
Benchmarking of Approaches for Gene Copy-Number Variation Analysis and Its Utility for Genetic Aberration Detection in High-Grade Serous Ovarian Carcinomas

 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Samples and Their Characterization
2.2. DNA Isolation
2.3. CoreExome Microarray-Based Genotyping
2.4. Estimation of the Gene Copy Number Using a Panel nCounter v2 Cancer CN Assay (NanoString CNV)
2.5. PCR Analysis
2.6. Biostatistics Data Analysis
3. Results
3.1. Comparison of Results of Gene Copy-Number Assessment
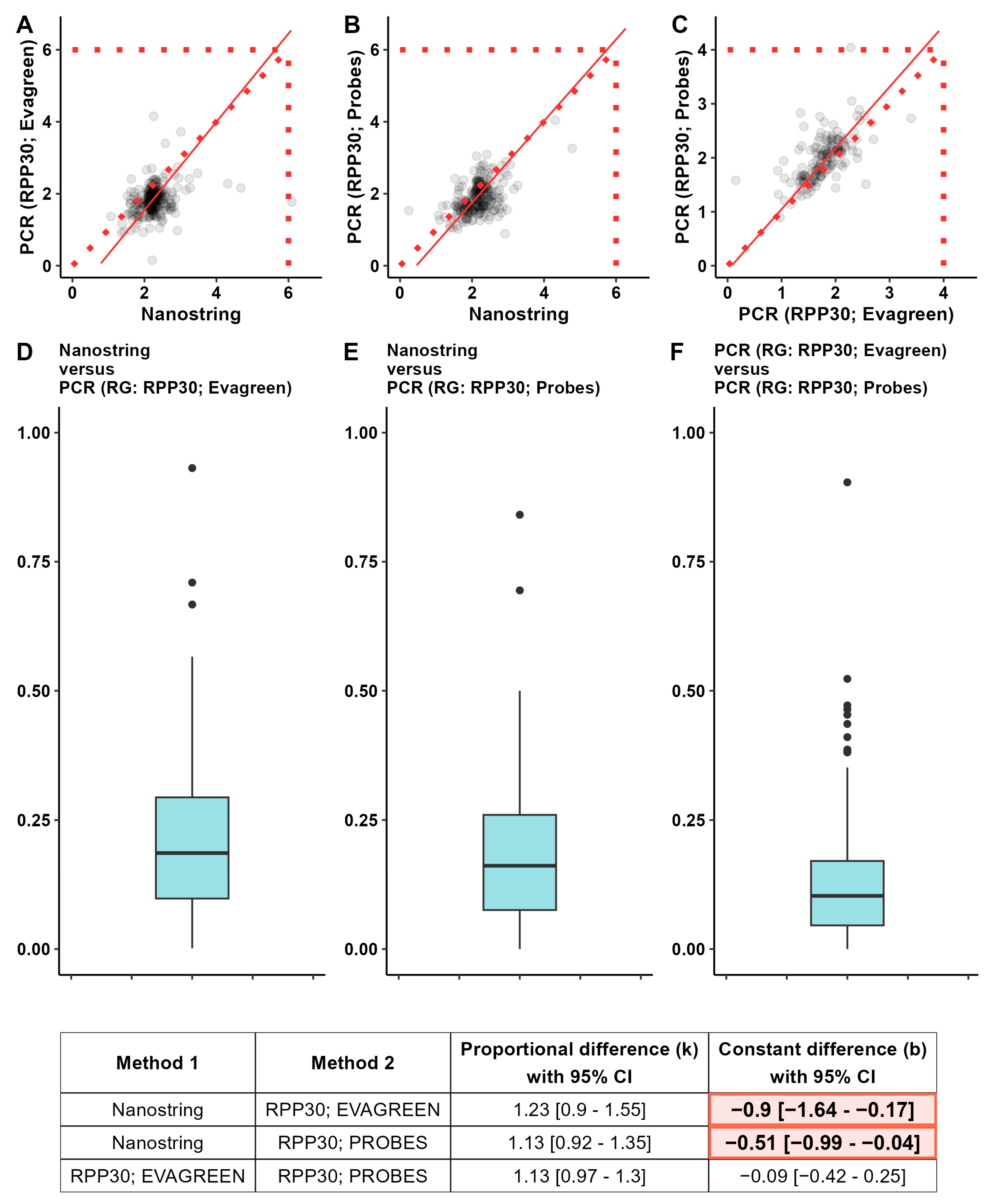
3.2. CNV Detection by Digital Droplet PCR
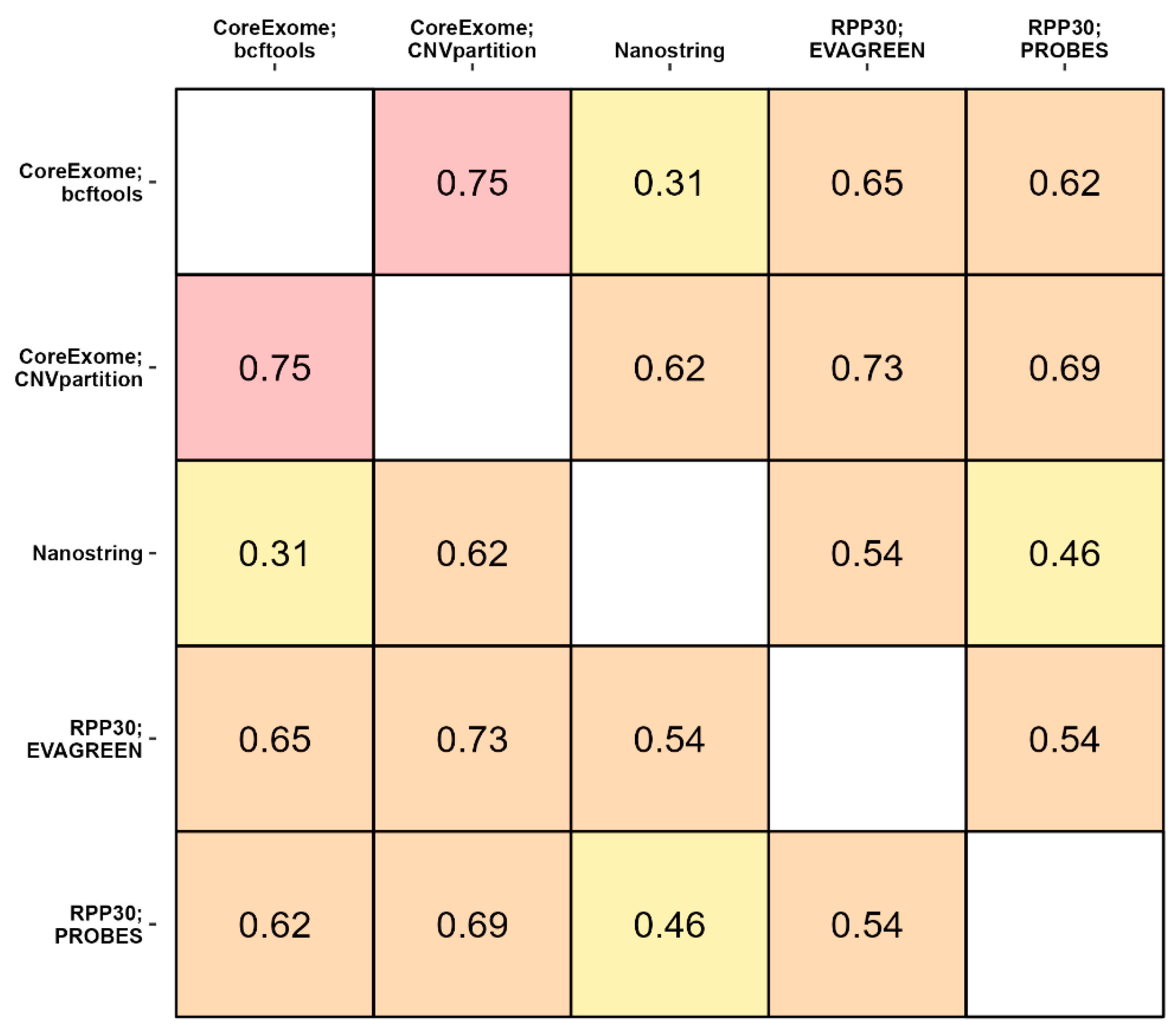
3.3. The Consistency between Three Methods of CNV Detection
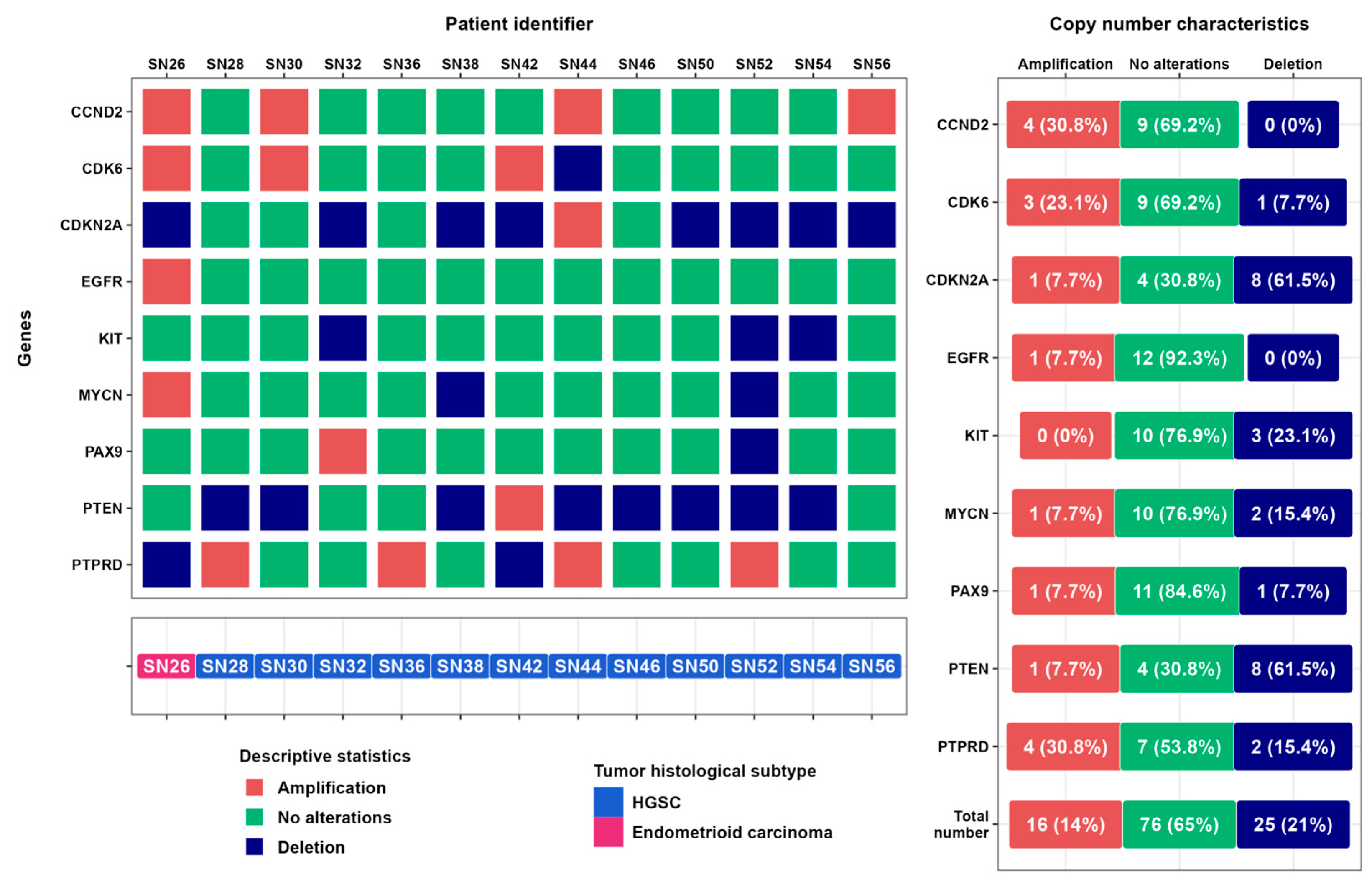
3.4. Results of CNV Detection in Ovarian Cancer Samples Based on ddPCR Data
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Age | Stage | TNM Classification | Histological Subtype | Immunohistochemical Markers | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| T | N | M | Ki-67 | p53 | PAX8 | WT1 | ||||
| SN26 | 45 | IIc | 2c | 0 | 0 | endometrioid carcinoma | high | mut type (+++) | 0 | 2 |
| SN28 | 58 | Ia | 1a | 0 | 0 | HGSC | high | mut type (−) | 1 | 3 |
| SN42 | 47 | IIIc | 3c | x | 0 | HGSC | high | mut type (+++) | 1 | 3 |
| SN56 | 43 | IIIc | 3c | x | 0 | HGSC | high | mut type (+++) | 3 | 3 |
| SN30 | 54 | IIIc | 3c | 1 | 0 | HGSC | high | mut type (+++) | 1 | 2 |
| SN32 | 60 | IIIc | 3c | 0 | 0 | HGSC | high | mut type (+++) | 3 | 0 |
| SN36 | 60 | IIIa | 3a | 1 | 0 | HGSC | high | mut type (+++) | 1 | 1 |
| SN38 | 49 | IVb | 3c | x | 1 | HGSC | high | mut type (+++) | 1 | 1 |
| SN44 | 41 | IIIc | 3c | x | 0 | HGSC | high | mut type (+++) | 1 | 2 |
| SN46 | 38 | IIIc | 3c | 1 | 0 | HGSC | high | mut type (−) | 3 | 1 |
| SN50 | 44 | IVb | 3c | x | 1 | HGSC | high | mut type (+++) | 2 | 3 |
| SN52 | 62 | IIa | 2a | 0 | 0 | HGSC | high | mut type (−) | 1 | 1 |
| SN54 | 66 | IIIc | 3c | x | 1 | HGSC | high | mut type (+++) | 1 | 0 |
| Immunohistochemical Markers | Gradations | |
|---|---|---|
| PAX8 and WT1 | negative | 0 |
| weak | 1 | |
| moderate | 2 | |
| strong | 3 | |
| p53 | strong | mut (+++) |
| negative | mut (−) | |
| weak | wild type (+) | |
| Ki-67 index | >50% | high |
| 15–49% | moderate | |
| <15% | low | |
Appendix B
| Gene | PCR-Product Length, bp | Tm, °C | Primer | Sequence 5′–3′ |
|---|---|---|---|---|
| C8orf4 (TCIM) | 191 | 62.7 | Forward | GAAAACTCTTCCGTCCCTGC |
| 63.7 | Reverse | CCTCCTCCCTTCCCCTATCT | ||
| 67.9 | Probe | [FAM]CACTTTGTCAGGCCCTAGGACTTAAATCG[BHQ1] | ||
| CCND2 | 164 | 62.7 | Forward | CAGCCGAGCAGTGAGAAATC |
| 63.1 | Reverse | CTCCCTTGAACACGCACAAA | ||
| 69.5 | Probe | [FAM]TGCAATGAAGACCTGGAAATCCCCGA[BHQ1] | ||
