
Epithelial–Mesenchymal Plasticity and Epigenetic Heterogeneity in Cancer


{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Epithelial–Mesenchymal Plasticity Contributes to Tumor Heterogeneity
3. Epigenetic Regulation of Epithelial–Mesenchymal Plasticity and Tumor Heterogeneity
4. Regulation of Epithelial–Mesenchymal Plasticity and Epigenetics by Mechanical Heterogeneity in the Tumor Microenvironment
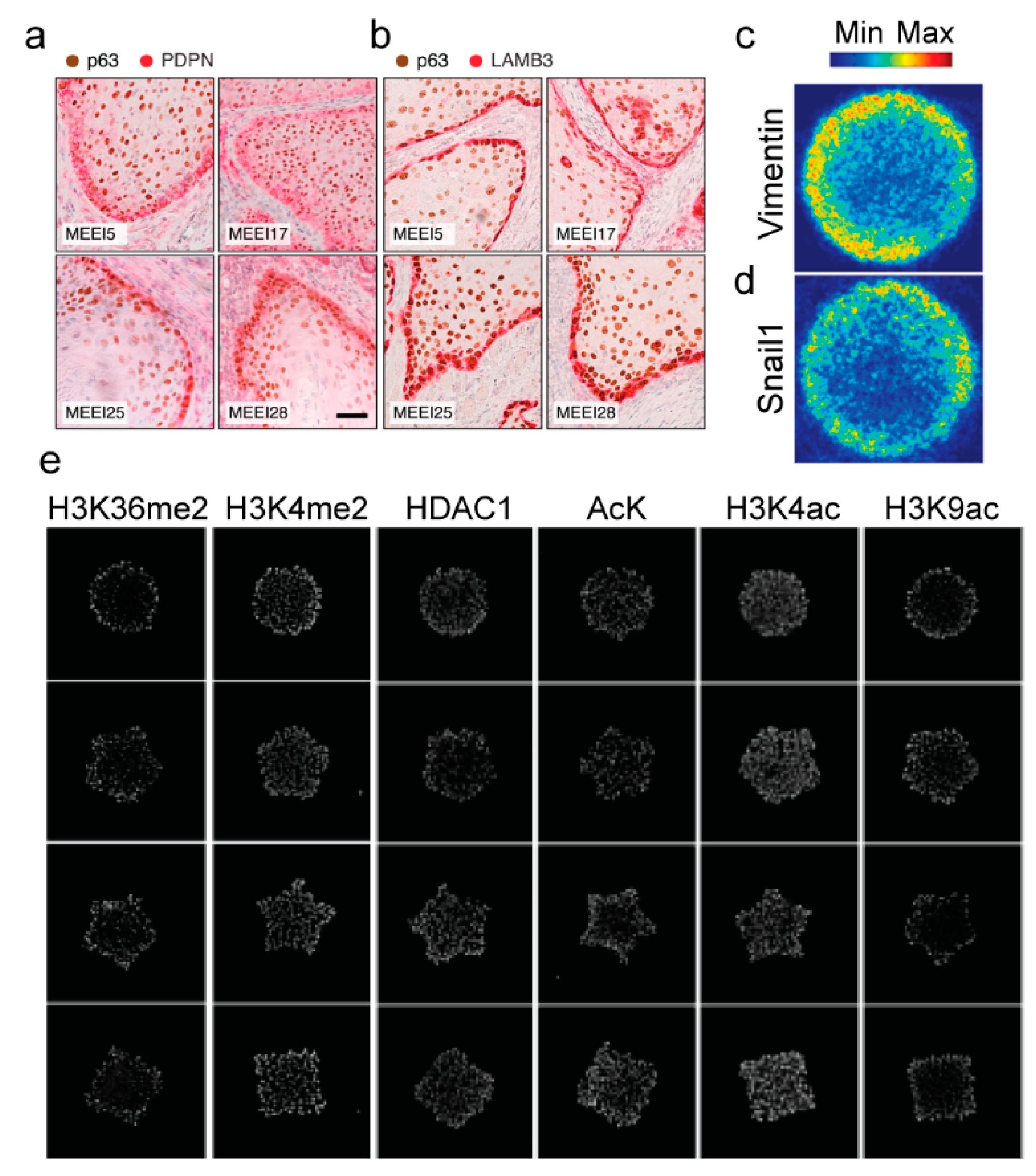
5. Therapeutic Approaches to Target Tumor Cell Heterogeneity and Tumor Recurrence
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Whiteside, T. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Rapsomaniki, M.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.; et al. A single-cell atlas of the tumor and immune ecosystem of human breast cancer. Cell 2019, 177, 1330–1345. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Morris, L.; Riaz, N.; Desrichard, A.; Senbabaoglu, Y.; Hakimi, A.; Makarov, V.; Reis-Filho, J.; Chan, T. Pan-cancer analysis of intratumor heterogeneity as a prognostic determinant of survival. Oncotarget 2016, 7, 10051–10063. [Google Scholar] [CrossRef]
- Joung, J.; Oh, B.; Hong, H.; Al-Khalidi, H.; Al-Alem, F.; Lee, H.; Bae, J.; Kim, J.; Cha, H.; Alotaibi, M.; et al. Tumor heterogeneity predicts metastatic potential in colorectal cancer. Clin. Cancer Res. 2017, 23, 7209–7216. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428, Erratum in J. Clin. Investig. 2010, 120, 1786.. [Google Scholar] [CrossRef]
- O’Connor, J.W.; Gomez, E.W. Biomechanics of TGFbeta-induced epithelial-mesenchymal transition: Implications for fibrosis and cancer. Clin. Transl. Med. 2014, 3, 23. [Google Scholar] [CrossRef]
- Ye, X.; Weinberg, R. Epithelial-mesenchymal plasticity: A central regulator of cancer progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef]
- Ramaekers, F.; Haag, D.; Kant, A.; Moesker, O.; Jap, P.; Vooijs, G. Coexpression of keratin- and vimentin-type intermediate filaments in human metastatic carcinoma cells. Proc. Natl. Acad. Sci. USA 1983, 80, 2618–2622. [Google Scholar] [CrossRef]
- Gonzalez, V.; Samusik, N.; Chen, T.; Savig, E.; Aghaeepour, N.; Quigley, D.; Huang, Y.; Giangarrà, V.; Borowsky, A.; Hubbard, N.; et al. Commonly occurring cell subsets in high-grade serious ovarian tumors identified by single-cell mass cytometry. Cell Rep. 2018, 22, 1875–1888. [Google Scholar] [CrossRef]
- Ruscetti, M.; Quach, B.; Dadashian, E.; Mulholland, D.; Wu, H. Tracking and functional characterization of epithelial-mesenchymal transition and mesenchymal tumor cells during prostate cancer metastasis. Cancer Res. 2015, 75, 2749–2759. [Google Scholar] [CrossRef] [PubMed]
- Tyler, M.; Tirosh, I. Decoupling epithelial-mesenchymal transitions from stromal profiles by integrative expression analysis. Nat. Commun. 2021, 12, 2592. [Google Scholar] [CrossRef] [PubMed]
- Colacino, J.; Azizi, E.; Brooks, M.; Harouaka, R.; Fouladdel, S.; McDermott, S.; Lee, M.; Hill, D.; Madden, J.; Boerner, J.; et al. Heterogeneity of human breast stem and progenitor cells as revealed by transcriptional profiling. Stem Cell Rep. 2018, 10, 1596–1609. [Google Scholar] [CrossRef] [PubMed]
- Kröger, C.; Afeyan, A.; Mraz, J.; Weinberg, R. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 7353–7362. [Google Scholar] [CrossRef]
- Boareto, M.; Jolly, M.; Goldman, A.; Pietilä, M.; Mani, S.; Sengupta, S.; Ben-Jacob, E.; Levine, H.; Onuchic, J. Notch-jagged signalling can give rise to clusters of cells exhibiting a hybrid epithelial/mesenchymal phenotype. J. R. Soc. Interface 2016, 13, 20151106. [Google Scholar] [CrossRef]
- Deshmukh, A.; Vasaikar, S.; Tomczak, K.; Mani, S. Identification of EMT signaling cross-talk and gene regulatory networks by single-cell RNA sequencing. Proc. Natl. Acad. Sci. USA 2021, 118, e2102050118. [Google Scholar] [CrossRef]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Ng Eaton, E.; Weinberg, R.A. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015, 525, 256–260. [Google Scholar] [CrossRef]
