

The Importance of Real-World Data in Evaluating the Safety of Biosimilars: A Descriptive Study of Clinical Practice in an Oncohematological Italian Population

 , , , , , and
, , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Objective
3. Methods
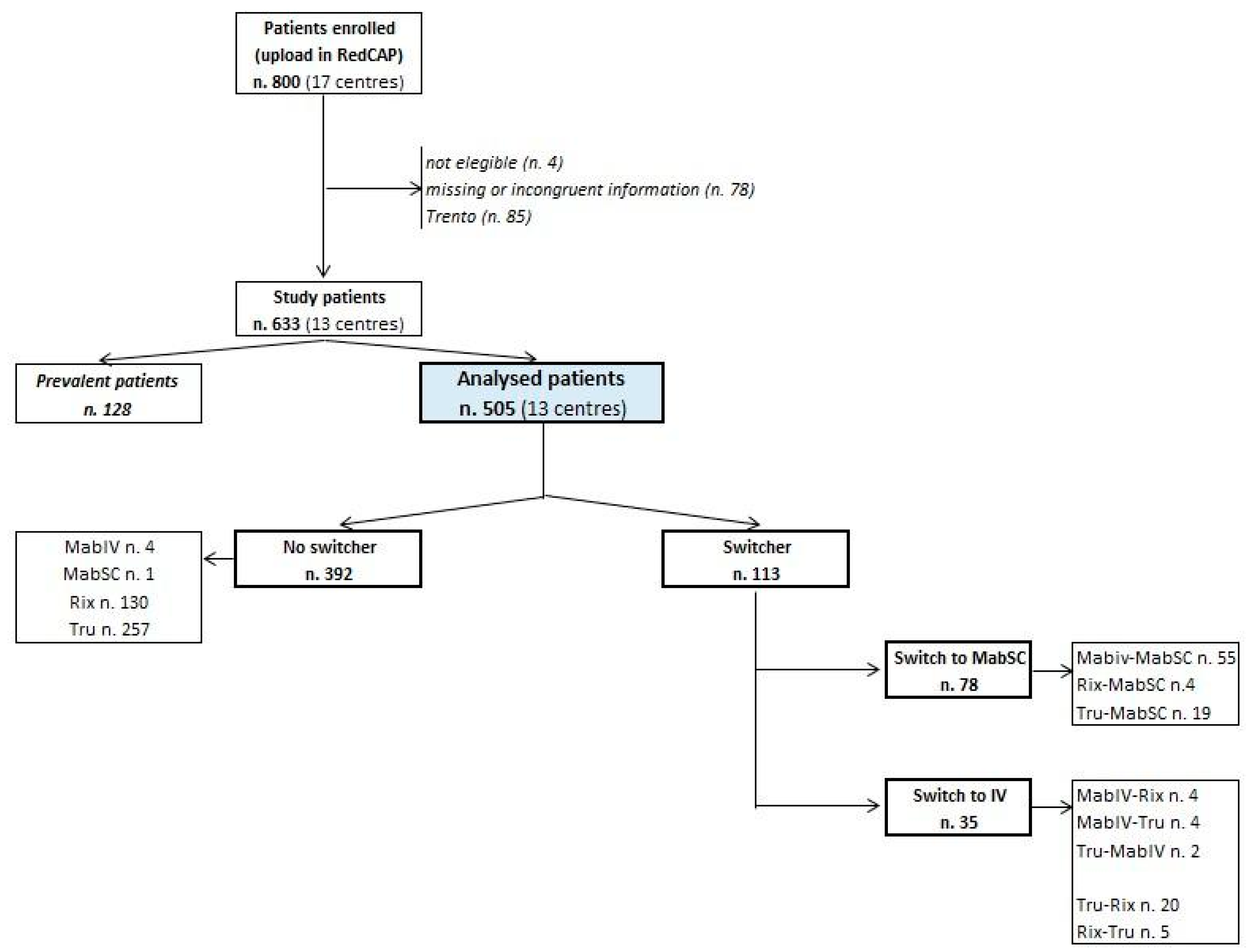
3.1. Study Population
3.2. Data Collection
3.3. Clinical Outcomes
3.4. Statistical Analysis
4. Results
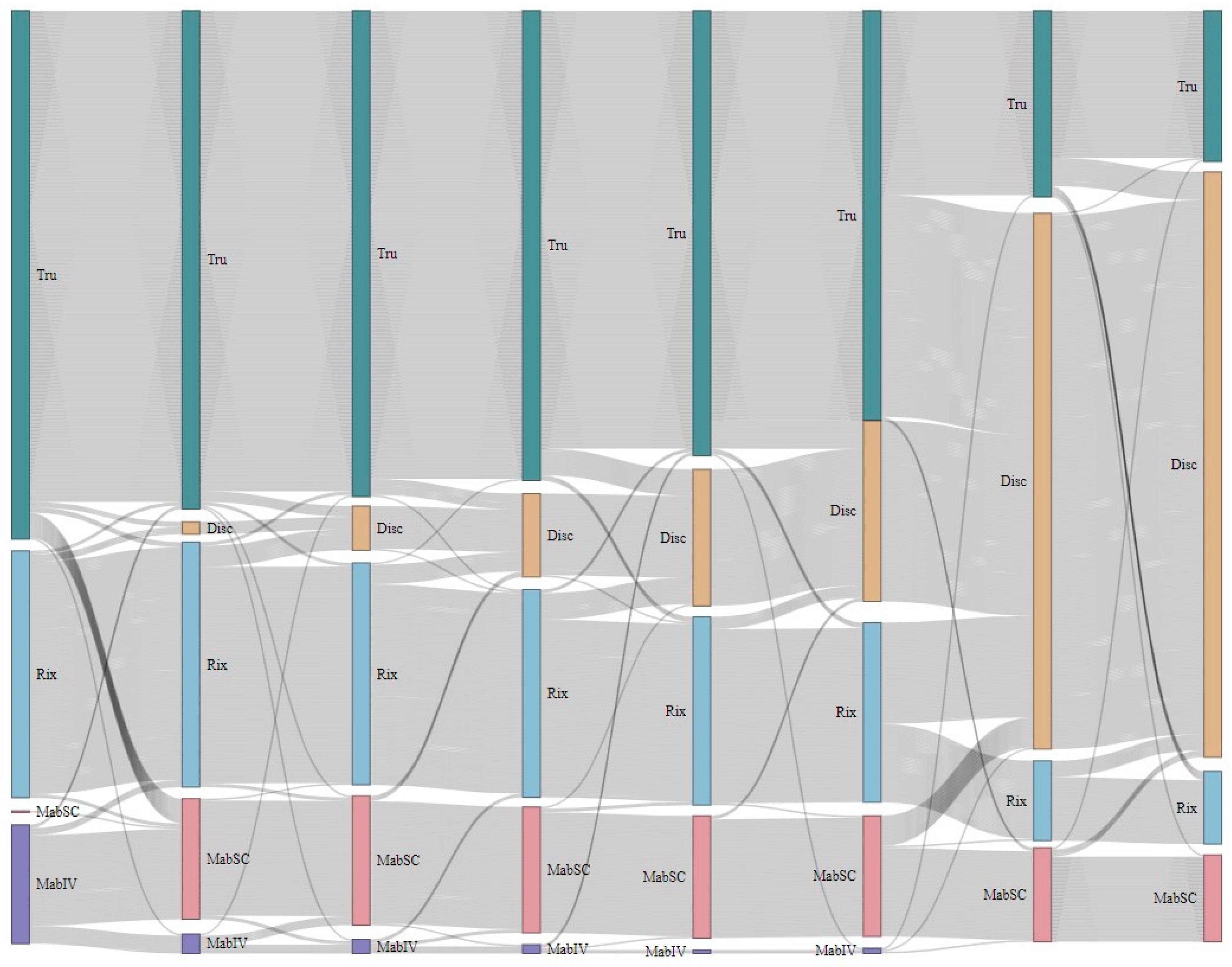
4.1. Rituximab Treatment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All Patients | n. RTX Infusions | Schedule * | ||||
|---|---|---|---|---|---|---|
| Mean ± sd | Median [IQR] | RTX + Chemotherapy | RTX Monotherapy | RTX + Chemotherapy/ RTX Monotherapy | ||
| No switch during the study period | 392 | 6.4 ± 2.7 | 6.0 [6.0–8.0] | 252 (64.3%) | 23 (5.9%) | 111 (28.3%) |
| Originator—Mabthera IV | 4 | 7.3 ± 3.9 | 6.0 [5.0–9.5] | 3 (75.0%) | 0 (0.0%) | 1 (25.0%) |
| Originator—Mabthera SC | 1 | 7.0 | 7.0 | 1 (100.0%) | 0 (0.0%) | 0 (0.0%) |
| Biosimilar—Rixathon | 130 | 5.8 ± 2.4 | 6.0 [4.0–7.0] | 101 (77.7%) | 3 (2.3%) | 25 (19.2%) |