| CDK6 | 161 | 63.3 | Forward | GTGGACATGGAACCTGGAGA |
| 63.1 | Reverse | GAAAGACACAACTGGCAGCA | ||
| 68.4 | Probe | [FAM]TCTCACCTATGACAGGGTTGAGCCA[BHQ1] | ||
| CDKN2A | 193 | 63.5 | Forward | TGACTCCCTCCCCATTTTCC |
| 63.3 | Reverse | TTTTGGAGAGTCGGACTGCT | ||
| 68.9 | Probe | [FAM]TTGCCCCAGACAGCCGTTTTACAC[BHQ1] | ||
| EGFR | 232 | 63.7 | Forward | ATCCTGAGATGGTTTGGGCA |
| 63.1 | Reverse | ATAGAAGTACCCGCCTGCTC | ||
| 69.2 | Probe | [FAM]CCCTCCAGACCTCTTTCCCCACC[BHQ1] | ||
| HMGA2 | 216 | 63.2 | Forward | GGGGAGAAGGAACACACAGA |
| 63.5 | Reverse | AGTTAGATGCAGCAGTGGGT | ||
| 68.9 | Probe | [FAM]AGCAGTAACAATGCTCCAAACCACACC[BHQ1] | ||
| KDR | 188 | 62 | Forward | AGGAAGAACTATCAGTTGACAGAAT |
| 63.2 | Reverse | GGGAGAGAGAACATAAGAGCTACC | ||
| 69.4 | Probe | [FAM]TCAACCCATGTTTTCCCCTTCTCATAGCAT[BHQ1] | ||
| KIT | 250 | 63.5 | Forward | AGCCCTACTGCATGTCAAGT |
| 63.3 | Reverse | CCTACTCCAGTGCCCAAGAA | ||
| 69 | Probe | [FAM]CTCCTGTTACTGTAGCTGGCCTGGG[BHQ1] | ||
| MET | 154 | 63.4 | Forward | TTGTCTTCCCATCCACCCTC |
| 63.6 | Reverse | TCCCCATTTCTTCCTTCCCC | ||
| 68.2 | Probe | [FAM]ACTATGAGCTGTGAGAGTCTGGTCATTGAT[BHQ1] | ||
| MYCN | 161 | 61.7 | Forward | ATGAGTTGTGAAAGTTTTGAGTAGAT |
| 61.6 | Reverse | ACTTTGCATTTACCCAGTTCTATG | ||
| 68.1 | Probe | [FAM]TGCCTTTTTCCTAGCCTGTTTCTTCCT[BHQ1] | ||
| PAX9 | 165 | 61.7 | Forward | CCAGTCTGGTGAGAAAATAGACT |
| 62.1 | Reverse | ACAGCCACCTAAAAACATTTGATAA | ||
| 69.5 | Probe | [FAM]AACCATACCATACAGGGACTCTCCTGTCA[BHQ1] | ||
| PTEN | 202 | 61.7 | Forward | GGTTTAATAGAGGTGAACTGTCTTTC |
| 62.2 | Reverse | AGACTGTACTCTAAAAGCATTTCCT | ||
| 67.2 | Probe | [FAM]CTTTTCTTTGTTGGTGGCATGAGTCCTAT[BHQ1] | ||
| PTPRD | 200 | 61.7 | Forward | CCATCAGTGGAAAATTAAGAGCTAC |
| 61.6 | Reverse | TATTAACCATCCAATTATGACAGTGAG | ||
| 67 | Probe | [FAM]AGATGAAGGGACATAGCATCTGATTATCGT[BHQ1] | ||
| YWHAZ | 247 | 63.4 | Forward | TCAGCGACAGGTCTCCAAAT |
| 63.3 | Reverse | TCTCCTCCCTTTTGTGGTCC | ||
| 68.8 | Probe | [FAM]AGGGTCTAAGGAGACCAATGCCCAG[BHQ1] | ||
| TP53 | 192 | 63.3 | Forward | GGACCTCTTAACCTGTGGCT |
| 63.4 | Reverse | AAAGCTGTTCCGTCCCAGTA | ||
| 69 | Probe | [FAM]CAGAAAGGACAAGGGTGGTTGGGAGTA[BHQ1] | ||
| RPP30 | 62 | 62.9 | Forward | GATTTGGACCTGCGAGCG |
| 64.2 | Reverse | GCGGCTGTCTCCACAAGT | ||
| 69.9 | Probe | [R6G] TCTGACCTGAAGGCTCTGCGCG[BHQ2] | ||
| ALB | 94 | 61.8 | Forward | GACTTGCCAAGACATATGAAACC |
| 61.6 | Reverse | TCCAACAATAAACCTACCACTTTG | ||
| 69.3 | Probe | [R6G] TGCTGTGCCGCTGCAGATCC[BHQ2] |
Appendix C
| Cycling Step | Temperature, °C | Time | Ramp Rate | Number of Cycles |
|---|---|---|---|---|
| Enzyme activation | 95 | 10 min | 2 °C/s | 1 |
| Denaturation | 94 | 30 s | 40 | |
| Annealing/extension | 58 | 1 min | ||
| Enzyme deactivation | 98 | 10 min | 1 | |