- Subbalakshmi, A.; Sahoo, S.; Biswas, K.; Jolly, M. A computational systems biology approach identifies SLUG as a mediator of partial epithelial-mesenchymal transition (EMT). Cells Tissues Organs 2022, 211, 689–702. [Google Scholar] [CrossRef]
- Voon, D.C.; Thiery, J.P. The Emerging Roles of RUNX Transcription Factors in Epithelial-Mesenchymal Transition. Adv. Exp. Med. Biol. 2017, 962, 471–489. [Google Scholar] [CrossRef]
- Brown, M.; Abdollahi, B.; Wilkins, O.; Lu, H.; Chakraborty, P.; Ognjenovic, N.; Muller, K.; Jolly, M.; Christensen, B.; Hassanpour, S.; et al. Phenotypic heterogeneity driven by plasticity of the intermediate EMT state governs disease progression and metastasis in breast cancer. Sci. Adv. 2022, 8, eabj8002. [Google Scholar] [CrossRef]
- Hebbar, A.; Moger, A.; Hari, K.; Jolly, M. Robustness in phenotypic plasticity and heterogeneity patterns enabled by EMT networks. Biophys. J. 2022, 121, 3600–3615. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Kleer, C.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.; Ghosh, D.; Sewalt, R.; Otte, A.; Hayes, D.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef]
- Varambally, S.; Dhanasekaran, S.; Zhou, M.; Barrette, T.; Kumar-Sinha, C.; Sanda, M.; Ghosh, D.; Pienta, K.; Sewalt, R.; Otte, A.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Wozniak, R.; Klimecki, W.; Lau, S.; Feinstein, Y.; Futscher, B. 5-Aza-2′-deoxycytidine-mediated reductions in G9A histone methyltransferase and H3 K9 di-methylation levels are linked to tumor suppressor gene reactivation. Oncogene 2007, 26, 77–90. [Google Scholar] [CrossRef]
- Spurling, C.; Godman, C.; Noonan, E.; Rasmussen, T.; Rosenberg, D.; Giardina, C. HDAC3 overexpression and colon cancer cell proliferation and differentiation. Mol. Carcinog. 2008, 47, 137–147. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Buitrago, D.; Labrador, M.; Arcon, J.; Lema, R.; Flores, O.; Esteve-Codina, A.; Blanc, J.; Villegas, N.; Bellido, D.; Gut, M.; et al. Impact of DNA methylation on 3D genome structure. Nat. Commun. 2021, 12, 3243. [Google Scholar] [CrossRef]
- Esteller, M.; Sanchez-Cespedes, M.; Rosell, R.; Sidransky, D.; Baylin, S.; Herman, J. Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non-small cell lung cancer patients. Cancer Res. 1999, 59, 67–70. [Google Scholar]
- Dulaimi, E.; Hillinck, J.; de Caceres, I.; Al-Saleem, T.; Cairns, P. Tumor suppressor gene promoter hypermethylation in serum of breast cancer patients. Clin. Cancer Res. 2004, 10, 6189–6193. [Google Scholar] [CrossRef] [PubMed]
- Yanagawa, N.; Tamura, G.; Oizumi, H.; Takahashi, N.; Shimazaki, Y.; Motoyama, T. Promoter hypermethylation of tumor suppressor and tumor-related genes in non-small cell lung cancers. Cancer Sci. 2003, 94, 589–592. [Google Scholar] [CrossRef] [PubMed]
- Quek, K.; Li, J.; Estecio, M.; Zhang, J.; Fujimoto, J.; Roarty, E.; Little, L.; Chow, C.; Song, X.; Behrens, C.; et al. DNA methylation intratumor heterogeneity in localized lung adenocarcinomas. Oncotarget 2017, 8, 21994–22002. [Google Scholar] [CrossRef] [PubMed]
- Rastetter, M.; Schagdarsurengin, U.; Lahtz, C.; Fiedler, E.; Marsch, W.; Damman, R.; Helmbold, P. Frequent intra-tumoral heterogeneity of promoter hypermethylation in malignant melanoma. Histol. Histopathol. 2007, 22, 1005–1015. [Google Scholar]
- Thienpont, B.; Steinbacher, J.; Zhao, H.; D’Anna, F.; Kuchnio, A.; Ploumakis, A.; Ghesquière, B.; Van Dyck, L.; Boeckx, B.; Schoonjans, L.; et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016, 537, 63–68. [Google Scholar] [CrossRef]
- van Zijl, F.; Mair, M.; Csiszar, A.; Schneller, D.; Zulehner, G.; Huber, H.; Eferl, R.; Beug, H.; Dolznig, H.; Mikulits, W. Hepatic tumor-stroma crosstalk guides epithelial to mesenchymal transition at the tumor edge. Oncogene 2009, 28, 4022–4033. [Google Scholar] [CrossRef]
- Sethi, S.; Macoska, J.; Chen, W.; Sarkar, F. Molecular signature of epithelial-mesenchymal transition (EMT) in human prostate cancer bone metastasis. Am. J. Transl. Res. 2011, 3, 90–99. [Google Scholar]
- Jolly, M.K.; Tripathi, S.; Jia, D.; Mooney, S.; Celiktas, M.; Hanash, S.; Mani, S.; Pienta, K.; Ben-Jacob, E.; Levine, H. Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget 2016, 7, 27067–27084. [Google Scholar] [CrossRef]
- Pistore, C.; Giannoni, E.; Colangelo, T.; Rizzo, F.; Magnani, E.; Muccillo, L.; Giurato, G.; Mancini, M.; Rizzo, S.; Riccardi, M.; et al. DNA methylation variations are required for epithelial-to-mesenchymal transition induced by cancer-associated fibroblasts in prostate cancer cells. Oncogene 2017, 36, 5551–5566. [Google Scholar] [CrossRef]
- Ye, G.; Sun, G.; Jiao, P.; Chen, C.; Liu, Q.; Huang, X.; Zhang, R.; Cai, W.; Li, S.; Wu, J.; et al. OVOL2, an inhibitor of Wnt signaling, reduces invasive activities of human and mouse cancer cells and is down-regulated in human colorectal tumors. Gastroenterology 2016, 150, 659–671.e16. [Google Scholar] [CrossRef]
- Qi, X.; Han, H.; Zhang, H.; Xu, M.; Li, L.; Chen, L.; Xiang, T.; Feng, Q.; Kang, T.; Qian, C.; et al. OVOL2 links stemness and metastasis via fine-tuning epithelial-mesenchymal transition in nasopharyngeal carcinoma. Theranostics 2018, 8, 2202–2216. [Google Scholar] [CrossRef] [PubMed]
- Minamiya, Y.; Ono, T.; Saito, H.; Takahashi, N.; Ito, M.; Motoyama, S.; Ogawa, J. Strong expression of HDAC3 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Tumor Biol. 2010, 31, 533–539. [Google Scholar] [CrossRef]
- Weichert, W.; Röske, A.; Gekeler, V.; Beckers, T.; Ebert, M.; Pross, M.; Dietel, M.; Denkert, C.; Röcken, C. Association of patterns of class 1 histone deacetylase expression with patient prognosis in gastric cancer: A retrospective analysis. Lancet Oncol. 2008, 9, 139–148. [Google Scholar] [CrossRef]
- Buurman, R.; Gürlevik, E.; Schäffer, V.; Eilers, M.; Sandbothe, M.; Kreipe, H.; Wilkens, L.; Schlegelberger, B.; Kühnel, F.; Skawran, B. Histone deacetylases activate hepatocyte growth factor signaling by repressing microRNA-449 in hepatocellular carcinoma cells. Gastroenterology 2012, 143, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Poyet, C.; Jentsch, B.; Hermanns, T.; Schweckendiek, D.; Seifert, H.; Schmidtpeter, M.; Sulser, T.; Moch, H.; Wild, P.; Kristiansen, G. Expression of histone deacetylases 1, 2, and 3 in urothelial bladder cancer. BMC Clin. Pathol. 2014, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hoshino, T.; Redner, R.; Kajigaya, S.; Liu, J. ETO, fusion partner in t (8; 21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. Proc. Natl. Acad. Sci. USA 1998, 95, 10860–10865. [Google Scholar] [CrossRef]
- Wang, F.; Zhange, W.; Song, Z.; Wang, M.; Wu, H.; Yang, Y.; Chen, R. A novel miRNA inhibits metastasis of prostate cancer via decreasing CREBBP-mediated histone acetylation. Cancer Res. 2020, 147, 469–480. [Google Scholar] [CrossRef]
- Wawruszak, A.; Gumbarewicz, E.; Okon, E.; Jeleniewicz, W.; Czapinski, J.; Halasa, M.; Okla, K.; Smok-Kalwat, J.; Bocian, A.; Rivero-Muller, A.; et al. Histone deacetylase inhibitors reinforce the phenotypical markers of breast epithelial or mesenchymal cancer cells but inhibit their migratory properties. Cancer Manag. Res. 2019, 11, 8345–8358. [Google Scholar] [CrossRef]
- Duan, R.; Du, W.; Guo, W. EZH2: A novel target for cancer treatment. J. Hematol. Oncol. 2020, 13, 104. [Google Scholar] [CrossRef]
- Simon, J.; Lange, C. Roles of EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2008, 647, 21–29. [Google Scholar] [CrossRef]
- Tsai, C.; Chien, M.; Chang, Y.; Lee, J.; Dai, S.; Cheng, S. Overexpression of histone H3 lysine 27 trimethylation is associated with aggressiveness and dedifferentiation of thyroid cancer. Endocr. Pathol. 2019, 30, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Padi, S.; Tindall, D.; Guo, B. Polycomb protein EZH2 suppresses apoptosis by silencing the proapoptotic miR-31. Cell Death Dis. 2014, 5, e1486. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Dhanasekaran, S.; Kim, J.; Mani, R.; Tomlins, S.; Mehra, R.; Laxman, B.; Cao, X.; Yu, J.; Kleer, C.; et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 2008, 27, 7274–7284. [Google Scholar] [CrossRef]
