, D.; Werner, H.; Beitner-Johnson, D. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr. Rev. 1995, 16, 143–163. [Google Scholar] [CrossRef]
- Sciacca, L.; Costantino, A.; Pandini, G.; Mineo, R.; Frasca, F.; Scalia, P.; Sbraccia, P.; Goldfine, I.D.; Vigneri, R.; Belfiore, A. Insulin receptor activation by IGF-II in breast cancers: Evidence for a new autocrine/paracrine mechanism. Oncogene 1999, 18, 2471–2479. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Capanu, M.; O’reilly, E.M.; Ma, J.; Chou, J.F.; Gansukh, B.; Shia, J.; Kalin, M.; Katz, S.; Abad, L.; et al. A phase II study of cixutumumab (IMC-A12, NSC742460) in advanced hepatocellular carcinoma. J. Hepatol. 2014, 60, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Langer, C.J.; Novello, S.; Park, K.; Krzakowski, M.; Karp, D.D.; Mok, T.; Benner, R.J.; Scranton, J.R.; Olszanski, A.J.; Jassem, J. randomized, phase III trial of first-line figitumumab in combination with paclitaxel and carboplatin versus paclitaxel and carboplatin alone in patients with advanced non–small-cell lung cancer. J. Clin. Oncol. 2014, 32, 2059–2066. [Google Scholar] [CrossRef]
- Frasca, F.; Pandini, G.; Scalia, P.; Sciacca, L.; Mineo, R.; Costantino, A.; Goldfine, I.D.; Belfiore, A.; Vigneri, R. Insulin Receptor Isoform A, a Newly Recognized, High-Affinity Insulin-Like Growth Factor II Receptor in Fetal and Cancer Cells. Mol. Cell. Biol. 1999, 19, 3278–3288. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Huang, F.; Macedo, L.F.; Harrington, S.C.; Reeves, K.A.; Greer, A.; Haluska, P. Dual IGF-1R/InsR inhibitor BMS-754807 synergizes with hormonal agents in treatment of estrogen-dependent breast cancer. Cancer Res. 2011, 71, 7597–7607. [Google Scholar] [CrossRef] [PubMed]
- Dayyani, F.; Parikh, N.U.; Varkaris, A.S.; Song, J.H.; Moorthy, S.; Chatterji, T.; Maity, S.N.; Wolfe, A.R.; Carboni, J.M.; Gottardis, M.M.; et al. Combined inhibition of IGF-1R/IR and Src family kinases enhances antitumor effects in prostate cancer by decreasing activated survival pathways. PLoS ONE 2012, 7, e51189. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Rubin, R.; Brodt, P. Enhanced invasion and liver colonization by lung carcinoma cells overexpressing the type 1 Insulin-like growth factor receptor. Exp. Cell Res. 1998, 238, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Saikali, Z.; Setya, H.; Singh, G.; Persad, S. Role of IGF-1/IGF-1R in regulation of invasion in DU145 prostate cancer cells. Cancer Cell Int. 2008, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.S.; Awasthi, N.; Ponna, S.; von Holzen, U. Nab-Paclitaxel in the Treatment of Gastrointestinal Cancers-Improvements in Clinical Efficacy and Safety. Biomedicines 2023, 11, 2000. [Google Scholar] [CrossRef] [PubMed]
- Wiedmann, M.; Mössner, J. New and emerging combination therapies for esophageal cancer. Cancer Manag. Res. 2013, 5, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- O’neill, B.T.; Lauritzen, H.P.; Hirshman, M.F.; Smyth, G.; Goodyear, L.J.; Kahn, C.R. Differential Role of Insulin/IGF-1 Receptor Signaling in Muscle Growth and Glucose Homeostasis. Cell Rep. 2015, 11, 1220–1235. [Google Scholar] [CrossRef] [PubMed]
- Clemmons, D.R. Role of insulin-like growth factor iin maintaining normal glucose homeostasis. Horm. Res. Paediatr. 2004, 62 (Suppl. S1), 77–82. [Google Scholar] [CrossRef]
- Desai, J.; Solomon, B.J.; Davis, I.D.; Lipton, L.R.; Hicks, R.; Scott, A.M.; Park, J.; Clemens, P.L.; Gestone, T.A.; Finckenstein, F.G. Phase I dose-escalation study of daily BMS-754807, an oral, dual IGF-1R/insulin receptor (IR) inhibitor in subjects with solid tumors. J. Clin. Oncol. 2010, 28 (Suppl. S15), 3104. [Google Scholar] [CrossRef]
- Tamura, Y.; Nokihara, H.; Yamamoto, N.; Wakui, H.; Honda, K.; Asahina, H.; Tamura, T. O1-079—Phase 1 Dose-Escalating Study of BMS-754807 in Japanese Patients with Advanced Solid Tumors. Ann. Oncol. 2012, 23, xi111–xi112. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, M.S.; Johnson, C.; Ponna, S.; Scofield, D.; Awasthi, N.; von Holzen, U. Inhibition of Insulin-like Growth Factor 1 Receptor/Insulin Receptor Signaling by Small-Molecule Inhibitor BMS-754807 Leads to Improved Survival in Experimental Esophageal Adenocarcinoma. Cancers 2024, 16, 3175. https://doi.org/10.3390/cancers16183175
Hassan MS, Johnson C, Ponna S, Scofield D, Awasthi N, von Holzen U. Inhibition of Insulin-like Growth Factor 1 Receptor/Insulin Receptor Signaling by Small-Molecule Inhibitor BMS-754807 Leads to Improved Survival in Experimental Esophageal Adenocarcinoma. Cancers. 2024; 16(18):3175. https://doi.org/10.3390/cancers16183175
Chicago/Turabian StyleHassan, Md Sazzad, Chloe Johnson, Saisantosh Ponna, Dimitri Scofield, Niranjan Awasthi, and Urs von Holzen. 2024. "Inhibition of Insulin-like Growth Factor 1 Receptor/Insulin Receptor Signaling by Small-Molecule Inhibitor BMS-754807 Leads to Improved Survival in Experimental Esophageal Adenocarcinoma" Cancers 16, no. 18: 3175. https://doi.org/10.3390/cancers16183175
APA StyleHassan, M. S., Johnson, C., Ponna, S., Scofield, D., Awasthi, N., & von Holzen, U. (2024). Inhibition of Insulin-like Growth Factor 1 Receptor/Insulin Receptor Signaling by Small-Molecule Inhibitor BMS-754807 Leads to Improved Survival in Experimental Esophageal Adenocarcinoma. Cancers, 16(18), 3175. https://doi.org/10.3390/cancers16183175