| Hold (optional) | 4 | ∞ | 1 °C/s | 1 |
| Cycling Step | Temperature, °C | Time | Ramp Rate | Number of Cycles |
|---|---|---|---|---|
| Enzyme activation | 95 | 5 min | 2 °C/s | 1 |
| Denaturation | 95 | 30 s | 40 | |
| Annealing/extension | 60 | 1 min | ||
| Signal stabilization | 4 | 5 min | 1 | |
| 90 | 5 min | 1 | ||
| Hold (optional) | 4 | ∞ | 1°C/s | 1 |
Appendix D
| Genes | Ref. Gene RPP30; EvaGreen * | Ref. Gene RPP30; Probes ** | Ref. Gene ALB; EvaGreen * | Ref. Gene ALB; Probes ** |
|---|---|---|---|---|
| CCND2 | + | + | + | + |
| CDK6 | + | + | + | + |
| CDKN2A | + | + | + | + |
| EGFR | + | + | + | + |
| KIT | + | + | + | + |
| MYCN | + | + | + | + |
| PAX9 | + | + | + | + |
| PTEN | + | + | + | + |
| PTPRD | + | + | + | + |
| YWHAZ | + | + | + | + |
| KDR | + | + | ||
| C8orf4 (TCIM) | + | + | ||
| HMGA2 | + | + | ||
| MET | + | + | ||
| TP53 | + | + |
Appendix E
| Genes | Pcr_RPP30 EvaGreen (Normal) | Pcr_RPP30 EvaGreen (Tumor) | Pcr_RPP30 Probes (Normal) | Pcr_RPP30 Probes (Tumor) | NanoString CNV (Normal) | NanoString CNV (Tumor) | p-Value Pcr EvaGreen | p-Value Pcr Probes | p-Value NanoString CNV | p-Value *** | p-Value **** |
|---|---|---|---|---|---|---|---|---|---|---|---|
| C8orf4 (TCIM) | 1.61 [1.55–1.73] # | 1.59 [1.51–1.81] | No data | No data | 2.16 [2.11–2.22] | 2.2 [2.18–2.26] | 0.972 | No data | 0.807 | 0.882 | 1 |
| CCND2 | No data | No data | 1.75 [1.66–1.85] | 2.05 [1.91–2.57] | 2.06 [1.94–2.17] | 2.27 [2.15–2.44] | No data | 0.003 | 0.092 | 0.006 | 0.042 |
| CDK6 | 2.05 [2.03–2.13] | 2.15 [2.02–2.41] | 2.16 [2.13–2.34] | 2.29 [2.13–2.48] | 2.23 [2.2–2.35] | 2.42 [2.21–2.51] | 0.497 | 0.216 | 1 | 0.393 | 1 |
| HMGA2 | 1.92 [1.82–1.96] | 2.03 [1.8–2.1] | No data | No data | 2.27 [2.17–2.31] | 2.33 [2.28–2.45] | 0.505 | No data | 0.168 | 0.252 | 1 |
| KDR | 1.78 [1.67–1.91] | 1.88 [1.7–1.98] | No data | No data | 2.19 [2.14–2.27] | 2.3 [2.12–2.37] | 0.685 | No data | 0.363 | 0.475 | 1 |
| MET | 1.98 [1.92–2.16] | 2.06 [1.84–2.21] | No data | No data | 2.28 [2.14–2.36] | 2.22 [2.2–2.28] | 0.893 | No data | 0.421 | 0.573 | 1 |
| PAX9 | 1.81 [1.68–1.9] | 1.84 [1.83–1.92] | 2.13 [2.03–2.19] | 2.09 [1.93–2.24] | 2.23 [2.15–2.36] | 2.35 [2.15–2.69] | 0.685 | 0.542 | 0.168 | 0.324 | 1 |
Appendix F

| Genes * | Frequency of CNVs ** | Oncogene ***/Chromosomal Localization | Potential Prognostic Value of a Gene | Known Drug Target | References |
|---|---|---|---|---|---|
| MET | Amplification, 6.7% | Oncogene/Chr 7th | Prognostic biomarker | Drug target | [79,80] |
| HMGA2 | Amplification, 3.6% | Oncogene/Chr 14th | Prognostic biomarker | Drug target | [81,82] |
| KDR | Amplification, 2.1% | Oncogene/Chr 12th | Prognostic biomarker | Not found | [83] |