| Biosimilar—Truxima | 257 | 6.7 ± 2.8 | 6.0 [6.0–8.0] | 147 (57.2%) | 20 (7.8%) | 85 (33.1%) |
| Switch during the study period | 113 | 10.5 ± 5.3 | 8.0 [6.0–15.0] | 36 (31.9%) | 2 (1.8%) | 74 (65.5%) |
| sw protocol | 57 | 10.4 ± 5.5 | 8.0 [6.0–15] | 17 (29.8%) | 0 (0.0%) | 40 (70.2%) |
| sw or | 2 | 13.0 ± 7.1 | 13.0 [8.0–18.0] | 0 (0.0%) | 0 (0.0%) | 2 (100.0%) |
| sw ob | 21 | 12.5 ± 5.4 | 13.0 [8.0–17.0] | 4 (19.0%) | 0 (0.0%) | 17 (81.0%) |
| sw bb | 25 | 8.9 ± 4.3 | 8.0 [6.0–10.0] | 10 (40.0%) | 2 (8.0%) | 12 (48.0%) |
| sw bo | 8 | 10 ± 5.4 | 8.5 [7.0–12.5] | 5 (62.5%) | 0 (0.0%) | 3 (37.5%) |
| Total | 505 | 7.3 ± 3.8 | 6.0 [6.0–8.0] | 288 (57.0%) | 25 (5.0%) | 185 (36.6%) |
4.2. Adverse Reactions Related to Rituximab
5. Discussion
Limits of This Study
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Espiritu, M.J.; Collier, A.C.; Bingham, J.P. A 21st-century approach to age-old problems: The ascension of biologics in clinical therapeutics. Drug Discov. Today 2014, 19, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Miller, H.I. Biotech’s defining moments. Trends Biotechnol. 2007, 25, 56–59. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency (EMA). Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins as Active Substance: Non-Clinical and Clinical Issues. 2013. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-2.pdf (accessed on 23 August 2024).
- World Health Organization (WHO). Guidelines on Evaluation of Biosimilars. Available online: https://www.who.int/publications/m/item/guidelines-on-evaluation-of-biosimilars (accessed on 23 August 2024).
- US Food and Drug Administration (FDA). Biosimilar Guidance. Available online: https://www.fda.gov/vaccines-blood-biologics/general-biologics-guidances/biosimilars-guidances (accessed on 23 August 2024).
- Urru, S.A.M.; Spila Alegiani, S.; Guella, A.; Traversa, G.; Campomori, A. Safety of switching between rituximab biosimilars in onco-hematology. Sci. Rep. 2021, 11, 5956. [Google Scholar] [CrossRef] [PubMed]
- Trifirò, G.; Isgrò, V.; Ingrasciotta, Y.; Ientile, V.; L’Abbate, L.; Foti, S.S.; Belleudi, V.; Poggi, F.; Fontana, A.; