
Combined PET Radiotracer Approach Reveals Insights into Stromal Cell-Induced Metabolic Changes in Pancreatic Cancer In Vitro and In Vivo

 , ,
, ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
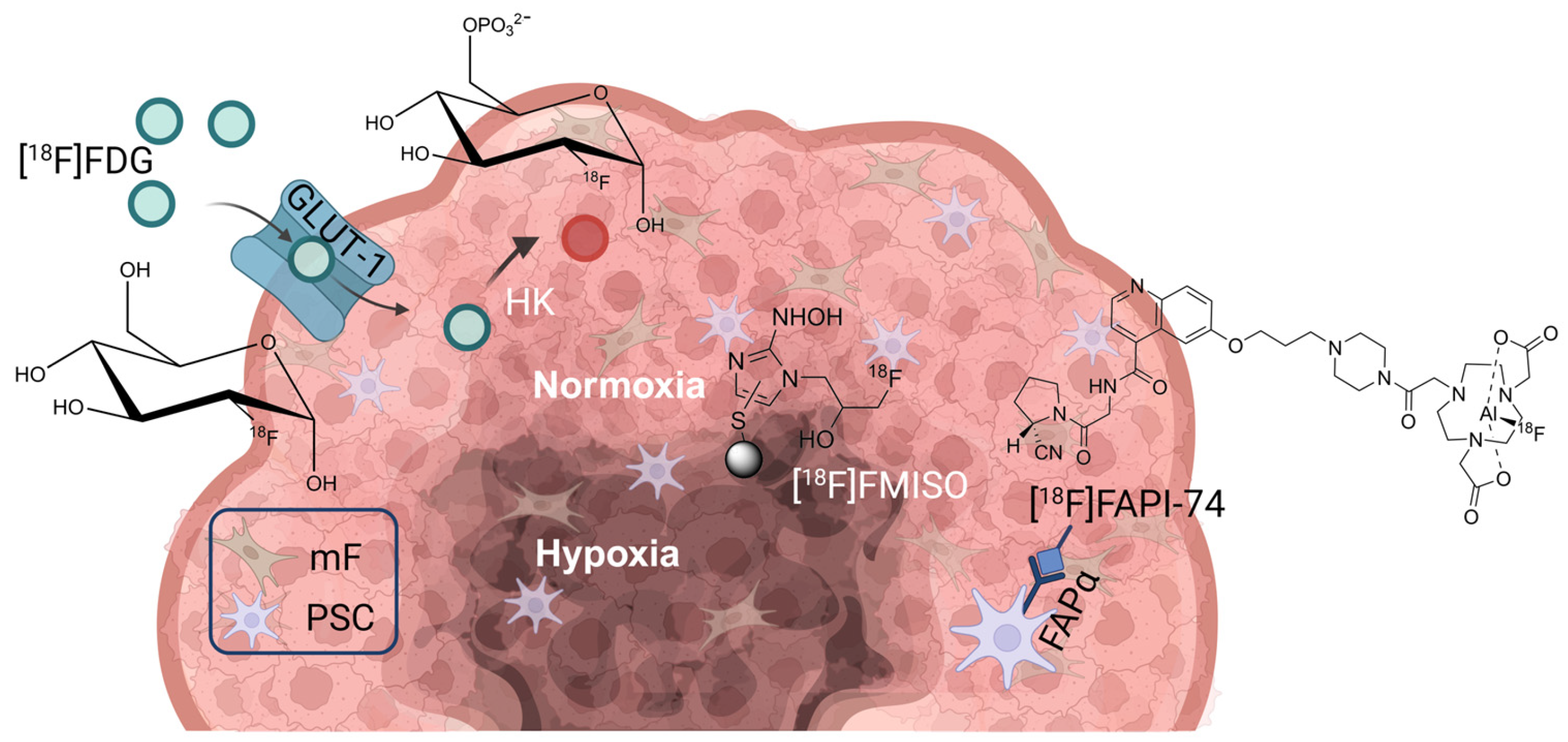
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Generation of Spheroids
2.3. Radiosynthesis of [18F]FAPI-74
2.4. Radiotracer Uptake In Vitro
2.5. Animal Experiments
2.6. Small Animal PET/CT
2.7. MRI
2.8. Immunohistochemistry
2.9. SDS-Page and Western Blotting
2.10. Statistical Analysis
3. Results
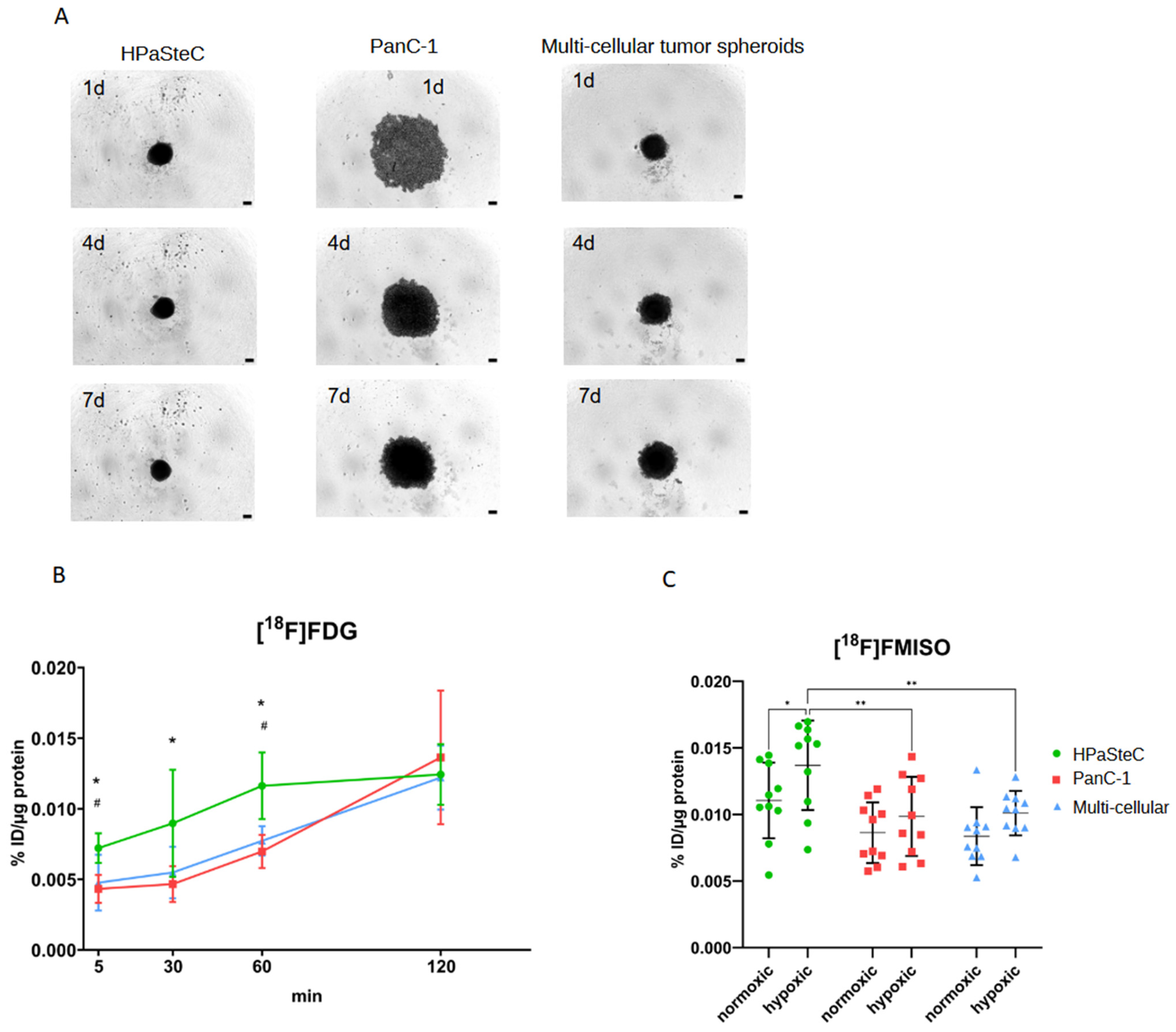
3.1. Characterization of Spheroid Models
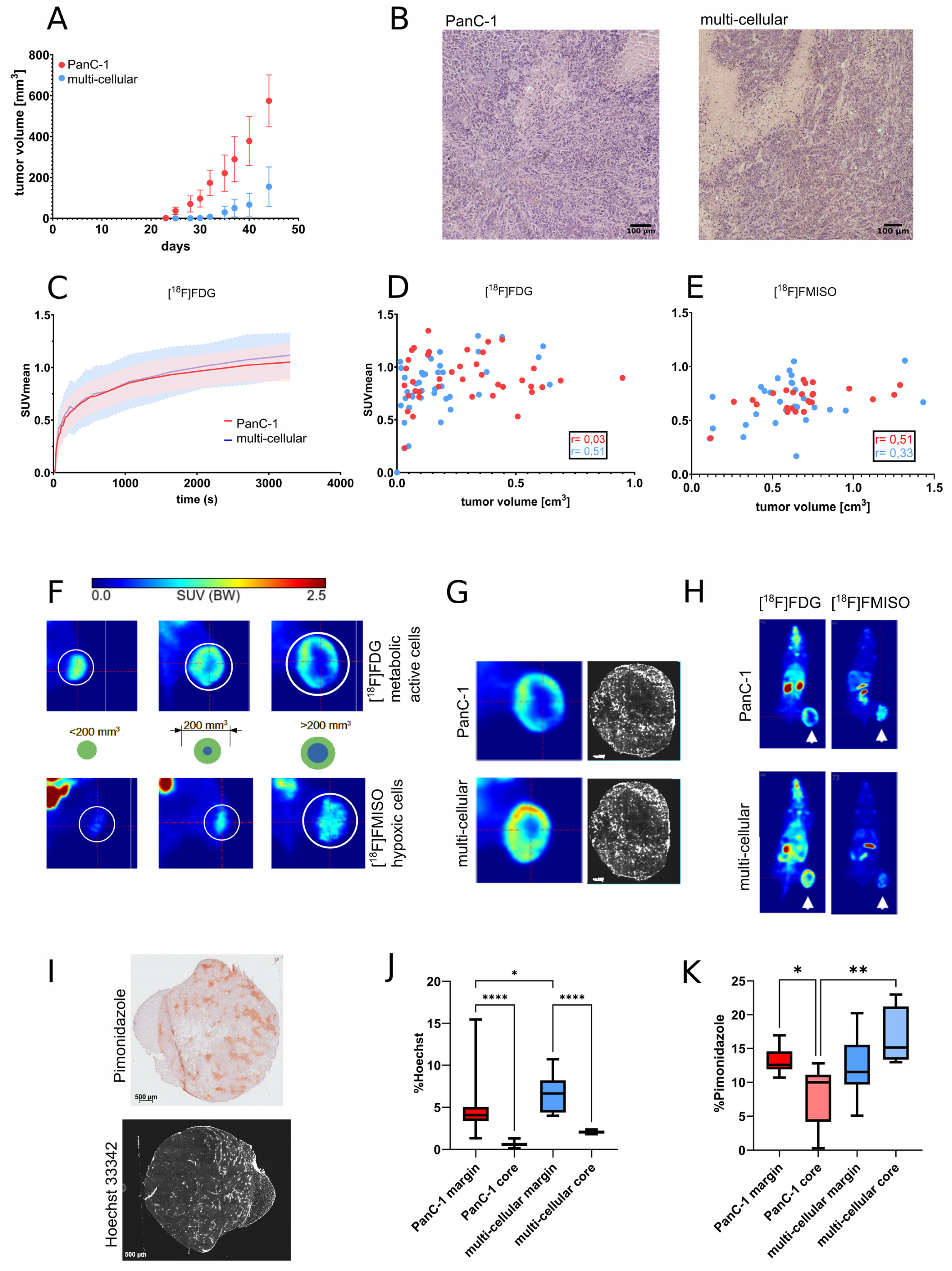
3.2. Growth and Radiopharmaceutical Characterization of Xenograft Tumors
3.3. MRI Imaging
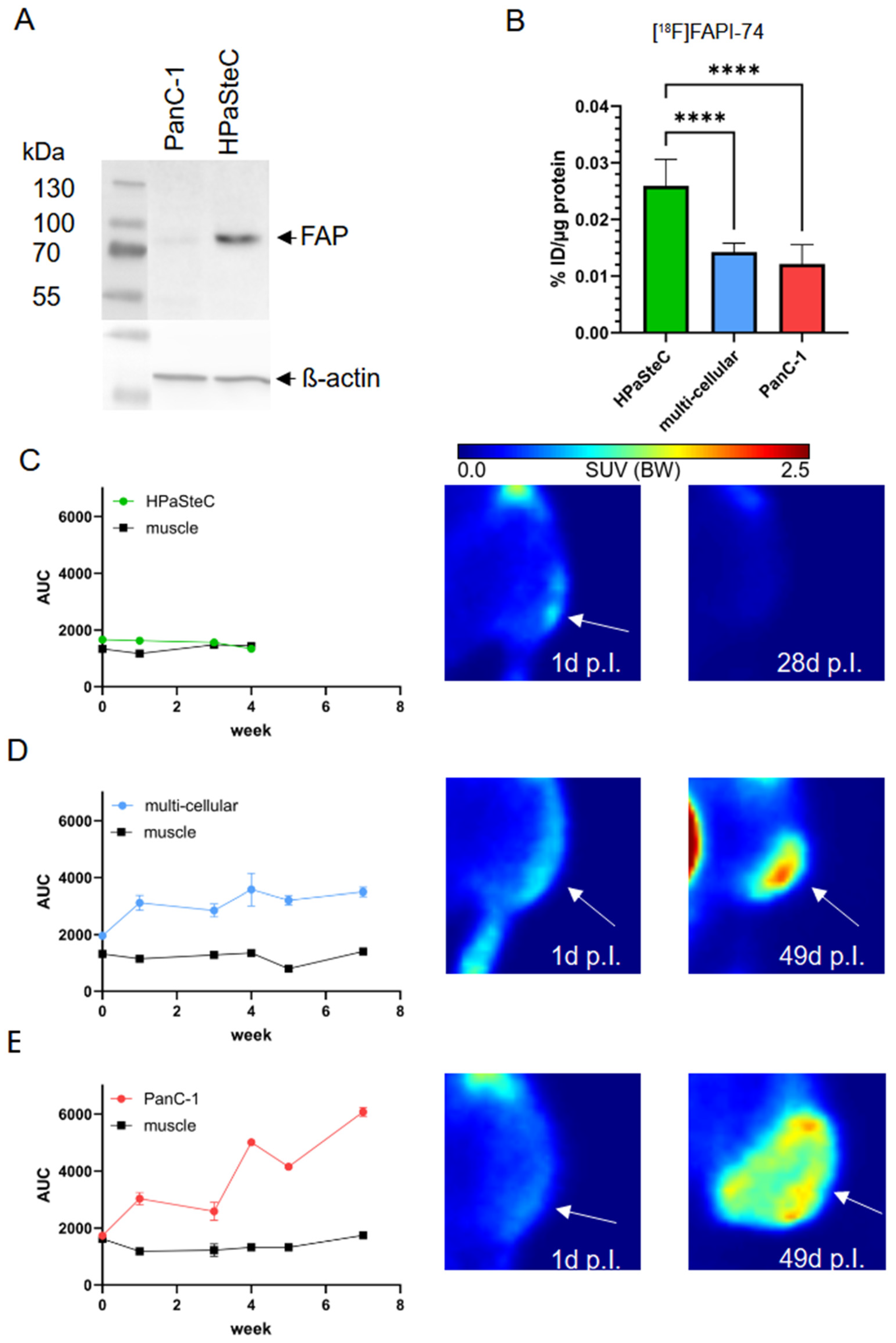
3.4. FAPα as Marker of PSCs
3.5. Immunohistological Staining
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, C.; Morvaridi, S.; Lam, G.; Chheda, C.; Kamata, Y.; Katsumata, M.; Edderkaoui, M.; Yuan, X.; Nissen, N.; Pandol, S.J.; et al. MSP-RON Signaling Is Activated in the Transition From Pancreatic Intraepithelial Neoplasia (PanIN) to Pancreatic Ductal Adenocarcinoma (PDAC). Front. Physiol. 2019, 10, 147. [Google Scholar] [CrossRef] [PubMed]
- Large, T.Y.S.L.; Bijlsma, M.F.; Kazemier, G.; Laarhoven, H.W.M.v.; Giovannetti, E.; Jimenez, C.R. Key biological processes driving metastatic spread of pancreatic cancer as identified by multi-omics studies. Semin. Cancer Biol. 2017, 44, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.-Z.; Cui, Z.-M.; Liu, X. Current developments, problems and solutions in the non-surgical treatment of pancreatic cancer. World J. Gastrointest. Oncol. 2013, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Vasseur, S.; Guillaumond, F. LDL Receptor: An open route to feed pancreatic tumor cells. Mol. Cell. Oncol. 2015, 3, e1033586. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells: A starring role in normal and diseased pancreas. Front. Physiol. 2012, 3, 344. [Google Scholar] [CrossRef] [PubMed]
- Phillips, P. Pancreatic stellate cells and fibrosis. In Pancreatic Cancer and Tumor Microenvironment; Grippo, P.J., Munshi, H.G., Eds.; Transworld Research Network: Trivandrum, India, 2012. [Google Scholar]
- McCarroll, J.A.; Naim, S.; Sharbeen, G.; Russia, N.; Lee, J.; Kavallaris, M.; Goldstein, D.; Phillips, P.A. Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Front. Physiol. 2014, 5, 141. [Google Scholar] [CrossRef]
- Erkan, M.; Adler, G.; Apte, M.V.; Bachem, M.G.; Buchholz, M.; Detlefsen, S.o.; Esposito, I.; Friess, H.; Gress, T.M.; Habisch, H.-J.; et al. StellaTUM: Current consensus and discussion on pancreatic stellate cell research. Gut 2011, 61, 172–178. [Google Scholar] [CrossRef]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Models Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef]
- Jo, Y.; Choi, N.; Kim, K.; Koo, H.-J.; Choi, J.; Kim, H.N. Chemoresistance of cancer cells: Requirements of tumor microenvironment-mimicking in vitro models in anti-cancer drug development. Theranostics 2018, 8, 5259. [Google Scholar] [CrossRef]