| C8orf4 (TCIM) | Amplification, 2.9% | No data/Chr 8th | Prognostic biomarker | Not found | [84] |
| PAX9 | Amplification, 1.4% | No data/Chr 14th | Prognostic biomarker | Not found | [85] |
| CDK6 | Amplification, 4.2% | Oncogene/Chr 21th | Not found | Drug target | [86] |
| CCND2 | Amplification, 11.6% | Oncogene/Chr13th | Not found | Drug target | [87] |
Appendix G

References
- PCAWG Transcriptome Core Group; Calabrese, C.; Davidson, N.R.; Demircioğlu, D.; Fonseca, N.A.; He, Y.; Kahles, A.; Lehmann, K.-V.; Liu, F.; Shiraishi, Y.; et al. Genomic Basis for RNA Alterations in Cancer. Nature 2020, 578, 129–136. [Google Scholar] [CrossRef]
- Steele, C.D.; Abbasi, A.; Islam, S.M.A.; Bowes, A.L.; Khandekar, A.; Haase, K.; Hames-Fathi, S.; Ajayi, D.; Verfaillie, A.; Dhami, P.; et al. Signatures of Copy Number Alterations in Human Cancer. Nature 2022, 606, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Harbers, L.; Agostini, F.; Nicos, M.; Poddighe, D.; Bienko, M.; Crosetto, N. Somatic Copy Number Alterations in Human Cancers: An Analysis of Publicly Available Data From The Cancer Genome Atlas. Front. Oncol. 2021, 11, 700568. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Li, J.; Jia, B.; Sang, H. CNV-MEANN: A Neural Network and Mind Evolutionary Algorithm-Based Detection of Copy Number Variations From Next-Generation Sequencing Data. Front. Genet. 2021, 12, 700874. [Google Scholar] [CrossRef]
- Andor, N.; Maley, C.C.; Ji, H.P. Genomic Instability in Cancer: Teetering on the Limit of Tolerance. Cancer Res. 2017, 77, 2179–2185. [Google Scholar] [CrossRef]
- Grist, E.; Friedrich, S.; Brawley, C.; Mendes, L.; Parry, M.; Ali, A.; Haran, A.; Hoyle, A.; Gilson, C.; Lall, S.; et al. Accumulation of Copy Number Alterations and Clinical Progression across Advanced Prostate Cancer. Genome Med. 2022, 14, 102. [Google Scholar] [CrossRef]
- Pan, X.; Hu, X.; Zhang, Y.-H.; Chen, L.; Zhu, L.; Wan, S.; Huang, T.; Cai, Y.-D. Identification of the Copy Number Variant Biomarkers for Breast Cancer Subtypes. Mol. Genet. Genom. 2019, 294, 95–110. [Google Scholar] [CrossRef]
- Dharanipragada, P.; Parekh, N. Genome-Wide Characterization of Copy Number Variations in Diffuse Large B-Cell Lymphoma with Implications in Targeted Therapy. Precis. Clin. Med. 2019, 2, 246–258. [Google Scholar] [CrossRef]
- Zhou, C.; Yuan, Z.; Ma, W.; Qi, L.; Mahavongtrakul, A.; Li, Y.; Li, H.; Gong, J.; Fan, R.R.; Li, J.; et al. Clinical Utility of Tumor Genomic Profiling in Patients with High Plasma Circulating Tumor DNA Burden or Metabolically Active Tumors. J. Hematol. Oncol. 2018, 11, 129. [Google Scholar] [CrossRef]
- Premarket Approval (PMA). Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P170019S017 (accessed on 30 August 2024).
