
The Molecular Mechanisms of Portal Vein Thrombosis in Hepatocellular Carcinoma

 , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
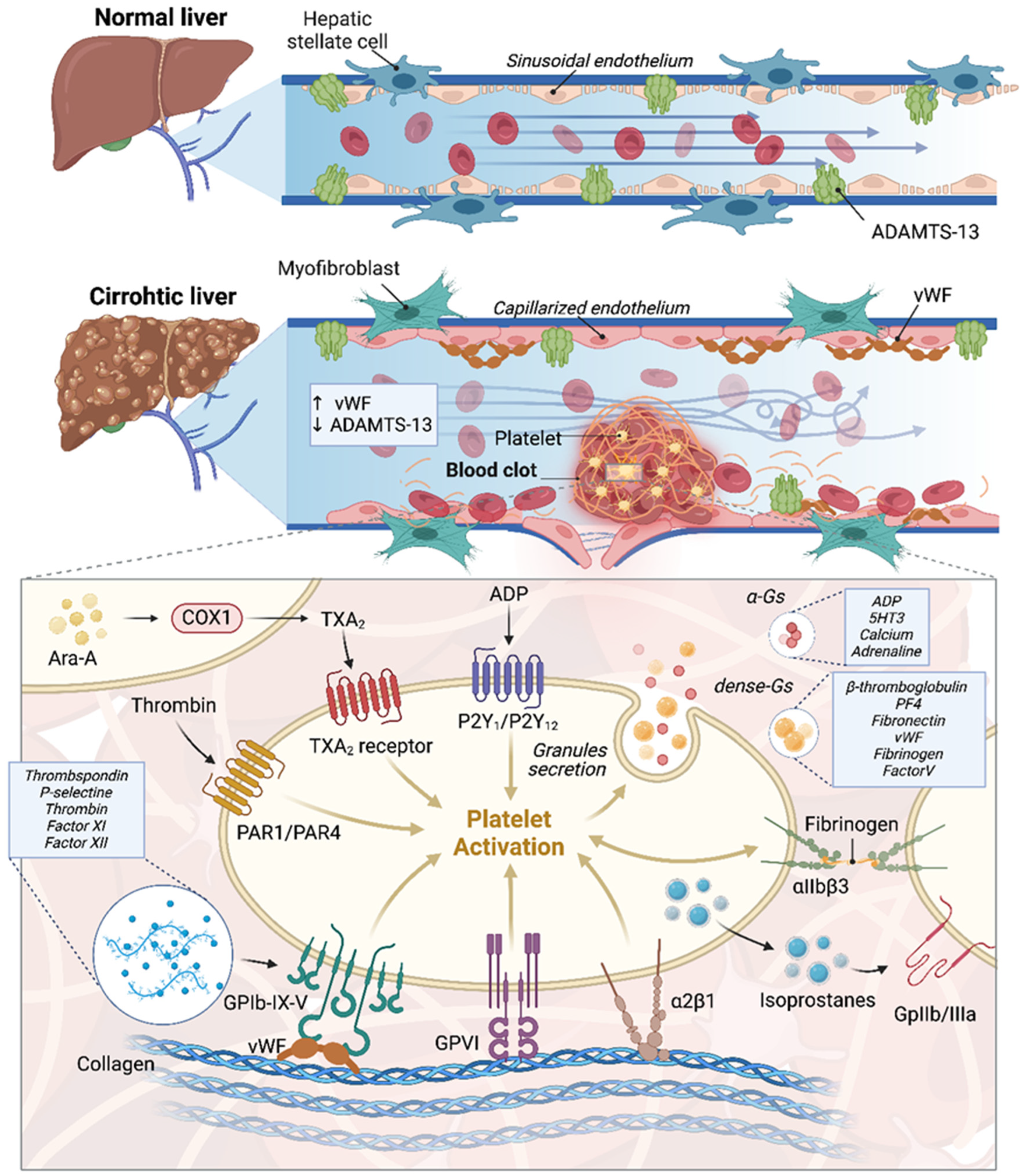
2. Endothelial Dysfunction and Portal Vein Thrombosis in Cirrhotic Patients with a Focus on HCC: Molecular Mechanisms and Pathophysiology
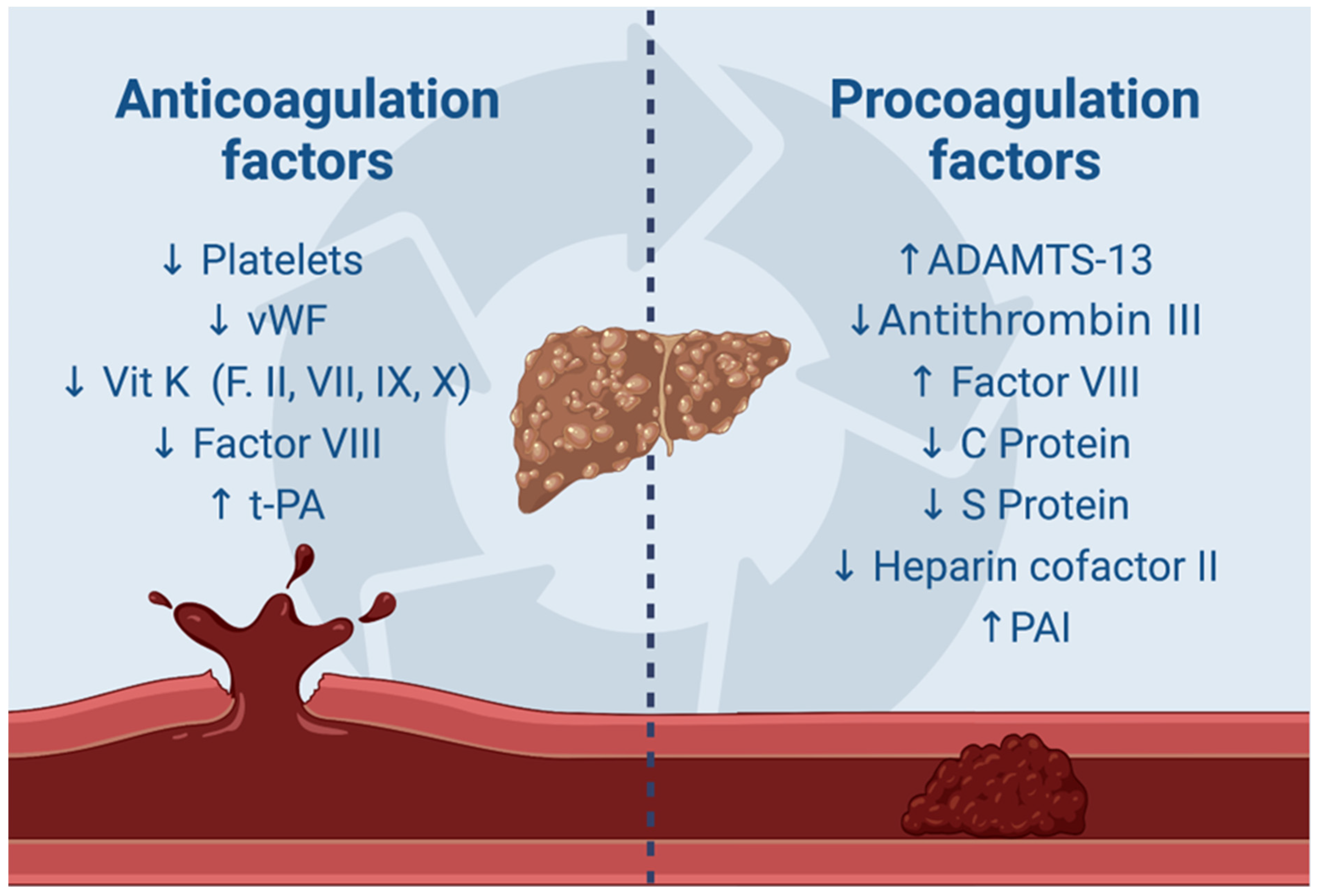
3. Molecular Insights into Coagulopathy and Thrombosis Mechanisms in Cirrhotic Patients with a Focus on HCC
4. Molecular Mechanisms of Blood Stasis and Thrombosis in Portal Vein Thrombosis in Cirrhosis
5. Gut–Liver Axis and Portal Vein Thrombosis in Cirrhotic and HCC Livers: The Role of Endotoxemia
6. Molecular Mechanisms of Microvesicles in Portal Vein Thrombosis in Cirrhotic Patients
7. A Direct Role of HCC Cells in Portal Vein Thrombosis: Molecular Mechanisms
8. The Role of Thrombophilia in the Genesis of Portal Vein Thrombosis
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ageno, W.; Dentali, F.; Pomero, F.; Fenoglio, L.; Squizzato, A.; Pagani, G.; Re, R.; Bonzini, M. Incidence rates and case fatality rates of portal vein thrombosis and Budd-Chiari Syndrome. Thromb. Haemost. 2017, 117, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Koumar, L.; Senthamizhselvan, K.; Barathi, D.; Verma, A.; Rao, P.; Selvaraj, J.; Sanker, V. Portal Vein Thrombosis in Patients With Cirrhosis of the Liver: Prevalence and Risk Factors. Cureus 2023, 15, e50134. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Okuda, K.; Ohnishi, K.; Kimura, K.; Matsutani, S.; Sumida, M.; Goto, N.; Musha, H.; Takashi, M.; Suzuki, N.; Shinagawa, T. Incidence of portal vein thrombosis in liver cirrhosis. An angiographic study in 708 patients. Gastroenterology 1985, 89, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Francoz, C.; Belghiti, J.; Vilgrain, V.; Sommacale, D.; Paradis, V.; Condat, B.; Denninger, M.H.; Sauvanet, A.; Valla, D.; Durand, F. Splanchnic vein thrombosis in candidates for liver transplantation: Usefulness of screening and anticoagulation. Gut 2005, 54, 691–697. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cerrito, L.; Annicchiarico, B.E.; Iezzi, R.; Gasbarrini, A.; Pompili, M.; Ponziani, F.R. Treatment of hepatocellular carcinoma in patients with portal vein tumor thrombosis: Beyond the known frontiers. World J. Gastroenterol. 2019, 25, 4360–4382. [Google Scholar] [CrossRef]
- Pan, J.; Wang, L.; Gao, F.; An, Y.; Yin, Y.; Guo, X.; Nery, F.G.; Yoshida, E.M.; Qi, X. Epidemiology of portal vein thrombosis in liver cirrhosis: A systematic review and meta-analysis. Eur. J. Intern. Med. 2022, 104, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Ponziani, F.R.; Zocco, M.A.; Garcovich, M.; D’Aversa, F.; Roccarina, D.; Gasbarrini, A. What we should know about portal vein thrombosis in cirrhotic patients: A changing perspective. World J. Gastroenterol. 2012, 28, 5014–5020. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Raja, K.; Jacob, M.; Asthana, S. Portal vein thrombosis in cirrhosis. J. Clin. Exp. Hepatol. 2014, 4, 320–331. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gavriilidis, P.; Pawlik, T.M.; Azoulay, D. Comprehensive review of hepatocellular carcinoma with portal vein tumor thrombus: State of art and future perspectives. Hepatobiliary Pancreat. Dis. Int. 2024, 23, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.A.; Ferry, D.R.; El-Gazzaz, G.; Mirza, D.F.; James, N.D.; McMaster, P.; Kerr, D.J. Hepatocellular carcinoma. Ann. Oncol. 2001, 12, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Quirk, M.; Kim, Y.H.; Saab, S.; Lee, E.W. Management of hepatocellular carcinoma with portal vein thrombosis. World J. Gastro-Enterol. 2015, 28, 3462–3471. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Quencer, K.B.; Friedman, T.; Sheth, R.; Oklu, R. Tumor thrombus: Incidence, imaging, prognosis and treatment. Cardiovasc. Diagn. Ther. 2017, 7 (Suppl. S3), S165–S177. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, L.; Chen, S.; Cao, H.; Feng, Z.; Yang, C. Efficacy of sorafenib plus transcatheter arterial chemoembolization in treating hepatocellular carcinoma with portal vein tumor thrombosis: A meta-analysis. Acta Pharm. 2024, 74, 405–422. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Chen, L.; Lei, Y.; Zhang, L.; Sun, T.; Liu, Y.; Zheng, C. Sorafenib plus transcatheter arterial chemoembolization with or without camrelizumab for the treatment of intermediate and advanced hepatocellular carcinoma. Br. J. Radiol. 2024, 97, 1320–1327. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sherman, C.B.; Behr, S.; Dodge, J.L.; Roberts, J.P.; Yao, F.Y.; Mehta, N. Distinguishing Tumor From Bland Portal Vein Thrombus in Liver Transplant Candidates With Hepatocellular Carcinoma: The A-VENA Criteria. Liver Transpl. 2019, 25, 207–216. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rossi, S.; Ghittoni, G.; Ravetta, V.; Torello Viera, F.; Rosa, L.; Serassi, M.; Scabini, M.; Vercelli, A.; Tinelli, C.; Dal Bello, B.; et al. Contrast-enhanced ultrasonography and spiral computed tomography in the detection and characterization of portal vein thrombosis complicating hepatocellular carcinoma. Eur. Radiol. 2008, 18, 1749–1756. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, Y.; Groszmann, R.J. Vascular endothelial dysfunction in cirrhosis. J. Hepatol. 