- Stylianopoulos, T.; Munn, L.L.; Jain, R.K. Reengineering the physical microenvironment of tumors to improve drug delivery and efficacy: From mathematical modeling to bench to bedside. Trends Cancer 2018, 4, 292–319. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.L.; Bankson, J.A.; Lemos, R., Jr.; Lai, S.Y.; Thittai, A.K.; He, Y.; Hostetter, G.; Demeure, M.J.; Von Hoff, D.D.; Powis, G. Radiosensitization and stromal imaging response correlates for the HIF-1 inhibitor PX-478 given with or without chemotherapy in pancreatic cancer. Mol. Cancer Ther. 2010, 9, 2057–2067. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Tashiro, K.; Dixit, A.; Soni, A.; Vogel, K.; Hall, B.; Shafqat, I.; Slaughter, J.; Param, N.; Le, A. Loss of HIF1A from pancreatic cancer cells increases expression of PPP1R1B and degradation of p53 to promote invasion and metastasis. Gastroenterology 2020, 159, 1882–1897.e5. [Google Scholar] [CrossRef]
- Schwartz, D.L.; Powis, G.; Thitai-Kumar, A.; He, Y.; Bankson, J.; Williams, R.; Lemos, R.; Oh, J.; Volgin, A.; Soghomonyan, S. The selective hypoxia inducible factor-1 inhibitor PX-478 provides in vivo radiosensitization through tumor stromal effects. Mol. Cancer Ther. 2009, 8, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Yang, G.; Zhou, W.; Qiu, J.; Chen, G.; Luo, W.; Zhao, F.; You, L.; Zheng, L.; Zhang, T. Targeting hypoxic tumor microenvironment in pancreatic cancer. J. Hematol. Oncol. 2021, 14, 14. [Google Scholar] [CrossRef]
- Sahani, D.V.; Bonaffini, P.A.; Catalano, O.A.; Guimaraes, A.R.; Blake, M.A. State-of-the-art PET/CT of the pancreas: Current role and emerging indications. Radiographics 2012, 32, 1133–1158. [Google Scholar] [CrossRef]
- Jha, P.; Bijan, B. PET/CT for pancreatic malignancy: Potential and pitfalls. J. Nucl. Med. Technol. 2015, 43, 92–97. [Google Scholar] [CrossRef]
- Lindner, T.; Loktev, A.; Giesel, F.; Kratochwil, C.; Altmann, A.; Haberkorn, U. Targeting of activated fibroblasts for imaging and therapy. EJNMMI Radiopharm. Chem. 2019, 4, 16. [Google Scholar] [CrossRef]
- Wen, Z.; Liu, Q.; Wu, J.; Xu, B.; Wang, J.; Liang, L.; Guo, Y.; Peng, M.; Zhao, Y.; Liao, Q. Fibroblast activation protein α-positive pancreatic stellate cells promote the migration and invasion of pancreatic cancer by CXCL1-mediated Akt phosphorylation. Ann. Transl. Med. 2019, 7, 532. [Google Scholar] [CrossRef]
- Suklabaidya, S.; Dash, P.; Das, B.; Suresh, V.; Sasmal, P.K.; Senapati, S. Experimental models of pancreatic cancer desmoplasia. Lab. Investig. 2018, 98, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Puré, E.; Blomberg, R. Pro-tumorigenic roles of fibroblast activation protein in cancer: Back to the basics. Oncogene 2018, 37, 4343–4357. [Google Scholar] [CrossRef] [PubMed]
- Teichgräber, V.; Monasterio, C.; Chaitanya, K.; Boger, R.; Gordon, K.; Dieterle, T.; Jäger, D.; Bauer, S. Specific inhibition of fibroblast activation protein (FAP)-alpha prevents tumor progression in vitro. Adv. Med. Sci. 2015, 60, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, B.; Lange, C.S. Cell-cell interactions in spheroids maintained in suspension. Acta Oncol. 2006, 45, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Breslin, S.; O’Driscoll, L. Three-dimensional cell culture: The missing link in drug discovery. Drug Discov. Today 2013, 18, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Doctor, A.; Seifert, V.; Ullrich, M.; Hauser, S.; Pietzsch, J. Three-Dimensional Cell Culture Systems in Radiopharmaceutical Cancer Research. Cancers 2020, 12, 2765. [Google Scholar] [CrossRef]
- Seifert, V.; Richter, S.; Bechmann, N.; Bachmann, M.; Ziegler, C.G.; Pietzsch, J.; Ullrich, M. HIF2alpha-Associated Pseudohypoxia Promotes Radioresistance in Pheochromocytoma: Insights from 3D Models. Cancers 2021, 13, 385. [Google Scholar] [CrossRef]
- Reissenweber, B.; Mosch, B.; Pietzsch, J. Experimental hypoxia does not influence gene expression and protein synthesis of Eph receptors and ephrin ligands in human melanoma cells in vitro. Melanoma Res. 2013, 23, 85–95. [Google Scholar] [CrossRef]
- Kpeglo, D.; Hughes, M.D.; Dougan, L.; Haddrick, M.; Knowles, M.A.; Evans, S.D.; Peyman, S.A. Modeling the mechanical stiffness of pancreatic ductal adenocarcinoma. Matrix Biol. Plus 2022, 14, 100109. [Google Scholar] [CrossRef]
- Nam, S.; Khawar, I.A.; Park, J.K.; Chang, S.; Kuh, H.-J. Cellular context-dependent interaction between cancer and stellate cells in hetero-type multicellular spheroids of pancreatic tumor. Biochem. Biophys. Res. Commun. 2019, 515, 183–189. [Google Scholar] [CrossRef]
- Kniess, T.; Zessin, J.; Mäding, P.; Kuchar, M.; Kiss, O.; Kopka, K. Synthesis of [18F] FMISO, a hypoxia-specific imaging probe for PET, an overview from a radiochemist’s perspective. EJNMMI Radiopharm. Chem. 2023, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Laube, M.; Pietzsch, J.; Kniess, T.; Kopka, K.; Neels, O. Optimization and automation of radiolabeling FAPI-74 using [F-18] AlF chemistry. In Proceedings of the European Journal of Nuclear Medicine and Molecular Imaging, New York, NY, USA, 1 September 2021; p. S221. [Google Scholar]
- Ullrich, M.; Bergmann, R.; Peitzsch, M.; Zenker, E.F.; Cartellieri, M.; Bachmann, M.; Ehrhart-Bornstein, M.; Block, N.L.; Schally, A.V.; Eisenhofer, G.; et al. Multimodal somatostatin receptor theranostics using [64Cu]Cu-/[177Lu]Lu-DOTA-(Tyr3)octreotate and AN-238 in a mouse pheochromocytoma model. Theranostics 2016, 6, 650–665. [Google Scholar] [CrossRef]
- Ullrich, M.; Richter, S.; Liers, J.; Drukewitz, S.; Friedemann, M.; Kotzerke, J.; Ziegler, C.G.; Nölting, S.; Kopka, K.; Pietzsch, J. Epigenetic drugs in somatostatin type 2 receptor radionuclide theranostics and radiation transcriptomics in mouse pheochromocytoma models. Theranostics 2023, 13, 278. [Google Scholar] [CrossRef] [PubMed]