- Lalonde, E.; Ewens, K.; Richards-Yutz, J.; Ebrahimzedeh, J.; Terai, M.; Gonsalves, C.F.; Sato, T.; Shields, C.L.; Ganguly, A. Improved Uveal Melanoma Copy Number Subtypes Including an Ultra–High-Risk Group. Ophthalmol. Sci. 2022, 2, 100121. [Google Scholar] [CrossRef]
- Tavares, I.; Martins, R.; Ribeiro, I.P.; Esteves, L.; Caramelo, F.; Abrantes, A.M.; Neves, R.; Caetano-Oliveira, R.; Botelho, M.F.; Barbosa De Melo, J.; et al. Development of a Genomic Predictive Model for Cholangiocarcinoma Using Copy Number Alteration Data. J. Clin. Pathol. 2022, 75, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Hatakeyama, K.; Nagashima, T.; Ohshima, K.; Urakami, K.; Yamaguchi, K.; Hirashima, Y. Characterization of Rare Histological Subtypes of Ovarian Cancer Based on Molecular Profiling. Cancer Med. 2023, 12, 387–395. [Google Scholar] [CrossRef]
- Zampaglione, E.; Kinde, B.; Place, E.M.; Navarro-Gomez, D.; Maher, M.; Jamshidi, F.; Nassiri, S.; Mazzone, J.A.; Finn, C.; Schlegel, D.; et al. Copy-Number Variation Contributes 9% of Pathogenicity in the Inherited Retinal Degenerations. Genet. Med. 2020, 22, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Van Roy, N.; Van Der Linden, M.; Menten, B.; Dheedene, A.; Vandeputte, C.; Van Dorpe, J.; Laureys, G.; Renard, M.; Sante, T.; Lammens, T.; et al. Shallow Whole Genome Sequencing on Circulating Cell-Free DNA Allows Reliable Noninvasive Copy-Number Profiling in Neuroblastoma Patients. Clin. Cancer Res. 2017, 23, 6305–6314. [Google Scholar] [CrossRef] [PubMed]
- Grond-Ginsbach, C.; Erhart, P.; Chen, B.; Kloss, M.; Engelter, S.T.; Cole, J.W. Copy Number Variation and Risk of Stroke. Stroke 2018, 49, 2549–2554. [Google Scholar] [CrossRef]
- Gordeeva, V.; Sharova, E.; Babalyan, K.; Sultanov, R.; Govorun, V.M.; Arapidi, G. Benchmarking Germline CNV Calling Tools from Exome Sequencing Data. Sci. Rep. 2021, 11, 14416. [Google Scholar] [CrossRef]
- Gabrielaite, M.; Torp, M.H.; Rasmussen, M.S.; Andreu-Sánchez, S.; Vieira, F.G.; Pedersen, C.B.; Kinalis, S.; Madsen, M.B.; Kodama, M.; Demircan, G.S.; et al. A Comparison of Tools for Copy-Number Variation Detection in Germline Whole Exome and Whole Genome Sequencing Data. Cancers 2021, 13, 6283. [Google Scholar] [CrossRef]
- Shen, W.; Szankasi, P.; Durtschi, J.; Kelley, T.W.; Xu, X. Genome-Wide Copy Number Variation Detection Using NGS: Data Analysis and Interpretation. In Tumor Profiling; Murray, S.S., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 1908, pp. 113–124. ISBN 978-1-4939-9002-3. [Google Scholar]
- Tsyganov, M.M.; Ibragimova, M.K.; Garbukov, E.Y.; Bragina, O.D.; Karchevskaya, A.A.; Usynin, E.A.; Litvyakov, N.V. Determination of BRCAness Phenotype in Breast Tumors for the Appointment of Neoadjuvant Chemotherapy Based on Platinum and Taxanes. Int. J. Mol. Sci. 2022, 24, 207. [Google Scholar] [CrossRef]
- Sagath, L.; Lehtokari, V.-L.; Pelin, K.; Kiiski, K. Array Comparative Genomic Hybridisation and Droplet Digital PCR Uncover Recurrent Copy Number Variation of the TTN Segmental Duplication Region. Genes 2022, 13, 905. [Google Scholar] [CrossRef]
- Sasa, K.; Saito, T.; Kurihara, T.; Hasegawa, N.; Sano, K.; Kubota, D.; Akaike, K.; Okubo, T.; Hayashi, T.; Takagi, T.; et al. Establishment of Rapid and Accurate Screening System for Molecular Target Therapy of Osteosarcoma. Technol. Cancer Res. Treat. 2022, 21, 153303382211382. [Google Scholar] [CrossRef]
- Norton, N.; Advani, P.P.; Serie, D.J.; Geiger, X.J.; Necela, B.M.; Axenfeld, B.C.; Kachergus, J.M.; Feathers, R.W.; Carr, J.M.; Crook, J.E.; et al. Assessment of Tumor Heterogeneity, as Evidenced by Gene Expression Profiles, Pathway Activation, and Gene Copy Number, in Patients with Multifocal Invasive Lobular Breast Tumors. PLoS ONE 2016, 11, e0153411. [Google Scholar] [CrossRef] [PubMed]
- O’Hern, K.; Barney, R.; Chambers, M.; Baker, C.; Stevanovic, M.; Tsongalis, G.J.; Hughes, E.; Sriharan, A. A Novel Method to Assess Copy Number Variation in Melanoma: Droplet Digital PCR for Precise Quantitation of the RREB1 Gene in Formalin-fixed, Paraffin-embedded Melanocytic Neoplasms, a proof-of-concept Study. J. Cutan. Pathol. 2023, 50, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Levsky, J.M.; Singer, R.H. Fluorescence in Situ Hybridization: Past, Present and Future. J. Cell Sci. 2003, 116, 2833–2838. [Google Scholar] [CrossRef] [PubMed]
- Schouten, J.P.; McElgunn, C.J.; Waaijer, R.; Zwijnenburg, D.; Diepvens, F.; Pals, G. Relative Quantification of 40 Nucleic Acid Sequences by Multiplex Ligation-Dependent Probe Amplification. Nucleic Acids Res. 2002, 30, e57. [Google Scholar] [CrossRef]
- Microarrays|Microarray Analysis Techniques and Products. Available online: https://emea.illumina.com/techniques/microarrays.html (accessed on 30 August 2024).
- Infinium Exome-24 Kit. Available online: https://emea.illumina.com/products/by-type/microarray-kits/infinium-exome.html#tabs-6c809a704c-item-8cfede5c7e-documentation (accessed on 30 August 2024).
- Rentas, S.; Abou Tayoun, A. Utility of Droplet Digital PCR and NGS-Based CNV Clinical Assays in Hearing Loss Diagnostics: Current Status and Future Prospects. Expert Rev. Mol. Diagn. 2021, 21, 213–221. [Google Scholar] [CrossRef]
- nCounter v2 Cancer CN Assay PRODUCT BULLETIN. Available online: https://nanostring.com/wp-content/uploads/v2-Cancer-CN-Assay.pdf (accessed on 30 August 2024).
- Kutasovic, J.R.; McCart Reed, A.E.; Males, R.; Sim, S.; Saunus, J.M.; Dalley, A.; McEvoy, C.R.; Dedina, L.; Miller, G.; Peyton, S.; et al. Breast Cancer Metastasis to Gynaecological Organs: A Clinico-pathological and Molecular Profiling Study. J. Pathol. Clin. Res. 2019, 5, 25–39. [Google Scholar] [CrossRef]
- Cusenza, V.Y.; Bisagni, A.; Rinaldini, M.; Cattani, C.; Frazzi, R. Copy Number Variation and Rearrangements Assessment in Cancer: Comparison of Droplet Digital PCR with the Current Approaches. Int. J. Mol. Sci. 2021, 22, 4732. [Google Scholar] [CrossRef]
- Sagath, L.; Lehtokari, V.-L.; Wallgren-Pettersson, C.; Pelin, K.; Kiiski, K. A Custom ddPCR Method for the Detection of Copy Number Variations in the Nebulin Triplicate Region. PLoS ONE 2022, 17, e0267793. [Google Scholar] [CrossRef]
- Danecek, P.; McCarthy, S.A.; HipSci Consortium; Durbin, R. A Method for Checking Genomic Integrity in Cultured Cell Lines from SNP Genotyping Data. PLoS ONE 2016, 11, e0155014. [Google Scholar] [CrossRef]