- Han, T.; Jiao, F.; Hu, H.; Yuan, C.; Wang, L.; Jin, Z.; Song, W.; Wang, L. EZH2 promotes cell migration and invasion but not alters cell proliferation by suppressing E-cadherin, partly through association with MALAT-1 in pancreatic cancer. Oncotarget 2016, 7, 11194–11207. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Zhang, R.; Mo, B.; Chen, L.; Liu, L.; Yu, Y.; Cao, W.; Fang, G.; Wan, Y.; Gu, Y.; et al. EZH2 as a novel therapeutic target for atrial fibrosis and atrial fibrillation. J. Mol. Cell. Cardiol. 2019, 135, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, Y.; Chen, X.; Wei, P.; Lin, Y.; Wu, Z.; Lin, Z.; Kang, D.; Ding, C. Single-cell RNA sequencing analysis reveals intra-tumoral heterogeneity of gliblastoma and a pro-tumor subset of tumor-associated macrophages characterized by EZH2 overexpression. Biochim. Biophys. Acta-Mol. Basis Dis. 2022, 1868, 166534. [Google Scholar] [CrossRef]
- Kampilafkos, P.; Melachrinou, M.; Kefalopoulou, Z.; Lakoumentas, J.; Sotiropoulou-Bonikou, G. Epigenetic modifications in cutaneous malignant melanoma: EZH2, H3K4me2, and H3K27me3 immunohistochemical expression is enhanced at the invasive front of tumor. Am. J. Dermatopathol. 2015, 37, 138–144. [Google Scholar] [CrossRef]
- Anwar, T.; Arellano-Garcia, C.; Ropa, J.; Chen, Y.; Kim, H.; Yoon, E.; Grigsby, S.; Basrur, V.; Nesvizhskii, A.; Muntean, A.; et al. p38-mediated phosphorylation at T367 induces EZH2 cytoplasmic localization to promote breast cancer metastasis. Nat. Commun. 2018, 9, 2801. [Google Scholar] [CrossRef]
- McMullen, E.; Skala, S.; Gonzalez, M.; Djomehri, S.; Chandrashekar, D.; Varambally, S.; Kleer, C. Subcellular localization of EZH2 phosphorylated at T367 stratifies metaplastic breast carcinoma subtypes. Breast Cancer 2021, 28, 496–505. [Google Scholar] [CrossRef]
- Yomtoubian, S.; Lee, S.; Verma, A.; Izzo, F.; Markowitz, G.; Choi, H.; Cerchietti, L.; Vahdat, L.; Brown, K.; Andreopoulou, E.; et al. Inhibition of EZH2 catalytic activity selectively targets a metastatic subpopulation in triple-negative breast cancer. Cell Rep. 2020, 30, 755–770.E6. [Google Scholar] [CrossRef]
- Bachmann, I.; Halvorsen, O.; Collett, K.; Stefansson, I.; Straume, O.; Haukaas, S.; Salvesen, H.; Otte, A.; Akslen, L. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. 2006, 24, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Zingg, D.; Debbache, J.; Schaefer, S.; Tuncer, E.; Frommel, S.; Cheng, P.; Arenas-Ramirez, N.; Haeusel, J.; Zhange, Y.; Bonalli, M.; et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat. Commun. 2015, 6, 6051. [Google Scholar] [CrossRef] [PubMed]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.; Zabierowski, S.; Brafford, P.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef]
- Jia, D.; Jolly, M.K.; Tripathi, S.C.; Den Hollander, P.; Huang, B.; Lu, M.; Celiktas, M.; Ramirez-Pena, E.; Ben-Jacob, E.; Onuchic, J.N.; et al. Distinguishing mechanisms underlying EMT tristability. Cancer Converg. 2017, 1, 2. [Google Scholar] [CrossRef]
- Jia, W.; Deshmukh, A.; Mani, S.; Jolly, M.; Levine, H. A possible role for epigenetic feedback regulation in the dynamics of epithelial-mesenchymal transition (EMT). Phys. Biol. 2019, 16, 066004. [Google Scholar] [CrossRef]
- Andriani, F.; Bertolini, G.; Facchinetti, F.; Baldoli, E.; Moro, M.; Casalini, P.; Caserini, R.; Milione, M.; Leone, G.; Pelosi, G.; et al. Conversion to stem-cell state in response to microenvironmental cues is regulated by balance between epithelial and mesenchymal features in lung cancer cells. Mol. Oncol. 2016, 10, 253–271. [Google Scholar] [CrossRef]
- Jain, P.; Corbo, S.; Mohammad, K.; Sahoo, S.; Ranganathan, S.; George, J.; Levine, H.; Taube, J.; Toneff, M.; Jolly, M. Epigenetic memory acquired during long-term EMT induction governs the recovery to the epithelial state. J. R. Soc. Interface 2023, 20, 20220627. [Google Scholar] [CrossRef]
- Jain, R.; Martin, J.; Stylianopoulos, T. The role of mechanical forces in tumor growth and therapy. Annu. Rev. Biomed. Eng. 2014, 16, 321–346. [Google Scholar] [CrossRef]