- Rothe, R.; Xu, Y.; Thomas, A.K.; Meister, S.; Zhang, Y.; Pietzsch, J.; Hauser, S. A modular, injectable, non-covalently assembled hydrogel system features widescale tunable degradability for controlled release and tissue integration. Biomaterials 2021, 269, 120637. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.; Haase-Kohn, C.; Lenk, J.; Hoppmann, S.; Bergmann, R.; Steinbach, J.; Pietzsch, J. Expression, purification and fluorine-18 radiolabeling of recombinant S100A4: A potential probe for molecular imaging of receptor for advanced glycation endproducts in vivo? Amino Acids 2011, 41, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Van der Slot, A.J.; Zuurmond, A.-M.; Bardoel, A.F.; Wijmenga, C.; Pruijs, H.E.; Sillence, D.O.; Brinckmann, J.; Abraham, D.J.; Black, C.M.; Verzijl, N. Identification of PLOD2 as telopeptide lysyl hydroxylase, an important enzyme in fibrosis. J. Biol. Chem. 2003, 278, 40967–40972. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, H.; Liu, T.; Chen, T.; Wang, D.; Tang, D. Heterogeneous pancreatic stellate cells are powerful contributors to the malignant progression of pancreatic cancer. Front. Cell Dev. Biol. 2021, 9, 783617. [Google Scholar] [CrossRef]
- Ogawa, Y.; Masugi, Y.; Abe, T.; Yamazaki, K.; Ueno, A.; Fujii-Nishimura, Y.; Hori, S.; Yagi, H.; Abe, Y.; Kitago, M. Three distinct stroma types in human pancreatic cancer identified by image analysis of fibroblast subpopulations and collagen. Clin. Cancer Res. 2021, 27, 107–119. [Google Scholar] [CrossRef]
- Hirschhaeuser, F.; Menne, H.; Dittfeld, C.; West, J.; Mueller-Klieser, W.; Kunz-Schughart, L.A. Multicellular tumor spheroids: An underestimated tool is catching up again. J. Biotechnol. 2010, 148, 3–15. [Google Scholar] [CrossRef]
- Ware, M.J.; Colbert, K.; Keshishian, V.; Ho, J.; Corr, S.J.; Curley, S.A.; Godin, B. Generation of Homogenous Three-Dimensional Pancreatic Cancer Cell Spheroids Using an Improved Hanging Drop Technique. Tissue Eng. Part C Methods 2016, 22, 312–321. [Google Scholar] [CrossRef]
- Dardousis, K.; Voolstra, C.; Roengvoraphoj, M.; Sekandarzad, A.; Mesghenna, S.; Winkler, J.; Ko, Y.; Hescheler, J.; Sachinidis, A. Identification of differentially expressed genes involved in the formation of multicellular tumor spheroids by HT-29 colon carcinoma cells. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Firuzi, O.; Che, P.P.; Hassouni, B.E.; Buijs, M.; Coppola, S.; Löhr, M.; Funel, N.; Heuchel, R.; Carnevale, I.; Schmidt, T.; et al. Role of c-MET Inhibitors in Overcoming Drug Resistance in Spheroid Models of Primary Human Pancreatic Cancer and Stellate Cells. Cancers 2019, 11, 638. [Google Scholar] [CrossRef] [PubMed]
- Kuen, J.; Darowski, D.; Kluge, T.; Majety, M. Pancreatic cancer cell/fibroblast co-culture induces M2 like macrophages that influence therapeutic response in a 3D model. PLoS ONE 2017, 12, e0182039. [Google Scholar] [CrossRef] [PubMed]
- Norberg, K.; Liu, X.; Fernández Moro, C.; Strell, C.; Nania, S.; Blümel, M.; Balboni, A.; Bozóky, B.; Heuchel, R.; Löhr, J. A novel pancreatic tumour and stellate cell 3D co-culture spheroid model. BMC Cancer 2020, 20, 475. [Google Scholar] [CrossRef] [PubMed]