- Heydt, C.; Becher, A.-K.; Wagener-Ryczek, S.; Ball, M.; Schultheis, A.M.; Schallenberg, S.; Rüsseler, V.; Büttner, R.; Merkelbach-Bruse, S. Comparison of in Situ and Extraction-Based Methods for the Detection of MET Amplifications in Solid Tumors. Comput. Struct. Biotechnol. J. 2019, 17, 1339–1347. [Google Scholar] [CrossRef]
- Peng, H.; Lu, L.; Zhou, Z.; Liu, J.; Zhang, D.; Nan, K.; Zhao, X.; Li, F.; Tian, L.; Dong, H.; et al. CNV Detection from Circulating Tumor DNA in Late Stage Non-Small Cell Lung Cancer Patients. Genes 2019, 10, 926. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Liu, X.Q.; Arreaza, G.; Chen, C.; Albright, A.; Qiu, P.; Marton, M.J. Concordance between Single-Nucleotide Polymorphism–Based Genomic Instability Assays and a next-Generation Sequencing–Based Homologous Recombination Deficiency Test. BMC Cancer 2022, 22, 1310. [Google Scholar] [CrossRef] [PubMed]
- Rafter, P.; Gormley, I.C.; Parnell, A.C.; Kearney, J.F.; Berry, D.P. Concordance Rate between Copy Number Variants Detected Using Either High- or Medium-Density Single Nucleotide Polymorphism Genotype Panels and the Potential of Imputing Copy Number Variants from Flanking High Density Single Nucleotide Polymorphism Haplotypes in Cattle. BMC Genom. 2020, 21, 205. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Li, X.; Ding, Y.; Liu, Y.; Yang, M. Differences between Complex Epithelial Neoplasms of the Ovary and High-Grade Serous Ovarian Cancer: A Retrospective Observational Cohort Study. J. Ovarian Res. 2022, 15, 125. [Google Scholar] [CrossRef]
- Khashaba, M.; Fawzy, M.; Abdel-Aziz, A.; Eladawei, G.; Nagib, R. Subtyping of High Grade Serous Ovarian Carcinoma: Histopathological and Immunohistochemical Approach. J. Egypt. Natl. Cancer Inst. 2022, 34, 6. [Google Scholar] [CrossRef]
- Ritch, S.J.; Noman, A.S.M.; Goyeneche, A.A.; Telleria, C.M. The Metastatic Capacity of High-Grade Serous Ovarian Cancer Cells Changes along Disease Progression: Inhibition by Mifepristone. Cancer Cell Int. 2022, 22, 397. [Google Scholar] [CrossRef]
- Graf, R.P.; Eskander, R.; Brueggeman, L.; Stupack, D.G. Association of Copy Number Variation Signature and Survival in Patients With Serous Ovarian Cancer. JAMA Netw. Open 2021, 4, e2114162. [Google Scholar] [CrossRef]
- Engler, D.A.; Gupta, S.; Growdon, W.B.; Drapkin, R.I.; Nitta, M.; Sergent, P.A.; Allred, S.F.; Gross, J.; Deavers, M.T.; Kuo, W.-L.; et al. Genome Wide DNA Copy Number Analysis of Serous Type Ovarian Carcinomas Identifies Genetic Markers Predictive of Clinical Outcome. PLoS ONE 2012, 7, e30996. [Google Scholar] [CrossRef]
- Genovese, G. Freeseek/Gtc2vcf 2024. Available online: https://github.com/freeseek/gtc2vcf (accessed on 30 August 2024).
- GenomicRanges. Available online: http://bioconductor.org/packages/GenomicRanges/ (accessed on 30 August 2024).
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2012, 41, D36–D42. [Google Scholar] [CrossRef]
- Oscorbin, I.; Kechin, A.; Boyarskikh, U.; Filipenko, M. Multiplex ddPCR Assay for Screening Copy Number Variations in BRCA1 Gene. Breast Cancer Res. Treat. 2019, 178, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Real-Time PCR Application Guide. Available online: https://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_5279.pdf (accessed on 30 August 2024).
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A Tool to Design Target-Specific Primers for Polymerase Chain Reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- OligoAnalyzer Tool—Primer Analysis and Tm Calculator|IDT. Available online: https://eu.idtdna.com/pages/tools/oligoanalyzer (accessed on 30 August 2024).
- Bell, A.D.; Usher, C.L.; McCarroll, S.A. Analyzing Copy Number Variation with Droplet Digital PCR. In Digital PCR; Karlin-Neumann, G., Bizouarn, F., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; Volume 1768, pp. 143–160. ISBN 978-1-4939-7776-5. [Google Scholar]
- Data Analysis Guidelines for Copy Number Variation (CNV). Available online: https://nanostring.com/wp-content/uploads/MAN-C0014-02_nCounter_CNV_Data_Analysis_Guidelines.pdf (accessed on 30 August 2024).
- McHugh, M.L. Interrater Reliability: The Kappa Statistic. Biochem. Medica 2012, 22, 276–282. [Google Scholar] [CrossRef]
- Byrt, T.; Bishop, J.; Carlin, J.B. Bias, Prevalence and Kappa. J. Clin. Epidemiol. 1993, 46, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, M.; Sergeant, E.; Heuer, C.; Nunes, T.; Heuer, C.; Marshall, J.; Sanchez, J.; Thornton, R.; Reiczigel, J.; Robison-Cox, J.; et al. epiR: Tools for the Analysis of Epidemiological Data; The Comprehensive R Archive Network: New York, NY, USA, 2024. [Google Scholar]
- Potapov, S.; Model, F.; Schuetzenmeister, A.; Manuilova, E.; Dufey, F.; Raymaekers, J.; Seshan, V.E. Roche Mcr: Method Comparison Regression; The Comprehensive R Archive Network: New York, NY, USA, 2023. [Google Scholar]
- Bilic-Zulle, L. Comparison of Methods: Passing and Bablok Regression. Biochem. Medica 2011, 21, 49–52. [Google Scholar] [CrossRef]
- Mayer, B.; Gaus, W.; Braisch, J.U. The Fallacy of the Passing-Bablok-Regression. Jökull J. 2016, 66, 95–106. [Google Scholar]
- Han, M.; Yamaguchi, S.; Onishi, M.; Fujii, T.; Hosoya, M.; Wen, X.; Kido, H.; Kato, S. The MDM2 and CDKN2A Copy-Number-Variation Influence the TP53 -Signature-Score in Wild-Type TP53 Luminal Type Breast Cancer. Anticancer Res. 2022, 42, 2277–2288. [Google Scholar] [CrossRef]
- Li, S.; Dou, X.; Gao, R.; Ge, X.; Qian, M.; Wan, L. A Remark on Copy Number Variation Detection Methods. PLoS ONE 2018, 13, e0196226. [Google Scholar] [CrossRef]
- Ahn, S.; Hong, M.; Van Vrancken, M.; Lyou, Y.J.; Kim, S.T.; Park, S.H.; Kang, W.K.; Park, Y.S.; Jung, S.-H.; Woo, M.; et al. A nCounter CNV Assay to Detect HER2 Amplification: A Correlation Study with Immunohistochemistry and In Situ Hybridization in Advanced Gastric Cancer. Mol. Diagn. Ther. 2016, 20, 375–383. [Google Scholar] [CrossRef]
- Kutilin, D.S.; Tsandekova, M.R.; Porkhanova, N.V. Features of the Copy Number Variation of Certain Genes in Tumor Cells in Patients with Serous Ovarian Adenocarcinoma. Bull. Exp. Biol. Med. 2021, 170, 332–339. [Google Scholar] [CrossRef]
- Cosmic Cancer Gene Census. Available online: http://cancer.sanger.ac.uk/census (accessed on 30 August 2024).
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Broad GDAC Firehose. Available online: https://gdac.broadinstitute.org/ (accessed on 30 August 2024).