- Sankhe, C.; Sacco, J.; Gomez, E. Biophysical Regulation of TGFβ Signaling in the Tumor Microenvironment. In Engineering and Physical Approaches to Cancer. Current Cancer Research; Wong, I., Dawson, M., Eds.; Springer: Cham, Switzerland, 2023; pp. 159–200. [Google Scholar]
- Martinez-Vidal, L.; Murdica, V.; Venegoni, C.; Pederzoli, F.; Bandini, M.; Necchi, A.; Salonia, A.; Alfano, M. Causal contributors to tissue stiffness and clinical relevance in urology. Commun. Biol. 2021, 4, 1011. [Google Scholar] [CrossRef]
- Zhou, Z.; Ji, C.; Xiao, H.; Zhao, H.; Cui, Y.; Bian, X. Reorganized collagen in the tumor microenvironment of gastric cancer and its association with prognosis. J. Cancer 2017, 8, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Gurrala, R.; Byrne, C.; Brown, L.; Tiongco, R.; Matossian, M.; Savoie, J.; Collins-Burow, B.; Burow, M.; Martin, E.; Lau, F. Quantifying breast cancer-driven fiber alignment and collagen deposition in primary human breast tissue. Front. Bioeng. Biotechnol. 2021, 9, 618448. [Google Scholar] [CrossRef] [PubMed]
- Sternlicht, M.; Lochter, A.; Sympson, C.; Huey, B.; Rougier, J.; Gray, J.; Pinkel, D.; Bissell, M.; Werb, Z. The stromal proteinase MMP3/Stromelysin-1 promotes mammary carcinogenesis. Cell 1999, 98, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Lochter, A.; Galosy, S.; Muschler, J.; Freedman, N.; Werb, Z.; Bissell, M. Matrix metalloproteinase Stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J. Cell Biol. 1997, 139, 1861–1872. [Google Scholar] [CrossRef] [PubMed]
- Rudolph-Owen, L.; Chan, R.; Muller, W.; Matrisian, L. The matrix metalloproteinase matrilysin influences early-stage mammary tumorigenesis. Cancer Res. 1998, 58, 5500–5506. [Google Scholar]
- Lopez, J.; Kang, I.; You, W.; McDonald, D.; Weaver, V. In Situ force mapping of mammary gland transformation. Integr. Biol. 2011, 3, 910–921. [Google Scholar] [CrossRef]
- Stashko, C.; Hayward, M.K.; Northey, J.J.; Pearson, N.; Ironside, A.J.; Lakins, J.N.; Oria, R.; Goyette, M.A.; Mayo, L.; Russnes, H.G.; et al. A convolutional neural network STIFMap reveals associations between stromal stiffness and EMT in breast cancer. Nat. Commun. 2023, 14, 3561. [Google Scholar] [CrossRef]
- Oft, M.; Heider, K.; Beug, H. TGFβ signaling is necessary for carcinoma cell invasiveness and metastasis. Curr. Biol. 1998, 8, 1243–1252. [Google Scholar] [CrossRef]
- Alessandri, K.; Sarangi, B.; Gurchenkov, V.; Kieβling, T.; Fetler, L.; Rico, F.; Scheuring, S.; Lamaze, C.; Simon, A.; Geraldo, S. Cellular capsules as a tool for multicellular spheroid production and for investigating the mechanics of tumor progression in vitro. Proc. Natl. Acad. Sci. USA 2013, 110, 14843–14848. [Google Scholar] [CrossRef]
- Puram, S.; Tirsoh, I.; Parikh, A.; Patel, A.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.; Mroz, E.; Emerick, K.; et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 2017, 171, 1611–1624. [Google Scholar] [CrossRef]
- Rice, A.; Cortes, E.; Lachowski, D.; Cheung, B.; Karim, S.; Morton, J.; del Río Hernández, A. Matrix stiffness induces-epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.W.; Riley, P.N.; Nalluri, S.M.; Ashar, P.K.; Gomez, E.W. Matrix Rigidity Mediates TGFbeta1-Induced Epithelial-Myofibroblast Transition by Controlling Cytoskeletal Organization and MRTF-A Localization. J. Cell. Physiol. 2015, 230, 1829–1839. [Google Scholar] [CrossRef] [PubMed]
- Rabie, E.M.; Zhang, S.X.; Dunn, C.E.; Nelson, C.M. Substratum stiffness signals through integrin-linked kinase and beta1-integrin to regulate midbody proteins and abscission during EMT. Mol. Biol. Cell 2021, 32, 1664–1676. [Google Scholar] [CrossRef] [PubMed]
- Ondeck, M.G.; Kumar, A.; Placone, J.K.; Plunkett, C.M.; Matte, B.F.; Wong, K.C.; Fattet, L.; Yang, J.; Engler, A.J. Dynamically stiffened matrix promotes malignant transformation of mammary epithelial cells via collective mechanical signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 3502–3507. [Google Scholar] [CrossRef] [PubMed]