- Lenggenhager, D.; Amrutkar, M.; Sántha, P.; Aasrum, M.; Löhr, J.-M.; Gladhaug, I.; Verbeke, C. Commonly Used Pancreatic Stellate Cell Cultures Differ Phenotypically and in Their Interactions with Pancreatic Cancer Cells. Cells 2019, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Vonlaufen, A.; Joshi, S.; Qu, C.; Phillips, P.A.; Xu, Z.; Parker, N.R.; Toi, C.S.; Pirola, R.C.; Wilson, J.S.; Goldstein, D.; et al. Pancreatic Stellate Cells: Partners in Crime with Pancreatic Cancer Cells. Cancer Res. 2008, 68, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Koong, A.C.; Mehta, V.K.; Le, Q.T.; Fisher, G.A.; Terris, D.J.; Brown, J.M.; Bastidas, A.J.; Vierra, M. Pancreatic tumors show high levels of hypoxia. Int. J. Radiat. Oncol. Biol. Phys. 2000, 48, 919–922. [Google Scholar] [CrossRef]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar]
- Masamune, A.; Kikuta, K.; Watanabe, T.; Satoh, K.; Hirota, M.; Shimosegawa, T. Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G709–G717. [Google Scholar] [CrossRef]
- Bynigeri, R.R.; Jakkampudi, A.; Jangala, R.; Subramanyam, C.; Sasikala, M.; Rao, G.V.; Reddy, D.N.; Talukdar, R. Pancreatic stellate cell: Pandora’s box for pancreatic disease biology. World J. Gastroenterol. 2017, 23, 382. [Google Scholar] [CrossRef]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic Reaction in Pancreatic Cancer: Role of Pancreatic Stellate Cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Sun, B.; Li, W.; Zhou, J.; Gao, F.; Wang, X.; Cai, M.; Sun, Z. Pancreatic Stellate Cells: A Rising Translational Physiology Star as a Potential Stem Cell Type for Beta Cell Neogenesis. Front. Physiol. 2019, 10, 218. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Wilson, J.S. Dangerous liaisons: Pancreatic stellate cells and pancreatic cancer cells. J. Gastroenterol. Hepatol. 2012, 27, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Habisch, H.; Zhou, S.; Siech, M.; Bachem, M.G. Interaction of stellate cells with pancreatic carcinoma cells. Cancers 2010, 2, 1661–1682. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Wagner, M.; Ellenrieder, V.; Bachem, M.; Gress, T.M.; Buchholz, M. Pancreatic Stellate Cells Potentiate Proinvasive Effects of SERPINE2 Expression in Pancreatic Cancer Xenograft Tumors. Pancreatology 2007, 7, 380–385. [Google Scholar] [CrossRef]
- Vonlaufen, A.; Phillips, P.A.; Xu, Z.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Pancreatic Stellate Cells and Pancreatic Cancer Cells: An Unholy Alliance. Cancer Res. 2008, 68, 7707–7710. [Google Scholar] [CrossRef]
- Hauge, A.; Wegner, C.S.; Gaustad, J.-V.; Simonsen, T.G.; Andersen, L.M.K.; Rofstad, E.K. Diffusion-weighted MRI-derived ADC values reflect collagen I content in PDX models of uterine cervical cancer. Oncotarget 2017, 8, 105682. [Google Scholar] [CrossRef]
- Wegner, C.S.; Gaustad, J.-V.; Andersen, L.M.K.; Simonsen, T.G.; Rofstad, E.K. Diffusion-weighted and dynamic contrast-enhanced MRI of pancreatic adenocarcinoma xenografts: Associations with tumor differentiation and collagen content. J. Transl. Med. 2016, 14, 161. [Google Scholar] [CrossRef]