- Uhlén, M.; Pontén, F.; Lindskog, C. Charting the Human Proteome: Understanding Disease Using a Tissue-Based Atlas. Science 2015, 347, 1274. [Google Scholar] [CrossRef]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID Database: A Comprehensive Biomedical Resource of Curated Protein, Genetic, and Chemical Interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Yang, Z.; Moffa, A.B.; Haddad, R.; Streicher, K.L.; Ethier, S.P. Transforming Properties of TC-1 in Human Breast Cancer: Interaction with FGFR2 and Β-catenin Signaling Pathways. Int. J. Cancer 2007, 121, 1265–1273. [Google Scholar] [CrossRef]
- Kim, B.; Koo, H.; Yang, S.; Bang, S.; Jung, Y.; Kim, Y.; Kim, J.; Park, J.; Moon, R.T.; Song, K.; et al. TC1(C8orf4) Correlates with Wnt/β-Catenin Target Genes and Aggressive Biological Behavior in Gastric Cancer. Clin. Cancer Res. 2006, 12, 3541–3548. [Google Scholar] [CrossRef]
- De Melo Martins, P.C.; Parise Junior, O.; Pereira Hors, C.; Villela Miguel, R.E.; Da Costa Andrade, V.C.; Garicochea, B. C8orf4/TC-1(Thyroid Cancer-1) Gene Expression in Thyroid Cancer and Goiter. ORL 2007, 69, 127–130. [Google Scholar] [CrossRef]
- Muratovska, A.; Zhou, C.; He, S.; Goodyer, P.; Eccles, M.R. Paired-Box Genes Are Frequently Expressed in Cancer and Often Required for Cancer Cell Survival. Oncogene 2003, 22, 7989–7997. [Google Scholar] [CrossRef]
- Tacha, D.; Zhou, D.; Cheng, L. Expression of PAX8 in Normal and Neoplastic Tissues: A Comprehensive Immunohistochemical Study. Appl. Immunohistochem. Mol. Morphol. 2011, 19, 293–299. [Google Scholar] [CrossRef]
- Zhao, Z.; Szczepanski, A.P.; Tsuboyama, N.; Abdala-Valencia, H.; Goo, Y.A.; Singer, B.D.; Bartom, E.T.; Yue, F.; Wang, L. PAX9 Determines Epigenetic State Transition and Cell Fate in Cancer. Cancer Res. 2021, 81, 4696–4708. [Google Scholar] [CrossRef]
- Jia, S.; Zhou, J.; D’Souza, R.N. Pax9’s Dual Roles in Modulating Wnt Signaling during Murine Palatogenesis. Dev. Dyn. 2020, 249, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Hume, S.; Dianov, G.L.; Ramadan, K. A Unified Model for the G1/S Cell Cycle Transition. Nucleic Acids Res. 2020, 48, 12483–12501. [Google Scholar] [CrossRef] [PubMed]
- Myant, K.; Sansom, O. Efficient Wnt Mediated Intestinal Hyperproliferation Requires the Cyclin D2-CDK4/6 Complex. Cell Div. 2011, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Bååth, M.; Jönsson, J.-M.; Westbom Fremer, S.; Martín de la Fuente, L.; Tran, L.; Malander, S.; Kannisto, P.; Måsbäck, A.; Honeth, G.; Hedenfalk, I. MET Expression and Cancer Stem Cell Networks Impact Outcome in High-Grade Serous Ovarian Cancer. Genes 2021, 12, 742. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xie, K.; Li, H.; Wang, R.; Xu, X.; Chen, K.; Gu, H.; Fang, J. Suppression of C-Met-Overexpressing Tumors by a Novel c-Met/CD3 Bispecific Antibody. Drug Des. Dev. Ther. 2020, 14, 3201–3214. [Google Scholar] [CrossRef]
- Song, Z.; Liao, C.; Yao, L.; Xu, X.; Shen, X.; Tian, S.; Wang, S.; Xing, F. miR-219-5p Attenuates Cisplatin Resistance of Ovarian Cancer by Inactivating Wnt/β-Catenin Signaling and Autophagy via Targeting HMGA2. Cancer Gene Ther. 2022, 30, 596–607. [Google Scholar] [CrossRef]
- Wei, J.-J. HMGA2: A Biomarker in Gynecologic Neoplasia. J. Clin. Transl. Pathol. 2022, 2, 3–7. [Google Scholar] [CrossRef]
- Si, M.; Zhang, J.; Cao, J.; Xie, Z.; Shu, S.; Zhu, Y.; Lang, J. Integrated Analysis To Identify Molecular Biomarkers Of High-Grade Serous Ovarian Cancer. OncoTargets Ther. 2019, 12, 10057–10075. [Google Scholar] [CrossRef]
- Xu, H.-T.; Liu, Y.; Liu, S.-L.; Miao, Y.; Li, Q.-C.; Wang, E.-H. TC-1 (C8orf4) Expression Is Correlated with Differentiation in Ovarian Carcinomas and Might Distinguish Metastatic Ovarian from Metastatic Colorectal Carcinomas. Virchows Arch. Int. J. Pathol. 2013, 462, 281–287. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y.; Paiboonrungruang, C.; Li, Y.; Peters, H.; Kist, R.; Xiong, Z. PAX9 in Cancer Development. Int. J. Mol. Sci. 2022, 23, 5589. [Google Scholar] [CrossRef]
- Dall’Acqua, A.; Bartoletti, M.; Masoudi-Khoram, N.; Sorio, R.; Puglisi, F.; Belletti, B.; Baldassarre, G. Inhibition of CDK4/6 as Therapeutic Approach for Ovarian Cancer Patients: Current Evidences and Future Perspectives. Cancers 2021, 13, 3035. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Yan, Y.; Qiu, X.; Ye, C.; Jiang, X.; Song, S.; Zhang, Y.; Chang, H.; Wang, L.; He, X.; et al. miR-93-5p Suppresses Ovarian Cancer Malignancy and Negatively Regulate CCND2 by Binding to Its 3′UTR Region. Discov. Oncol. 2022, 13, 15. [Google Scholar] [CrossRef]
- De Marco, C.; Zoppoli, P.; Rinaldo, N.; Morganella, S.; Morello, M.; Zuccalà, V.; Carriero, M.V.; Malanga, D.; Chirillo, R.; Bruni, P.; et al. Genome-Wide Analysis of Copy Number Alterations Led to the Characterisation of PDCD10 as Oncogene in Ovarian Cancer. Transl. Oncol. 2021, 14, 101013. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Demeulemeester, J.; Wedge, D.C.; Vollan, H.K.M.; Pitt, J.J.; Russnes, H.G.; Pandey, B.P.; Nilsen, G.; Nord, S.; Bignell, G.R.; et al. Pan-Cancer Analysis of Homozygous Deletions in Primary Tumours Uncovers Rare Tumour Suppressors. Nat. Commun. 2017, 8, 1221. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Petrucci, E.; Pasquini, L.; Castelli, G.; Pelosi, E. Ovarian Cancers: Genetic Abnormalities, Tumor Heterogeneity and Progression, Clonal Evolution and Cancer Stem Cells. Medicines 2018, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.A.; Gibson, W.J.; Brown, M.S.; Kosmicki, J.A.; Busanovich, J.P.; Wei, H.; Urbanski, L.M.; Curimjee, N.; Berger, A.C.; Gao, G.F.; et al. Loss of Heterozygosity of Essential Genes Represents a Widespread Class of Potential Cancer Vulnerabilities. Nat. Commun. 2020, 11, 2517. [Google Scholar] [CrossRef]
- Skirnisdottir, I.; Mayrhofer, M.; Rydåker, M.; Åkerud, H.; Isaksson, A. Loss-of-Heterozygosity on Chromosome 19q in Early-Stage Serous Ovarian Cancer Is Associated with Recurrent Disease. BMC Cancer 2012, 12, 407. [Google Scholar] [CrossRef]
- Zhai, Y.; Wu, R.; Kuick, R.; Sessine, M.S.; Schulman, S.; Green, M.; Fearon, E.R.; Cho, K.R. High-grade Serous Carcinomas Arise in the Mouse Oviduct via Defects Linked to the Human Disease. J. Pathol. 2017, 243, 16–25. [Google Scholar] [CrossRef]
- Szabova, L.; Yin, C.; Bupp, S.; Guerin, T.M.; Schlomer, J.J.; Householder, D.B.; Baran, M.L.; Yi, M.; Song, Y.; Sun, W.; et al. Perturbation of Rb, P53, and Brca1 or Brca2 Cooperate in Inducing Metastatic Serous Epithelial Ovarian Cancer. Cancer Res. 2012, 72, 4141–4153. [Google Scholar] [CrossRef]
- Ryland, G.L.; Doyle, M.A.; Goode, D.; Boyle, S.E.; Choong, D.Y.H.; Rowley, S.M.; Li, J.; Australian Ovarian Cancer Study Group; Bowtell, D.D.; Tothill, R.W.; et al. Loss of Heterozygosity: What Is It Good For? BMC Med. Genom. 2015, 8, 45. [Google Scholar] [CrossRef]
- Zhang, K.; Lin, G.; Han, D.; Han, Y.; Peng, R.; Li, J. Adaptation of ACMG-ClinGen Technical Standards for Copy Number Variant Interpretation Concordance. Front. Genet. 2022, 13, 829728. [Google Scholar] [CrossRef] [PubMed]
- cBioPortal for Cancer Genomics. Available online: https://www.cbioportal.org/ (accessed on 30 August 2024).