- Sankhe, C.; Sacco, J.; Lawton, J.; Fair, R.; Soares, D.; Aldahdooh, M.; Gomez, E.; Gomez, E. Breast cancer cells exhibit mesenchymal-epithelial plasticity following dynamic modulation of matrix stiffness. Adv. Biol. 2024, 8, 2400087. [Google Scholar] [CrossRef]
- Sacco, J.; Vaneman, Z.; Gomez, E. Extracellular matrix viscoelasticity regulates TGFβ1-induced epithelial-mesenchymal transition and apoptosis via integrin linked kinase. J. Cell. Physiol. 2024, 239, e31165. [Google Scholar] [CrossRef]
- Wei, S.; Fattet, L.; Tsai, J.; Guo, Y.; Pai, V.; Majeski, H.; Chen, A.; Sah, R.; Taylor, S.; Engler, A.; et al. Matrix stiffness drives epithelial-mesenchymal transition and tumor metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat. Cell Biol. 2015, 17, 678–688. [Google Scholar] [CrossRef]
- Shou, Y.; Teo, X.; Li, X.; Zhicheng, L.; Liu, L.; Sun, X.; Jonhson, W.; Ding, J.; Lim, C.; Tay, A. Dynamic magneto-softening of 3D hydrogel reverses malignant transformation of cancer cells and enhances drug efficacy. ACS Nano 2023, 17, 2851–2867. [Google Scholar] [CrossRef]
- Pol, M.; Gao, H.; Zhang, H.; George, O.; Fox, J.; Jia, X. Dynamic modulation of matrix adhesiveness induces epithelial-to-mesenchymal transition in prostate cancer cells in 3D. Biomaterials 2023, 299, 122180. [Google Scholar] [CrossRef]
- Leggett, S.; Brennan, M.; Martinez, S.; Tien, J.; Nelson, C. Relatively rare populations of invasive cells drive progression of heterogeneous tumors. Cell. Mol. Bioeng. 2024, 17, 7–24. [Google Scholar] [CrossRef]
- Torab, P.; Yan, Y.; Yamashita, H.; Warrick, J.I.; Raman, J.D.; DeGraff, D.J.; Wong, P.K. Three-Dimensional Microtumors for Probing Heterogeneity of Invasive Bladder Cancer. Anal. Chem. 2020, 92, 8768–8775. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Muralidharan, A.; Saateh, A.; Ding, Z.; Ten Dijke, P.; Boukany, P.E. A Programmable Multifunctional 3D Cancer Cell Invasion Micro Platform. Small 2022, 18, e2107757. [Google Scholar] [CrossRef] [PubMed]
- Selvaggio, G.; Canato, S.; Pawar, A.; Monteiro, P.; Guerreiro, P.; Brás, M.; Janody, F.; Chaouiya, C. Hybrid epithelial-mesenchymal phenotypes are controlled by microenvironmental factors. Cancer Res. 2020, 80, 2407–2420. [Google Scholar] [CrossRef] [PubMed]
- Gomez, E.; Chen, Q.; Gjorevski, N.; Nelson, C. Tissue geometry patterns epithelial-mesenchymal transition via intercellular mechanotransduction. J. Cell. Biochem. 2010, 110, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Zhou, Y.; Duan, X.; Fang, X.; Zhang, Q.; Zhang, Y.; Wang, P.; Huang, J. Spontaneous formation and spatial self-organization of mechanically induced mesenchymal-like cells within geometrically confined cancer cell monolayers. Biomaterials 2022, 281, 121337. [Google Scholar] [CrossRef]
- Nelson, C.; VanDuijn, M.; Inman, J.; Fletcher, D.; Bissell, M. Tissue geometry determines sites of mammary branching morphogenesis in organotypic cultures. Science 2006, 314, 298–300. [Google Scholar] [CrossRef]
- Nelson, C.; Jean, R.; Tan, J.; Liu, W.; Sniadecki, N.; Spector, A.; Chen, C. Emergent patterns of growth controlled by multicellular form and mechanics. Proc. Natl. Acad. Sci. USA 2005, 102, 11594–11599. [Google Scholar] [CrossRef]
- Lee, J.; Abdeen, A.; Wycislo, K.; Fan, T.; Kilian, K. Interfacial geometry dictates cancer cell tumorigenicity. Nat. Mater. 2016, 15, 856–862. [Google Scholar] [CrossRef]
- Lee, J.; Molley, T.; Seward, C.; Abdeen, A.; Zhang, H.; Wang, X.; Gandhi, H.; Yang, J.; Gaus, K.; Kilian, K. Geometric regulation of histone state directs melanoma reprogramming. Commun. Biol. 2020, 3, 341. [Google Scholar] [CrossRef]
- Hensel, J.; Flaig, T.; Theodorescu, D. Clinical opportunities and challenges in targeting tumour dormancy. Nat. Rev. Clin. Oncol. 2013, 10, 41. [Google Scholar] [CrossRef]