2007, 46, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Premont, R.T.; Kontos, C.D.; Huang, J.; Rockey, D.C. Endothelin-1 activates endothelial cell nitric-oxide synthase via het-erotrimeric G-protein betagamma subunit signaling to protein jinase B/Akt. J. Biol. Chem. 2003, 278, 49929–49935. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Okanoue, T.; Sawa, Y.; Hori, N.; Ohta, M.; Kagawa, K. Defenestration of the sinusoidal endothelial cell in a rat model of cirrhosis. Hepatology 1993, 17, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Bhunchet, E.; Fujieda, K. Capillarization and venularization of hepatic sinusoids in porcine serum-induced rat liver fibrosis: A mechanism to maintain liver blood flow. Hepatology 1993, 18, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Premont, R.T.; Kontos, C.D.; Zhu, S.; Rockey, D.C. A crucial role for GRK2 in regulation of endothelial cell nitric oxide synthase function in portal hypertension. Nat. Med. 2005, 11, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Karaa, A.; Kamoun, W.S.; Xu, H.; Zhang, J.; Clemens, M.G. Differential effects of oxidative stress on hepatic endothelial and Kupffer cell eicosanoid release in response to endothelin-1. Microcirculation 2006, 13, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Abraldes, J.G.; Iwakiri, Y.; Loureiro-Silva, M.; Haq, O.; Sessa, W.C.; Groszmann, R.J. Mild increases in portal pressure upregulate vascular endothelial growth factor and endothelial nitric oxide synthase in the intestinal microcirculatory bed, leading to a hyperdynamic state. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.; Mejias, M.; Angermayr, B.; Garcia-Pagan, J.C.; Rodés, J.; Bosch, J. Inhibition of VEGF receptor-2 decreases the de-velopment of hyperdynamic splanchnic circulation and portal-systemic collateral vessels in portal hypertensive rats. J. Hepatol. 2005, 43, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Graupera, M.; García-Pagán, J.C.; Abraldes, J.G.; Peralta, C.; Bragulat, M.; Corominola, H.; Bosch, J.; Rodés, J. Cyclooxygenase-derived products modulate the increased intrahepatic resistance of cirrhotic rat livers. Hepatology 2003, 37, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Furuta, K.; Islam, S.; Caporarello, N.; Kostallari, E.; Dielis, K.; Tschumperlin, D.J.; Hirsova, P.; Ibrahim, S.H. Liver sinusoidal endothelial cell expressed vascular cell adhesion molecule 1 promotes liver fibrosis. Front. Immunol. 2022, 13, 983255. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wei, Y.; Chen, X.; Shen, H.; Wu, W.; Cao, G.; Chen, W.; Wang, Y.; Shen, H.; Yu, S.; Zhang, J. P-Selectin Level at First and Third Day After Portal Hypertensive Splenectomy for Early Prediction of Portal Vein Thrombosis in Patients With Cirrhosis. Clin. Appl. Thromb. Hemost. 2018, 24, 76S–83S. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Purdy, M.; Obi, A.; Myers, D.; Wakefield, T. P- and E- selectin in venous thrombosis and non-venous pathologies. J. Thromb. Haemost. 2022, 20, 1056–1066. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhou, X.H.; Li, J.R.; Zheng, T.H.; Chen, H.; Cai, C.; Ye, S.L.; Gao, B.; Xue, T.C. Portal vein tumor thrombosis in hepatocellular carcinoma: Molecular mechanism and therapy. Clin. Exp. Metastasis 2023, 40, 5–32. [Google Scholar] [CrossRef] [PubMed]
- Pilard, M.; Ollivier, E.L.; Gourdou-Latyszenok, V.; Couturaud, F.; Lemarié, C.A. Endothelial Cell Phenotype, a Major Determinant of Venous Thrombo-Inflammation. Front. Cardiovasc. Med. 2022, 9, 864735. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Laridan, E.; Martinod, K.; De Meyer, S.F. Neutrophil Extracellular Traps in Arterial and Venous Thrombosis. Semin. Thromb. Hemost. 2019, 45, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Xu, S.; Zhang, Y.; Wang, L.; Yan, C.; Xu, Z.; Zhao, Q.; Qi, X. Neutrophil extracellular traps formation may be involved in the association of propranolol with the development of portal vein thrombosis. Thromb. Res. 2024, 238, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 18, 1952–1961. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vogel, S.; Bodenstein, R.; Chen, Q.; Feil, S.; Feil, R.; Rheinlaender, J.; Schäffer, T.E.; Bohn, E.; Frick, J.S.; Borst, O.; et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J. Clin. Investig. 2015, 125, 4638–4654. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pieters, M.; Wolberg, A.S. Fibrinogen and fibrin: An illustrated review. Res. Pract. Thromb. Haemost. 2019, 3, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Tadie, J.M.; Bae, H.B.; Jiang, S.; Park, D.W.; Bell, C.P.; Yang, H.; Pittet, J.F.; Tracey, K.; Thannickal, V.J.; Abraham, E.; et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2013, 1, 342–349. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Puricelli, C.; Boggio, E.; Gigliotti, C.L.; Stoppa, I.; Sutti, S.; Giordano, M.; Dianzani, U.; Rolla, R. Platelets, Protean Cells with All-Around Functions and Multifaceted Pharmacological Applications. Int. J. Mol. Sci. 2023, 26, 4565. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kim, S.W.; Lee, J.K. Role of HMGB1 in the Interplay between NETosis and Thrombosis in Ischemic Stroke: A Review. Cells 2020, 28, 1794. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Galasso, L.; Cerrito, L.; Maccauro, V.; Termite, F.; Ainora, M.E.; Gasbarrini, A.; Zocco, M.A. Hepatocellular Carcinoma and the Mul-tifaceted Relationship with Its Microenvironment: Attacking the Hepatocellular Carcinoma Defensive Fortress. Cancers 2024, 11, 1837. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, Z.; Zhao, M.; Qi, X.; Tang, Y.; Cheng, S. Mechanisms of portal vein tumour thrombus formation and development in patients with hepatocellular carcinoma. J. Cell. Mol. Med. 2023, 27, 2103–2111. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wen, W.; Ding, J.; Sun, W.; Fu, J.; Chen, Y.; Wu, K.; Ning, B.; Han, T.; Huang, L.; Chen, C.; et al. Cyclin G1-mediated epithelial-mesenchymal transition via phosphoinositide 3-kinase/Akt signaling facilitates liver cancer progression. Hepatology 2012, 55, 1787–1798. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.D.; Mucha, S.R.; Lindenmeyer, C.C. Cirrhotic coagulopathy: A rebalanced hemostasis. Clevel. Clin. J. Med. 2022, 1, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Scridon, A. Platelets and Their Role in Hemostasis and Thrombosis-From Physiology to Pathophysiology and Therapeutic Implications. Int. J. Mol. Sci. 2022, 23, 12772. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wallace, E.L.; Smyth, S.S. Targeting platelet thrombin receptor signaling to prevent thrombosis. Pharmaceuticals 2013, 2, 915–928. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Saad, J.; Asuka, E.; Schoenberger, L. Physiology, Platelet Activation. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar] [PubMed]
- Zou, J.; Swieringa, F.; de Laat, B.; de Groot, P.G.; Roest, M.; Heemskerk, J.W.M. Reversible Platelet Integrin αIIbβ3 Activation and Thrombus Instability. Int. J. Mol. Sci. 2022, 19, 12512. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sigal, S.H.; Sherman, Z.; Jesudian, A. Clinical Implications of Thrombocytopenia for the Cirrhotic Patient. Hepatic Med. Evid. Res. 2020, 14, 49–60. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ramadori, P.; Klag, T.; Malek, N.P.; Heikenwalder, M. Platelets in chronic liver disease, from bench to bedside. JHEP Rep. 2019, 25, 448–459. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Suzuki, J.; Namisaki, T.; Takya, H.; Kaji, K.; Nishimura, N.; Shibamoto, A.; Asada, S.; Kubo, T.; Iwai, S.; Tomooka, F.; et al. ADAMTS-13: A Prognostic Biomarker for Portal Vein Thrombosis in Japanese Patients with Liver Cirrhosis. Int. J. Mol. Sci. 2024, 26, 2678. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pan, J.; Dinh, T.T.; Rajaraman, A.; Lee, M.; Scholz, A.; Czupalla, C.J.; Kiefel, H.; Zhu, L.; Xia, L.; Morser, J.; et al. Patterns of expression of factor VIII and von Willebrand factor by endothelial cell subsets in vivo. Blood 2016, 7, 104–109. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lopes da Silva, M.; Cutler, D.F. von Willebrand factor multimerization and the polarity of secretory pathways in endothelial cells. Blood 2016, 14, 277–285. [Google Scholar] [CrossRef]
- Simbrunner, B.; Villesen, I.F.; Scheiner, B.; Paternostro, R.; Schwabl, P.; Stättermayer, A.F.; Marculescu, R.; Pinter, M.; Quehenberger, P.; Trauner, M.; et al. Von Willebrand factor processing in patients with advanced chronic liver disease and its relation to portal hypertension and clinical outcome. Hepatol. Int. 2023, 17, 1532–1544. [Google Scholar] [CrossRef] [PubMed]
- Koch, P.S.; Lee, K.H.; Goerdt, S.; Augustin, H.G. Angiodiversity and organotypic functions of sinusoidal endothelial cells. Angiogenesis 2021, 24, 289–310. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Khomich, O.; Ivanov, A.V.; Bartosch, B. Metabolic Hallmarks of Hepatic Stellate Cells in Liver Fibrosis. Cells 2019, 20, 24. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Islam, R.; Kundu, S.; Jha, S.B.; Rivera, A.P.; Flores Monar, G.V.; Islam, H.; Puttagunta, S.M.; Sange, I. Cirrhosis and Coagulopathy: Mechanisms of Hemostasis Changes in Liver Failure and Their Management. Cureus 2022, 3, e23785. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Senzolo, M.; Burra, P.; Cholongitas, E.; Burroughs, A.K. New insights into the coagulopathy of liver disease and liver transplan-tation. World J. Gastroenterol. 2006, 28, 7725–7736. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jiang, S.; Ai, Y.; Fan, X.; Huang, X.; Wu, L.; Ni, L.; Li, F.; Chen, S. Increased Factor VIII Activity Is Predictive of the Occurrence of Portal Vein Thrombosis in Cirrhosis. Thromb. Haemost. 2023, 123, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, A.; Primignani, M.; Lemma, L.; Chantarangkul, V.; Mannucci, P.M. Evidence that low protein C contributes to the pro-coagulant imbalance in cirrhosis. J. Hepatol. 2013, 59, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Hao, J.; Yang, N. Protein C and D-dimer are related to portal vein thrombosis in patients with liver cirrhosis. J. Gas-Troenterol. Hepatol. 2010, 25, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Zeb, F.; Ullah, H.; Harikrishna, A.; Khalaf, W.; Salih, N.; Waheed, A.; Amin, R.U. Portal Vein Thrombosis (PVT) Secondary to Protein C Deficiency in a Young Male. Cureus 2023, 29, e49688. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hung, H.C.; Lee, J.C.; Cheng, C.H.; Wang, Y.C.; Wu, T.H.; Lee, C.F.; Wu, T.J.; Chou, H.S.; Chan, K.M.; Lee, W.C. Protein S for Portal Vein Thrombosis in Cirrhotic Patients Waiting for Liver Transplantation. J. Clin. Med. 2020, 20, 1181. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, D.L.; Hao, J.Y.; Yang, N. Value of D-dimer and protein S for diagnosis of portal vein thrombosis in patients with liver cirrhosis. J. Int. Med. Res. 2013, 41, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, A.; Salerno, F.; Chantarangkul, V.; Clerici, M.; Cazzaniga, M.; Primignani, M.; Mannuccio Mannucci, P. Evidence of normal thrombin generation in cirrhosis despite abnormal conventional coagulation tests. Hepatology 2005, 41, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Sinegre, T.; Duron, C.; Lecompte, T.; Pereira, B.; Massoulier, S.; Lamblin, G.; Abergel, A.; Lebreton, A. Increased factor VIII plays a significant role in plasma hypercoagulability phenotype of patients with cirrhosis. J. Thromb. Haemost. 2018, 16, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Depasse, F.; Binder, N.B.; Mueller, J.; Wissel, T.; Schwers, S.; Germer, M.; Hermes, B.; Turecek, P.L. Thrombin generation assays are versatile tools in blood coagulation analysis: A review of technical features, and applications from research to laboratory routine. J. Thromb. Haemost. 2021, 19, 2907–2917. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zanetto, A.; Campello, E.; Bulato, C.; Willems, R.; Konings, J.; Roest, M.; Gavasso, S.; Nuozzi, G.; Toffanin, S.; Zanaga, P.; et al. Whole blood thrombin generation shows a significant hypocoagulable state in patients with decompensated cirrhosis. J. Thromb. Haemost. 2024, 22, 480–492. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Roberts, L.N.; Hendrix, W.; Konings, J.; Ow, T.W.; Rabinowich, L.; Barbouti, O.; de Laat, B.; Arya, R.; Patel, V.C.; et al. Whole blood thrombin generation profiles of patients with cirrhosis explored with a near patient assay. J. Thromb. Haemost. 2020, 18, 834–843. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Amiral, J.; Seghatchian, J. Revisiting the activated protein C-protein S-thrombomodulin ternary pathway: Impact of new un-derstanding on its laboratory investigation. Transfus. Apher. Sci. 2019, 58, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Katz, J.M.; Tadi, P. Physiology, Plasminogen Activation. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar] [PubMed]
- Pavlovic, N.; Rani, B.; Gerwins, P.; Heindryckx, F. Platelets as Key Factors in Hepatocellular Carcinoma. Cancers 2019, 20, 1022. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zanetto, A.; Senzolo, M.; Campello, E.; Bulato, C.; Gavasso, S.; Shalaby, S.; Gambato, M.; Vitale, A.; Cillo, U.; Farinati, F.; et al. Influence of Hepatocellular Carcinoma on Platelet Aggregation in Cirrhosis. Cancers 2021, 8, 1150. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhu, W.L.; Fan, B.L.; Liu, D.L.; Zhu, W.X. Abnormal expression of fibrinogen gamma (FGG) and plasma level of fibrinogen in patients with hepatocellular carcinoma. Anticancer Res. 2009, 29, 2531–2534. [Google Scholar]
- Zanetto, A.; Senzolo, M.; Vitale, A.; Cillo, U.; Radu, C.; Sartorello, F.; Spiezia, L.; Campello, E.; Rodriguez-Castro, K.; Ferrarese, A.; et al. Thromboelastometry hypercoagulable profiles and portal vein thrombosis in cirrhotic patients with hepatocellular carcinoma. Dig. Liver Dis. 2017, 49, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Zanetto, A.; Campello, E.; Bulato, C.; Gavasso, S.; Saggiorato, G.; Shalaby, S.; Spiezia, L.; Cillo, U.; Farinati, F.; Russo, F.P.; et al. More pronounced hyper-coagulable state and hypofibrinolysis in patients with cirrhosis with versus without HCC. Hepatol. Commun. 2021, 5, 1987–2000. [Google Scholar] [CrossRef] [PubMed]
- Poon, R.T.; Lau, C.P.; Ho, J.W.; Yu, W.C.; Fan, S.T.; Wong, J. Tissue factor expression correlates with tumor angiogenesis and invasiveness in human hepatocellular carcinoma. Clin. Cancer Res. 2003, 9, 5339–5345. [Google Scholar] [PubMed]
- Huang, S.Z.; Wei, M.N.; Huang, J.R.; Zhang, Z.J.; Zhang, W.J.; Jiang, Q.W.; Yang, Y.; Wang, H.Y.; Jin, H.L.; Wang, K.; et al. Targeting TF-AKT/ERK-EGFR Pathway Suppresses the Growth of Hepatocellular Carcinoma. Front. Oncol. 2019, 15, 150. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ahmadi, S.E.; Shabannezhad, A.; Kahrizi, A.; Akbar, A.; Safdari, S.M.; Hoseinnezhad, T.; Zahedi, M.; Sadeghi, S.; Mojarrad, M.G.; Safa, M. Tissue factor (coagulation factor III): A potential double-edge molecule to be targeted and re-targeted toward cancer. Biomark. Res. 2023, 6, 60. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zuñiga-Aguilar, E.; Ramírez-Fernández, O. Fibrosis and hepatic regeneration mechanism. Transl. Gastroenterol. Hepatol. 2022, 25, 9. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Genovesi, S.; Giussani, M.; Orlando, A.; Lieti, G.; Viazzi, F.; Parati, G. Relationship between endothelin and nitric oxide pathways in the onset and maintenance of hypertension in children and adolescents. Pediatr. Nephrol. 2022, 37, 537–545. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bosch, J.; Abraldes, J.G.; Fernández, M.; García-Pagán, J.C. Hepatic endothelial dysfunction and abnormal angiogenesis: New targets in the treatment of portal hypertension. J. Hepatol. 2010, 53, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Abhinand, C.S.; Raju, R.; Soumya, S.J.; Arya, P.S.; Sudhakaran, P.R. VEGF-A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. J. Cell Commun. Signal. 2016, 10, 347–354. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Martell, M.; Coll, M.; Ezkurdia, N.; Raurell, I.; Genescà, J. Physiopathology of splanchnic vasodilation in portal hypertension. World J. Hepatol. 2010, 27, 208–220. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Najafi, M.; Farhood, B.; Mortezaee, K. Extracellular matrix (ECM) stiffness and degradation as cancer drivers. J. Cell. Biochem. 2019, 120, 2782–2790. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Yang, S.; Wu, C.; Wang, Y.; Jiang, J.; Lu, Z. Protein expression of hypoxia-inducible factor-1 alpha and hepatocellular car-cinoma: A systematic review with meta-analysis. Clin. Res. Hepatol. Gastroenterol. 2014, 38, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Huang, X.Q.; Zhu, Y.L.; Ding, H.; Li, F.; Chen, S.Y. Increased portal vein diameter is predictive of portal vein thrombosis development in patients with liver cirrhosis. Ann. Transl. Med. 2021, 9, 289. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stine, J.G.; Wang, J.; Shah, P.M.; Argo, C.K.; Intagliata, N.; Uflacker, A.; Caldwell, S.H.; Northup, P.G. Decreased portal vein velocity is predictive of the development of portal vein thrombosis: A matched case-control study. Liver Int. 2018, 38, 94–101. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zocco, M.A.; Di Stasio, E.; De Cristofaro, R.; Novi, M.; Ainora, M.E.; Ponziani, F.; Riccardi, L.; Lancellotti, S.; Santoliquido, A.; Flore, R.; et al. Thrombotic risk factors in patients with liver cirrhosis: Correlation with MELD scoring system and portal vein thrombosis development. J. Hepatol. 2009, 51, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, S.G.; Mendoza, Y.P.; Bosch, J. Beta-blockers in cirrhosis: Evidence-based indications and limitations. JHEP Rep. 2019, 2, 100063. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, X.; Guo, X.; De Stefano, V.; Silva-Junior, G.; Goyal, H.; Bai, Z.; Zhao, Q.; Qi, X. Nonselective beta-blockers and development of portal vein thrombosis in liver cirrhosis: A systematic review and meta-analysis. Hepatol. Int. 2019, 13, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Bruns, T.; Zimmermann, H.W.; Stallmach, A. Risk factors and outcome of bacterial infections in cirrhosis. World J. Gastroenterol. 2014, 14, 2542–2554. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Irvine, K.M.; Ratnasekera, I.; Powell, E.E.; Hume, D.A. Causes and Consequences of Innate Immune Dysfunction in Cirrhosis. Front. Immunol. 2019, 25, 293, Erratum in: Front Immunol. 2019, 10, 818. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zanetto, A.; Campello, E.; Bulato, C.; Gavasso, S.; Saggiorato, G.; Shalaby, S.; Burra, P.; Angeli, P.; Senzolo, M.; Simioni, P. Global hemostatic profiling in patients with decompensated cirrhosis and bacterial infections. JHEP Rep. 2022, 20, 100493. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- El-Hady, H.A.; Mahmoud Abd-Elwahab, E.S.; Mostafa-Hedeab, G.; Shawky Elfarargy, M. Portal vein thrombosis in patients with COVID-19: A systematic review. Asian J. Surg. 2023, 46, 3017–3026. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hassnine, A.A.; Elsayed, A.M. COVID-19 in Cirrhotic Patients: Is Portal Vein Thrombosis a Potential Complication? Can. J. Gastroenterol. Hepatol. 2022, 26, 5900468. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dalbeni, A.; Cattazzo, F.; De Marco, L.; Bevilacqua, M.; Zoncapè, M.; Lombardi, R.; Stupia, R.; Mantovani, A.; Sacerdoti, D. Bacterial infections as a risk factor for non-neoplastic portal vein thrombosis development in cirrhotic patients. Dig. Liver Dis. 2024, 56, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.Y.; Zhang, Y.H.; Yi, S.Y.; Lei, L.; Ma, T.; Huang, R.; Yang, L.; Li, Z.M.; Zhang, D. Potential contribution of the gut microbiota to the development of portal vein thrombosis in liver cirrhosis. Front. Microbiol. 2023, 27, 1217338. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kolios, G.; Valatas, V.; Kouroumalis, E. Role of Kupffer cells in the pathogenesis of liver disease. World J. Gastroenterol. 2006, 14, 7413–7420. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yang, X.; Cheng, X.; Tang, Y.; Qiu, X.; Wang, Y.; Kang, H.; Wu, J.; Wang, Z.; Liu, Y.; Chen, F.; et al. Bacterial Endotoxin Activates the Coagulation Cascade through Gasdermin D-Dependent Phosphatidylserine Exposure. Immunity 2019, 17, 983–996.e6. [Google Scholar] [CrossRef] [PubMed]