- Loktev, A.; Lindner, T.; Mier, W.; Debus, J.; Altmann, A.; Jäger, D.; Giesel, F.; Kratochwil, C.; Barthe, P.; Roumestand, C. A tumor-imaging method targeting cancer-associated fibroblasts. J. Nucl. Med. 2018, 59, 1423–1429. [Google Scholar] [CrossRef]
- Mori, Y.; Dendl, K.; Cardinale, J.; Kratochwil, C.; Giesel, F.L.; Haberkorn, U. FAPI PET: Fibroblast activation protein inhibitor use in oncologic and nononcologic disease. Radiology 2023, 306, e220749. [Google Scholar] [CrossRef]
- Mori, Y.; Haberkorn, U.; Giesel, F.L. 68Ga- or 18F-FAPI PET/CT—What it can and cannot. Eur. Radiol. 2023, 33, 7877–7878. [Google Scholar] [CrossRef] [PubMed]
- Erkan, M.; Michalski, C.W.; Rieder, S.; Reiser–Erkan, C.; Abiatari, I.; Kolb, A.; Giese, N.A.; Esposito, I.; Friess, H.; Kleeff, J. The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 2008, 6, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Kishi, S.; Takeyama, Y.; Ueda, T.; Yasuda, T.; Shinzeki, M.; Kuroda, Y.; Yokozaki, H. Pancreatic duct obstruction itself induces expression of α smooth muscle actin in pancreatic stellate cells. J. Surg. Res. 2003, 114, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Vonlaufen, A.; Phillips, P.A.; Yanga, L.; Xua, Z.; Fiala-Beera, E.; Zhanga, X.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Isolation of quiescent human pancreatic stellate cells: A promising in vitro tool for studies of human pancreatic stellate cell biology. Pancreatology 2010, 10, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Schneiderhan, W.; Diaz, F.; Fundel, M.; Zhou, S.; Siech, M.; Hasel, C.; Moller, P.; Gschwend, J.E.; Seufferlein, T.; Gress, T.; et al. Pancreatic stellate cells are an important source of MMP-2 in human pancreatic cancer and accelerate tumor progression in a murine xenograft model and CAM assay. J. Cell Sci. 2007, 120, 512–519. [Google Scholar] [CrossRef]
- Biswas, C.; Bloch, K.J.; Gross, J. Collagenolytic activity of rabbit V2 carcinoma implanted in the nude mouse. J. Natl. Cancer Inst. 1982, 69, 1329–1336. [Google Scholar]
- Olsen, C.J.; Moreira, J.; Lukanidin, E.M.; Ambartsumian, N.S. Human mammary fibroblasts stimulate invasion of breast cancer cells in a three-dimensional culture and increase stroma development in mouse xenografts. BMC Cancer 2010, 10, 444. [Google Scholar] [CrossRef]
- Scarlett, C.J.; Colvin, E.K.; Pinese, M.; Chang, D.K.; Morey, A.L.; Musgrove, E.A.; Pajic, M.; Apte, M.; Henshall, S.M.; Sutherland, R.L. Recruitment and activation of pancreatic stellate cells from the bone marrow in pancreatic cancer: A model of tumor-host interaction. PLoS ONE 2011, 6, e26088. [Google Scholar] [CrossRef]
- Krüger, J.; Fischer, A.; Breunig, M.; Allgöwer, C.; Schulte, L.; Merkle, J.; Mulaw, M.A.; Okeke, N.; Melzer, M.K.; Morgenstern, C. DNA methylation-associated allelic inactivation regulates Keratin 19 gene expression during pancreatic development and carcinogenesis. J. Pathol. 2023, 261, 139–155. [Google Scholar] [CrossRef]
- Yao, H.; Yang, Z.; Liu, Z.; Miao, X.; Yang, L.; Li, D.; Zou, Q.; Yuan, Y. Glypican-3 and KRT19 are markers associating with metastasis and poor prognosis of pancreatic ductal adenocarcinoma. Cancer Biomark. 2016, 17, 397–404. [Google Scholar] [CrossRef]