- Harper, K.; Sosa, M.; Entenberg, D.; Hosseini, H.; Cheung, J.; Nobre, R.; Avivar-Valderas, A.; Nagi, C.; Girnius, N.; Davis, R.; et al. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature 2016, 540, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Aouad, P.; Zhang, Y.; De Martino, F.; Stibolt, C.; Ail, S.; Ambrosini, G.; Mani, S.; Maggs, K.; Quinn, H.; Sflomos, G.; et al. Epithelial-mesenchymal plasticity determines estrogen receptor positive breast cancer dormancy and epithelial reconversion drives recurrence. Nat. Commun. 2022, 13, 4975. [Google Scholar] [CrossRef] [PubMed]
- Netti, P.; Berk, D.; Swartz, M.; Grodzinsky, A.; Jain, R. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000, 60, 2497–2503. [Google Scholar] [PubMed]
- Trédan, O.; Galmarini, C.; Patel, K.; Tannock, I. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, Y.; Wu, J.; Duncan, R.; Strohalm, J.; Ulbrich, K.; Akaike, T.; Maeda, H. Early phase tumor accumulation of macromolecules: A great difference in clearance rate between tumor and normal tissues. Jpn. J. Cancer Res. 1998, 89, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent Smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Swenson, C.; Bolcsak, L.; Batist, G.; Guthrie, T., Jr.; Tkaczuk, K.; Boxenbaum, H.; Welles, L.; Chow, S.; Bhamra, R.; Chaikin, P. Pharmacokinetics of doxorubicin administered iv as Myocet (TLC D-99; liposome-encapsulated doxorubicin citrate) compared with conventional doxorubicin when given in combination with cyclophosphamide in patients with metastatic breast cancer. Anti-Cancer Drugs 2003, 14, 239–246. [Google Scholar] [CrossRef]
- Gordon, A.; Fleagle, J.; Guthrie, D.; Parkin, D.; Gore, M.; Lacave, A. Recurrent epithelial ovarian carcinoma: A randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J. Clin. Oncol. 2001, 19, 3312–3322. [Google Scholar] [CrossRef]
- Qin, X.; Lv, X.; Li, P.; Yang, R.; Xia, Q.; Chen, Y.; Peng, Y.; Li, L.; Li, S.; Li, T.; et al. Matrix stiffness modulates ILK-mediated YAP activation to control the drug resistance of breast cancer cells. Biochem. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165625. [Google Scholar] [CrossRef]
- Anlaş, A.; Nelson, C. Soft microenvironments induce chemoresistance by increasing autophagy downstream of integrin-linked kinase. Cancer Res. 2020, 80, 4103–4113. [Google Scholar] [CrossRef]
- Hinohara, K.; Wu, H.; Vigneau, S.; McDonald, T.; Igarashi, K.; Yamamoto, K.; Madsen, T.; Fassl, A.; Egri, S.; Papanastasiou, M.; et al. KDM5 histone demethylase activity links cellular transcriptomic heterogeneity to therapeutic response. Cancer Cell 2018, 34, 939–953.E9. [Google Scholar] [CrossRef] [PubMed]
- Liau, B.; Sievers, C.; Donohue, L.; Gillespie, S.; Flavahan, W.; Miller, T.; Venteicher, A.; Hebert, C.; Carey, C.; Rodig, S.; et al. Adaptive chromatin remodeling drives glioblastoma stem cell plasticitiy and drug tolerance. Cell Stem Cell 2017, 20, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Marsolier, J.; Prompsy, P.; Durand, A.; Lyne, A.; Landragin, C.; Trouchet, A.; Bento, S.; Eisele, A.; Foulon, S.; Baudre, L.; et al. H3K27me3 conditions chemotolerance in triple-negative breast cancer. Nat. Genet. 2022, 54, 459–468. [Google Scholar] [CrossRef]
- Hirukawa, A.; Smith, H.; Zuo, D.; Dufour, C.; Savage, P.; Bertos, N.; Johnson, R.; Bui, T.; Bourque, G.; Basik, M.; et al. Targeting EZH2 reactivates a breast cancer subtype-specific anti-metastatic transcriptional program. Nat. Commun. 2018, 9, 2547. [Google Scholar] [CrossRef]
- Lehmann, B.; Colaprico, A.; Silva, T.; Chen, J.; An, H.; Ban, Y.; Huang, H.; Wang, L.; James, J.; Balko, J.; et al. Multi-omics analysis identifies therapeutic vulnerabilities in triple-negative breast cancer subtypes. Nat. Commun. 2021, 12, 6276. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.; Döhner, H.; Pocock, C.; Montesinos, P.; Afanasyev, B.; Dombret, H.; Ravandi, F.; Sayar, H.; Jang, J.; Porkka, K.; et al. Oral azacitidine maintenance therapy for acute myeloid leukemia in first remission. N. Engl. J. Med. 2020, 383, 2526–2537. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, O.; Horwitz, S.; Masszi, T.; Van Hoof, A.; Brown, P.; Doordujin, J.; Hess, G.; Jurczak, W.; Chawla, S.; Bhat, G. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: Results of the pivotal phase II BELIEF (CLN-19) study. J. Clin. Oncol. 2015, 33, 2492–2499. [Google Scholar] [CrossRef]