- Chopyk, D.M.; Grakoui, A. Contribution of the Intestinal Microbiome and Gut Barrier to Hepatic Disorders. Gastroenterology 2020, 159, 849–863. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Di Tommaso, N.; Gasbarrini, A.; Ponziani, F.R. Intestinal Barrier in Human Health and Disease. Int. J. Environ. Res. Public Health 2021, 6, 12836. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Queck, A.; Carnevale, R.; Uschner, F.E.; Schierwagen, R.; Klein, S.; Jansen, C.; Meyer, C.; Praktiknjo, M.; Thomas, D.; Strassburg, C.; et al. Role of portal venous platelet activation in patients with decompensated cirrhosis and TIPS. Gut 2020, 69, 1535–1536. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, M.; Feng, J.; Zhou, D.; Wang, J. Bacterial lipopolysaccharide-induced endothelial activation and dysfunction: A new pre-dictive and therapeutic paradigm for sepsis. Eur. J. Med. Res. 2023, 12, 339. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jäckel, S.; Kiouptsi, K.; Lillich, M.; Hendrikx, T.; Khandagale, A.; Kollar, B.; Hörmann, N.; Reiss, C.; Subramaniam, S.; Wilms, E.; et al. Gut microbiota regulate hepatic von Willebrand factor synthesis and arterial thrombus formation via Toll-like receptor-2. Blood 2017, 27, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, R.; Raparelli, V.; Nocella, C.; Bartimoccia, S.; Novo, M.; Severino, A.; De Falco, E.; Cammisotto, V.; Pasquale, C.; Crescioli, C.; et al. Gut-derived endotoxin stimulates factor VIII secretion from endothelial cells. Implications for hypercoagulability in cirrhosis. J. Hepatol. 2017, 67, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Hally, K.; Fauteux-Daniel, S.; Hamzeh-Cognasse, H.; Larsen, P.; Cognasse, F. Revisiting Platelets and Toll-Like Receptors (TLRs): At the Interface of Vascular Immunity and Thrombosis. Int. J. Mol. Sci. 2020, 26, 6150. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Duttaroy, A.K. Role of Gut Microbiota and Their Metabolites on Atherosclerosis, Hypertension and Human Blood Platelet Function: A Review. Nutrients 2021, 3, 144. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rolling, C.C.; Barrett, T.J.; Berger, J.S. Platelet-monocyte aggregates: Molecular mediators of thromboinflammation. Front. Cardiovasc. Med. 2023, 15, 960398. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ren, Z.; Li, A.; Jiang, J.; Zhou, L.; Yu, Z.; Lu, H.; Xie, H.; Chen, X.; Shao, L.; Zhang, R.; et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut 2019, 68, 1014–1023. [Google Scholar] [CrossRef]
- Huang, H.; Ren, Z.; Gao, X.; Hu, X.; Zhou, Y.; Jiang, J.; Lu, H.; Yin, S.; Ji, J.; Zhou, L.; et al. Integrated analysis of microbiome and host transcriptome reveals correlations between gut microbiota and clinical outcomes in HBV-related hepatocellular carcinoma. Genome Med. 2020, 12, 102. [Google Scholar] [CrossRef]
- Gabr, M.A.; Bessa, S.S.; El-Zamarani, E.A. Portal vein thrombosis in Egyptian patients with liver cirrhosis: Role of methylenetetrahydrofolate reductase C677T gene mutation. Hepatol. Res. 2010, 40, 486–493. [Google Scholar] [CrossRef]
- Pasta, L.; Pasta, F.; D’Amico, M. PAI-1 4G-4G, MTHFR 677TT, V Leiden 506Q, and Prothrombin 20210A in Splanchnic Vein Thrombosis: Analysis of Individual Patient Data From Three Prospective Studies. J. Clin. Exp. Hepatol. 2016, 6, 10–14. [Google Scholar] [CrossRef]
- Jinato, T.; Anuntakarun, S.; Satthawiwat, N.; Chuaypen, N.; Tangkijvanich, P. Distinct alterations of gut microbiota between viraland non-viral-related hepatocellular carcinoma. Appl. Microbiol. Biotechnol. 2024, 108, 34. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Paroni Sterbini, F.; Petito, V.; et al. Hepatocellular Carcinoma Is Associated with Gut Microbiota Profile and Inflammation in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Buzas, E.I. The roles of extracellular vesicles in the immune system. Nat. Rev. Immunol. 2023, 23, 236–250. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nail, H.M.; Chiu, C.C.; Leung, C.H.; Ahmed, M.M.M.; Wang, H.D. Exosomal miRNA-mediated intercellular communications and immunomodulatory effects in tumor microenvironments. J. Biomed. Sci. 2023, 21, 69. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chaudhary, P.K.; Kim, S.; Kim, S. Shedding Light on the Cell Biology of Platelet-Derived Extracellular Vesicles and Their Bio-medical Applications. Life 2023, 16, 1403. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Eustes, A.S.; Dayal, S. The Role of Platelet-Derived Extracellular Vesicles in Immune-Mediated Thrombosis. Int. J. Mol. Sci. 2022, 16, 7837. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, W.; Zuo, B.; Wang, Y.; Li, X.; Weng, Z.; Zhai, J.; Wu, Q.; He, Y. Megakaryocyte- and Platelet-Derived Microparticles as Novel Diagnostic and Prognostic Biomarkers for Immune Thrombocytopenia. J. Clin. Med. 2022, 16, 6776. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Witek, R.P.; Yang, L.; Liu, R.; Jung, Y.; Omenetti, A.; Syn, W.K.; Choi, S.S.; Cheong, Y.; Fearing, C.M.; Agboola, K.M.; et al. Liver cell-derived microparticles activate hedgehog signaling and alter gene expression in hepatic endothelial cells. Gastroenterology 2009, 136, 320–330.e2. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lemoinne, S.; Cadoret, A.; Rautou, P.E.; El Mourabit, H.; Ratziu, V.; Corpechot, C.; Rey, C.; Bosselut, N.; Barbu, V.; Wendum, D.; et al. Portal myofibroblasts promote vascular remodeling underlying cirrhosis formation through the release of microparticles. Hepatology 2015, 61, 1041–1055. [Google Scholar] [CrossRef] [PubMed]