- Shang, M.; Zhang, L.; Chen, X.; Zheng, S. Identification of hub genes and regulators associated with pancreatic ductal adenocarcinoma based on integrated gene expression profile analysis. Discov. Med. 2019, 28, 159–172. [Google Scholar] [PubMed]
- Saha, S.K.; Kim, K.; Yang, G.-M.; Choi, H.Y.; Cho, S.-G. Cytokeratin 19 (KRT19) has a role in the reprogramming of cancer stem cell-like cells to less aggressive and more drug-sensitive cells. Int. J. Mol. Sci. 2018, 19, 1423. [Google Scholar] [CrossRef]
- Kuony, A.; Michon, F. Epithelial markers aSMA, Krt14, and Krt19 unveil elements of murine lacrimal gland morphogenesis and maturation. Front. Physiol. 2017, 8, 286958. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Pang, M.; Hou, X.; Yuan, S.; Sun, L. PLOD2 in cancer research. Biomed. Pharmacother. 2017, 90, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Laube, M.; Wodtke, R.; Kopka, K.; Kniess, T.; Pietzsch, J. 18F-Chemistry in HPLC vials - a microliter scale radiofluorination approach. Nucl. Med. Biol. 2021, 96-97, S61. [Google Scholar] [CrossRef]
- McBride, W.J.; Sharkey, R.M.; Goldenberg, D.M. Radiofluorination using aluminum-fluoride (Al18F). EJNMMI Res. 2013, 3, 36. [Google Scholar] [CrossRef]
- McBride, W.J.; Sharkey, R.M.; Karacay, H.; D’Souza, C.A.; Rossi, E.A.; Laverman, P.; Chang, C.-H.; Boerman, O.C.; Goldenberg, D.M. A novel method of 18F radiolabeling for PET. J. Nucl. Med. 2009, 50, 991–998. [Google Scholar] [CrossRef]
- Laverman, P.; McBride, W.J.; Sharkey, R.M.; Goldenberg, D.M.; Boerman, O.C. Al18F labeling of peptides and proteins. J. Labelled Compd. Rad. 2014, 57, 219–223. [Google Scholar] [CrossRef]
- Kumar, K.; Ghosh, A. 18F-AlF labeled peptide and protein conjugates as positron emission tomography imaging pharmaceuticals. Bioconjugate Chem. 2018, 29, 953–975. [Google Scholar] [CrossRef]
- Fersing, C.; Bouhlel, A.; Cantelli, C.; Garrigue, P.; Lisowski, V.; Guillet, B. A comprehensive review of non-covalent radiofluorination approaches using aluminum [18F] fluoride: Will [18F] AlF replace 68Ga for metal chelate labeling? Molecules 2019, 24, 2866. [Google Scholar] [CrossRef]
- Allott, L.; Da Pieve, C.; Turton, D.; Smith, G. A general [18 F] AlF radiochemistry procedure on two automated synthesis platforms. React. Chem. Eng. 2017, 2, 68–74. [Google Scholar] [CrossRef]
- Kersemans, K.; De Man, K.; Courtyn, J.; Van Royen, T.; Piron, S.; Moerman, L.; Brans, B.; De Vos, F. Automated radiosynthesis of Al [18F] PSMA-11 for large scale routine use. Appl. Radiat. Isotopes 2018, 135, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Giglio, J.; Zeni, M.; Savio, E.; Engler, H. Synthesis of an Al18F radiofluorinated GLU-UREA-LYS(AHX)-HBED-CC PSMA ligand in an automated synthesis platform. EJNMMI Radiopharm. Chem. 2018, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tshibangu, T.; Cawthorne, C.; Serdons, K.; Pauwels, E.; Gsell, W.; Bormans, G.; Deroose, C.M.; Cleeren, F. Automated GMP compliant production of [18 F] AlF-NOTA-octreotide. EJNMMI Radiopharm. Chem. 2020, 5, 1–23. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, X.; Shen, T.; Yao, Y.; Chen, M.; Li, Z.; Li, X.; Shen, J.; Kou, Y.; Chen, S. FAPI-04 PET/CT using [18F] AlF labeling strategy: Automatic synthesis, quality control, and in vivo assessment in patient. Front. Oncol. 2021, 11, 649148. [Google Scholar] [CrossRef]