- Filì, C.; Candoni, A.; Zannier, M.; Olivieri, J.; Imbergamo, S.; Caizzi, M.; Nadali, G.; Di Bona, E.; Ermacora, A.; Gottardi, M.; et al. Efficacy and toxicity of Decitabine in patients with acute myeloid leukemia (AML): A multicenter real-world experience. Leuk. Res. 2019, 76, 33–38. [Google Scholar] [CrossRef]
- Morel, D.; Jeffery, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S. Combining epigenetic drugs with other therapies for solid tumours—Past lessons and future promise. Nat. Rev. Clin. Oncol. 2020, 17, 91–107. [Google Scholar] [CrossRef]
- Wang, N.; Ma, T.; Yu, B. Targeting epigenetic regulators to overcome drug resistance in cancers. Signal Transduct. Target. Ther. 2023, 8, 69. [Google Scholar] [CrossRef]
- Jiang, Z.; Li, W.; Xichun, H.; Zhang, Q.; Sun, T.; Cui, S.; Wang, S.; Ouyang, Q.; Yin, Y.; Geng, C.; et al. Tucidinostat plus exemestane for postmenepausal patients with advanced, hormone receptor-positive breast cancer (ACE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, W.; Hu, X.; Sun, T.; Cui, S.; Wang, S.; Ouyang, Q.; Yin, Y.; Geng, C.; Tong, Z.; et al. Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer: A long-term safety and overall survival update from the randomised, double-blind, placebo-controlled, phase 3 trial. Transl. Breast Cancer Res. 2023, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Wang, B.; Lin, C.; Chien, P.; Wu, Y.; Ko, J.; Chen, J. Chidamide alleviates TGF-β-induced epithelial–mesenchymal transition in lung cancer cell lines. Mol. Biol. Rep. 2016, 43, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Gau, Y.; Sabnis, G. Histone deacetylase inhibitor entinostat reverses epithelial to mesenchymal transition of breast cancer cells by reversing the repression of E-cadherin. Breast Cancer Res. Treat. 2014, 143, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Yardley, D.; Ismali-Khan, R.; Melichar, B.; Lichinitser, M.; Munster, P.; Klein, P.; Cruickshank, S.; Miller, K.; Lee, M.; Trepel, J. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J. Clin. Oncol. 2013, 31, 2128–2135. [Google Scholar]
- Connolly, R.; Zhao, F.; Miller, K.; Lee, M.; Piekarz, R.; Smith, K.; Brown-Glaberman, U.; Winn, J.; Faller, B.; Onitilo, A.; et al. E2112: Randomized Phase III Trial of Endocrine Therapy Plus Entinostat or Placebo in Hormone Receptor-Positive Advanced Breast Cancer. A Trial of the ECOG-ACRIN Cancer Research Group. J. Clin. Oncol. 2021, 39, 3171–3181. [Google Scholar] [CrossRef]
- Sakamoto, T.; Kobayashi, S.; Yamada, D.; Nagano, H.; Tomokuni, A.; Tomimaru, Y.; Noda, T.; Gotch, K.; Asaoka, T.; Wada, H.; et al. A Histone Deacetylase Inhibitor Suppresses Epithelial-Mesenchymal Transition and Attenuates Chemoresistance in Biliary Tract Cancer. PLoS ONE 2016, 11, e0145985. [Google Scholar] [CrossRef]
- Teknos, T.; Grecula, J.; Agrawal, A.; Old, M.; Ozer, E.; Carrau, R.; Kang, S.; Rocco, J.; Blakaj, D.; Diavolitsis, V.; et al. A phase 1 trial of Vorinostat in combination with concurrent chemoradiation therapy in the treatment of advanced staged head and neck squamous cell carcinoma. Investig. New Drugs 2019, 37, 702–710. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sacco, J.L.; Gomez, E.W. Epithelial–Mesenchymal Plasticity and Epigenetic Heterogeneity in Cancer. Cancers 2024, 16, 3289. https://doi.org/10.3390/cancers16193289
Sacco JL, Gomez EW. Epithelial–Mesenchymal Plasticity and Epigenetic Heterogeneity in Cancer. Cancers. 2024; 16(19):3289. https://doi.org/10.3390/cancers16193289
Chicago/Turabian StyleSacco, Jessica L., and Esther W. Gomez. 2024. "Epithelial–Mesenchymal Plasticity and Epigenetic Heterogeneity in Cancer" Cancers 16, no. 19: 3289. https://doi.org/10.3390/cancers16193289
APA StyleSacco, J. L., & Gomez, E. W. (2024). Epithelial–Mesenchymal Plasticity and Epigenetic Heterogeneity in Cancer. Cancers, 16(19), 3289. https://doi.org/10.3390/cancers16193289