- Weil, D.; Di Martino, V.; Mourey, G.; Biichle, S.; Renaudin, A.; Laheurte, C.; Cypriani, B.; Delabrousse, E.; Grandclément, E.; Thévenot, T.; et al. Small Annexin V-Positive Platelet-Derived Microvesicles Affect Prognosis in Cirrhosis: A Longitudinal Study. Clin. Transl. Gastroenterol. 2021, 28, e00333. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, X.; Yao, Z.; Zhao, L.; Zhang, Y.; Cao, M.; Li, T.; Ding, W.; Liu, Y.; Deng, R.; Dong, Z.; et al. Phosphatidylserine on blood cells and endothelial cells contributes to the hypercoagulable state in cirrhosis. Liver Int. 2016, 36, 1800–1810. [Google Scholar] [CrossRef] [PubMed]
- Campello, E.; Radu, C.M.; Zanetto, A.; Bulato, C.; Shalaby, S.; Spiezia, L.; Franceschet, E.; Burra, P.; Russo, F.P.; Simioni, P. Changes in plasma circulating microvesicles in patients with HCV-related cirrhosis after treatment with direct-acting antivirals. Liver Int. 2020, 40, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Campello, E.; Zanetto, A.; Spiezia, L.; Radu, C.M.; Gavasso, S.; Ferrarese, A.; Farinati, F.; Senzolo, M.; Simioni, P. Hypercoagulability detected by circulating microparticles in patients with hepatocellular carcinoma and cirrhosis. Thromb. Res. 2016, 143, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, S.V.; Facciuto, M.E.; Heydt, D.; Chen, J.; Islam, H.K.; Kajstura, M.; Ramaswamy, G.; Aguero-Rosenfeld, M. Dynamics of circulating microparticles in liver transplant patients. J. Gastrointest. Liver Dis. 2008, 17, 261–268. [Google Scholar]
- Taleb, R.S.Z.; Moez, P.; Younan, D.; Eisenacher, M.; Tenbusch, M.; Sitek, B.; Bracht, T. Quantitative proteome analysis of plasma microparticles for the characterization of HCV-induced hepatic cirrhosis and hepatocellular carcinoma. Proteom. Clin. Appl. 2017, 11, 1700014. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, B. The functional role of exosome in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2018, 144, 2085–2095. [Google Scholar] [CrossRef] [PubMed]
- van der Windt, D.J.; Sud, V.; Zhang, H.; Varley, P.R.; Goswami, J.; Yazdani, H.O.; Tohme, S.; Loughran, P.; O’Doherty, R.M.; Minervini, M.I.; et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology 2018, 68, 1347–1360. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Abdelgawad, I.A. Epithelial Cell Adhesion Molecule mRNA Can be a Potential Marker to Predict Metastasis in Hepatocellular Carcinoma Patients. Asian Pac. J. Cancer Prev. 2020, 1, 861–866. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhong, C.; Wu, J.D.; Fang, M.M.; Pu, L.Y. Clinicopathological significance and prognostic value of the expression of the cancer stem cell marker CD133 in hepatocellular carcinoma: A meta-analysis. Tumor Biol. 2015, 36, 7623–7630. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Pan, Y.F.; Ding, Z.W.; Yang, G.Z.; Tan, Y.X.; Yang, C.; Jiang, T.Y.; Liu, L.J.; Zhang, B.; Han, T.; et al. RMP promotes venous metastases of hepatocellular carcinoma through promoting IL-6 transcription. Oncogene 2015, 19, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Liu, S.; Cheng, Y.; Lu, L.; Shi, J.; Xu, G.; Li, N.; Cheng, K.; Wu, M.; Cheng, S.; et al. ICAM-1-Related Noncoding RNA in Cancer Stem Cells Maintains ICAM-1 Expression in Hepatocellular Carcinoma. Clin. Cancer Res. 2016, 15, 2041–2050. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Lin, Z.; Mai, P.; Zhang, E.; Peng, L. Identification of prognostic biomarkers associated with the occurrence of portal vein tumor thrombus in hepatocellular carcinoma. Aging 2021, 13, 11786–11807. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Hu, H.S.; Feng, Y.X.; Shi, J.; Li, N.; Guo, W.X.; Xue, J.; Xie, D.; Liu, S.R.; Wu, M.C.; et al. Characterisation of a novel cell line (CSQT-2) with high metastatic activity derived from portal vein tumour thrombus of hepatocellular carcinoma. Br. J. Cancer 2010, 25, 1618–1626. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guo, W.; Xue, J.; Shi, J.; Li, N.; Shao, Y.; Yu, X.; Shen, F.; Wu, M.; Liu, S.; Cheng, S. Proteomics analysis of distinct portal vein tumor thrombi in hepatocellular carcinoma patients. J. Proteome Res. 2010, 6, 4170–4175. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Li, Y.; Yi, X.; Chen, G.; Jin, S.; Dai, Y.; Cui, B.; Dai, B.; Lin, H.; Zhou, D. Epigenome-wide DNA methylation profiling of portal vein tumor thrombosis (PVTT) tissues in hepatocellular carcinoma patients. Neoplasia 2020, 22, 630–643. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liao, W.; Liu, W.; Liu, X.; Yuan, Q.; Ou, Y.; Qi, Y.; Huang, W.; Wang, Y.; Huang, J. Upregulation of FAM83D affects the proliferation and invasion of hepatocellular carcinoma. Oncotarget 2015, 15, 24132–24147. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, F.; Zhang, L.; Xu, Y.; Song, D.; He, W.; Ji, X.; Shao, J. Hypermethylation of SCAND3 and Myo1g Gene Are Potential Diagnostic Biomarkers for Hepatocellular Carcinoma. Cancers 2020, 18, 2332. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, J.; Wu, S.; Huang, T. Expression and role of VEGFA and miR-381 in portal vein tumor thrombi in patients with hepato-cellular carcinoma. Exp. Ther. Med. 2018, 15, 5450–5456. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, M.; Wei, L.; Liu, P.Y.; Zhang, X.M.; Liu, F.; Yang, F.; Hu, X.S.; Mo, Z.C. Lnc-ATG9B-4 aggravates progress of hepatocellular carcinoma through cell proliferation and migration by upregulating CDK5. Exp. Biol. Med. 2021, 246, 177–186. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Song, L.N.; Qiao, G.L.; Yu, J.; Yang, C.M.; Chen, Y.; Deng, Z.F.; Song, L.H.; Ma, L.J.; Yan, H.L. Hsa_circ_0003998 promotes epithelial to mesenchymal transition of hepatocellular carcinoma by sponging miR-143-3p and PCBP1. J. Exp. Clin. Cancer Res. 2020, 17, 114. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lewis, C.S.; Bari, K.; Xie, C.; Sherman, K.E.; Vasse, M.; Van Dreden, P.; Bogdanov, V.Y. Potential utility of a multi-component coagulation factor panel to calculate MELD scores and assess the risk of portal vein thrombosis in chronic liver disease. BMC Gastroenterol. 2023, 9, 65. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nie, G.L.; Yan, J.; Li, Y.; Zhang, H.L.; Xie, D.N.; Zhu, X.W.; Li, X. Predictive model for non-malignant portal vein thrombosis associated with cirrhosis based on inflammatory biomarkers. World J. Gastrointest. Oncol. 2024, 15, 1213–1226. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fukui, S.; Wada, H.; Ikeda, K.; Kobayashi, M.; Shimada, Y.; Nakazawa, Y.; Mizutani, H.; Ichikawa, Y.; Nishiura, Y.; Moritani, I.; et al. Detection of a Prethrombotic State in Patients with Hepatocellular Carcinoma, Using a Clot Waveform Analysis. Clin. Appl. Thromb. Hemost. 2024, 30, 10760296241246002. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, T.; Yu, Y.; Liu, J.; Tian, X.; Kong, M.; Wu, L.; Tang, S.; Gu, S.; Zhao, J.; Cui, Y.; et al. PIVKA-II level is correlated to development of portal vein tumor thrombus in patients with HBV-related hepatocellular carcinoma. Infect. Agent. Cancer 2019, 14, 13. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tong, M.; Fung, T.M.; Luk, S.T.; Ng, K.Y.; Lee, T.K.; Lin, C.H.; Yam, J.W.; Chan, K.W.; Ng, F.; Zheng, B.J.; et al. ANXA3/JNK Signaling Promotes Self-Renewal and Tumor Growth, and Its Blockade Provides a Therapeutic Target for Hepatocellular Carcinoma. Stem Cell Rep. 2015, 14, 45–59. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]


{kind=link}
{kind=link}
{kind=link}
| Factor | Role in Portal Vein Thrombosis in HCC | References |
|---|---|---|
| Endothelial Damage and Inflammation | HCC cells secrete IL-6 and TNF-α, contributing to endothelial damage and increasing susceptibility to thrombosis. | [34] |
| Endothelial-to-Mesenchymal Transition (EMT) | HCC induces EMT in vascular endothelial cells via the Cyclin G1/PI3K/Akt/GSK-3/Snail pathway, promoting portal thrombosis. | [35,36] |
| Platelet Activation | HCC increases platelet count and induces hyperactivation, enhancing platelet aggregation and thrombotic risk. | [65,66] |
| Fibrinogen levels | Elevated fibrinogen levels in HCC patients correlate with increased risk of portal thrombosis. | [67,68] |
| Fibrinolysis Impairment | HCC decreases fibrinolysis, further exacerbating the pro-thrombotic state. | [69] |
| Tissue Factor (TF) Expression | HCC cells increase TF synthesis, initiating the coagulation cascade and promoting thrombosis. | [70,71,72] |
| ECM Remodeling and Hypoxia | HCC-related ECM remodeling and hypoxia increase VEGF and HIF-1α, correlating with greater vascular invasion and thrombotic risk. | [34,78,79] |
| Study | Results | Mechanism/Cell Type |
|---|---|---|
| Abdelgawad IA et al. [122] | EpCAM-expressing CSCs exclusively in HCC patients linked to portal vein thrombosis and metastasis. | CSCs |
| Zhong C. et al. [123] | CD133-expressing CSCs associated with portal vein thrombosis, independent of cirrhosis. | CSCs |
| Zhang et al. et al. [124] | RNA-mediated IL-6 transcription in CSCs stimulating metastasis and portal vein thrombosis. | CSCs |
| Guo W. et al. [125] | Non-coding RNA linked to ICAM-1 expression in HCC CSCs and portal vein thrombosis. | CSCs |
| Wang T. et al. [127] | Portal vein thrombosis originating from circulating HCC cells (CSQT-2 cell line). | Circulating tumor cells |
| Fan X et al. [129] Liao W et al. [130] Xu F et al. [131] | DNA methylation changes (TNFRSF10A, FAM83D, SCAND3) and vascular invasion in HCC. | Epigenetics |
| Wang J. et al. [132] | miR-381 downregulation and VEGFA expression promoting angiogenesis and portal vein thrombosis. | miRNA |
| Li M. et al. [133] Song LN et al. [134] | lncRNA role in HCC vascular invasion via CDK5, FOSL2, and CD44v6 activation. | lncRNA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galasso, L.; Cerrito, L.; Termite, F.; Mignini, I.; Esposto, G.; Borriello, R.; Ainora, M.E.; Gasbarrini, A.; Zocco, M.A. The Molecular Mechanisms of Portal Vein Thrombosis in Hepatocellular Carcinoma. Cancers 2024, 16, 3247. https://doi.org/10.3390/cancers16193247
Galasso L, Cerrito L, Termite F, Mignini I, Esposto G, Borriello R, Ainora ME, Gasbarrini A, Zocco MA. The Molecular Mechanisms of Portal Vein Thrombosis in Hepatocellular Carcinoma. Cancers. 2024; 16(19):3247. https://doi.org/10.3390/cancers16193247
Chicago/Turabian StyleGalasso, Linda, Lucia Cerrito, Fabrizio Termite, Irene Mignini, Giorgio Esposto, Raffaele Borriello, Maria Elena Ainora, Antonio Gasbarrini, and Maria Assunta Zocco. 2024. "The Molecular Mechanisms of Portal Vein Thrombosis in Hepatocellular Carcinoma" Cancers 16, no. 19: 3247. https://doi.org/10.3390/cancers16193247