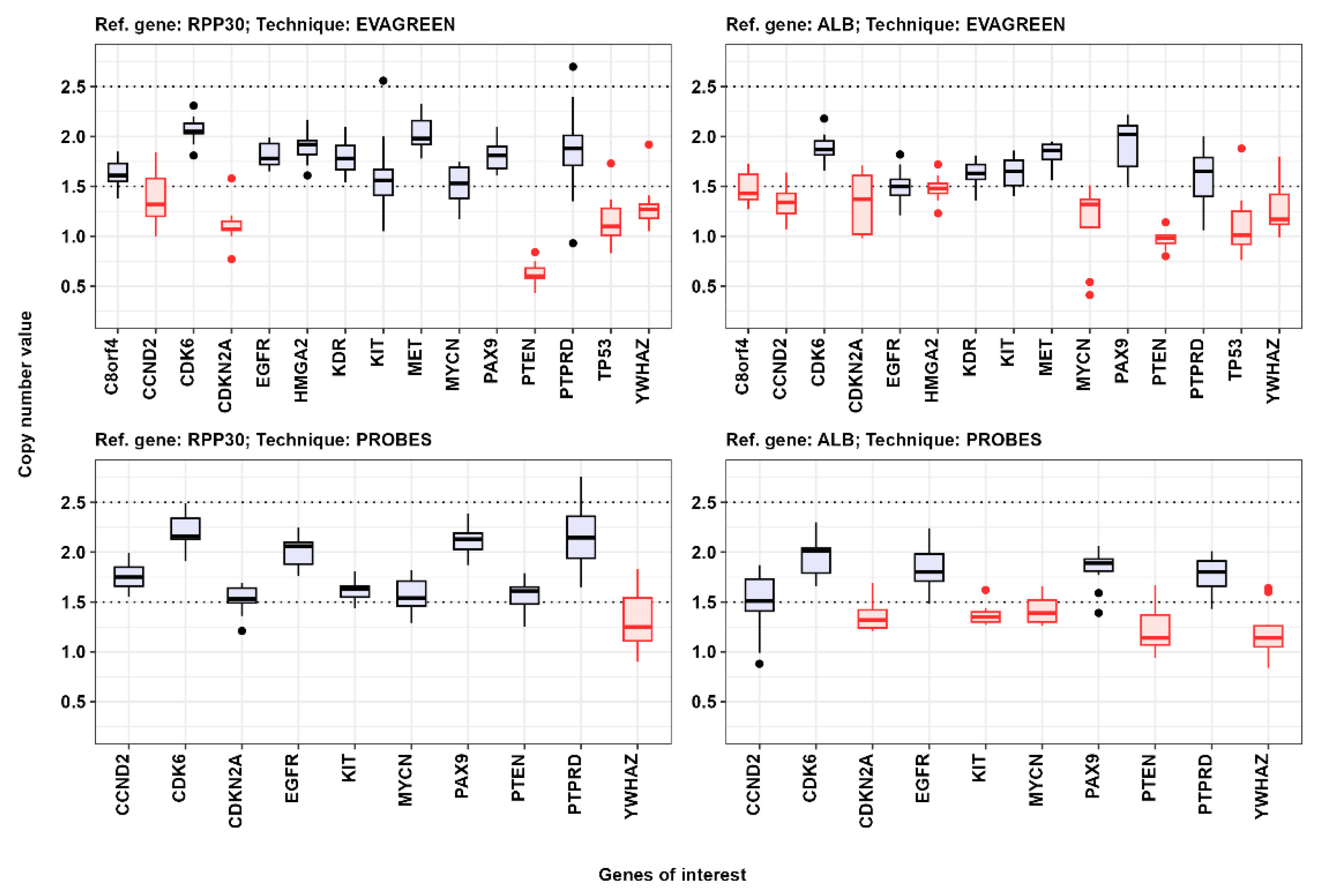
| Genes | p-Value PCR EvaGreen | p-Value PCR Probes | p-Value NanoString CNV | p-Value | p-Value (Corrected) |
|---|---|---|---|---|---|
| C8orf4 | 0.972 | No data | 0.807 | 0.882 | 1 |
| CCND2 | No data | 0.003 | 0.092 | 0.006 | 0.042 |
| CDK6 | 0.497 | 0.216 | 1 | 0.393 | 1 |
| HMGA2 | 0.505 | No data | 0.168 | 0.252 | 1 |
| KDR | 0.685 | No data | 0.363 | 0.475 | 1 |
| MET | 0.893 | No data | 0.421 | 0.573 | 1 |
| PAX9 | 0.685 | 0.542 | 0.168 | 0.324 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grebnev, P.A.; Meshkov, I.O.; Ershov, P.V.; Makhotenko, A.V.; Azarian, V.B.; Erokhina, M.V.; Galeta, A.A.; Zakubanskiy, A.V.; Shingalieva, O.S.; Tregubova, A.V.; et al. Benchmarking of Approaches for Gene Copy-Number Variation Analysis and Its Utility for Genetic Aberration Detection in High-Grade Serous Ovarian Carcinomas. Cancers 2024, 16, 3252. https://doi.org/10.3390/cancers16193252
Grebnev PA, Meshkov IO, Ershov PV, Makhotenko AV, Azarian VB, Erokhina MV, Galeta AA, Zakubanskiy AV, Shingalieva OS, Tregubova AV, et al. Benchmarking of Approaches for Gene Copy-Number Variation Analysis and Its Utility for Genetic Aberration Detection in High-Grade Serous Ovarian Carcinomas. Cancers. 2024; 16(19):3252. https://doi.org/10.3390/cancers16193252
Chicago/Turabian StyleGrebnev, Pavel Alekseevich, Ivan Olegovich Meshkov, Pavel Viktorovich Ershov, Antonida Viktorovna Makhotenko, Valentina Bogdanovna Azarian, Marina Vyacheslavovna Erokhina, Anastasiya Aleksandrovna Galeta, Aleksandr Vladimirovich Zakubanskiy, Olga Sergeevna Shingalieva, Anna Vasilevna Tregubova, and et al. 2024. "Benchmarking of Approaches for Gene Copy-Number Variation Analysis and Its Utility for Genetic Aberration Detection in High-Grade Serous Ovarian Carcinomas" Cancers 16, no. 19: 3252. https://doi.org/10.3390/cancers16193252