- Hu, K.; Li, J.; Wang, L.; Huang, Y.; Li, L.; Ye, S.; Han, Y.; Huang, S.; Wu, H.; Su, J. Preclinical evaluation and pilot clinical study of [18F] AlF-labeled FAPI-tracer for PET imaging of cancer associated fibroblasts. Acta Pharm. Sin. B 2022, 12, 867–875. [Google Scholar] [CrossRef]
- Giesel, F.L.; Adeberg, S.; Syed, M.; Lindner, T.; Jiménez-Franco, L.D.; Mavriopoulou, E.; Staudinger, F.; Tonndorf-Martini, E.; Regnery, S.; Rieken, S. FAPI-74 PET/CT using either 18F-AlF or cold-kit 68Ga labeling: Biodistribution, radiation dosimetry, and tumor delineation in lung cancer patients. . Nucl. Med. 2021, 62, 201–207. [Google Scholar] [CrossRef]
- Naka, S.; Watabe, T.; Lindner, T.; Cardinale, J.; Kurimoto, K.; Moore, M.; Tatsumi, M.; Mori, Y.; Shimosegawa, E.; Valla Jr, F. One-pot and one-step automated radio-synthesis of [18F] AlF-FAPI-74 using a multi purpose synthesizer: A proof-of-concept experiment. EJNMMI Radiopharm. Chem. 2021, 6, 28. [Google Scholar] [CrossRef]
- D’Souza, C.A.; McBride, W.J.; Sharkey, R.M.; Todaro, L.J.; Goldenberg, D.M. High-yielding aqueous 18F-labeling of peptides via Al18F chelation. Bioconjugate Chem. 2011, 22, 1793–1803. [Google Scholar] [CrossRef]
- Laverman, P.; D’souza, C.A.; Eek, A.; McBride, W.J.; Sharkey, R.M.; Oyen, W.J.G.; Goldenberg, D.M.; Boerman, O.C. Optimized labeling of NOTA-conjugated octreotide with F-18. Tumor Biol. 2011, 33, 427–434. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doctor, A.; Laube, M.; Meister, S.; Kiss, O.C.; Kopka, K.; Hauser, S.; Pietzsch, J. Combined PET Radiotracer Approach Reveals Insights into Stromal Cell-Induced Metabolic Changes in Pancreatic Cancer In Vitro and In Vivo. Cancers 2024, 16, 3393. https://doi.org/10.3390/cancers16193393
Doctor A, Laube M, Meister S, Kiss OC, Kopka K, Hauser S, Pietzsch J. Combined PET Radiotracer Approach Reveals Insights into Stromal Cell-Induced Metabolic Changes in Pancreatic Cancer In Vitro and In Vivo. Cancers. 2024; 16(19):3393. https://doi.org/10.3390/cancers16193393
Chicago/Turabian StyleDoctor, Alina, Markus Laube, Sebastian Meister, Oliver C. Kiss, Klaus Kopka, Sandra Hauser, and Jens Pietzsch. 2024. "Combined PET Radiotracer Approach Reveals Insights into Stromal Cell-Induced Metabolic Changes in Pancreatic Cancer In Vitro and In Vivo" Cancers 16, no. 19: 3393. https://doi.org/10.3390/cancers16193393
APA StyleDoctor, A., Laube, M., Meister, S., Kiss, O. C., Kopka, K., Hauser, S., & Pietzsch, J. (2024). Combined PET Radiotracer Approach Reveals Insights into Stromal Cell-Induced Metabolic Changes in Pancreatic Cancer In Vitro and In Vivo. Cancers, 16(19), 3393. https://doi.org/10.3390/cancers16193393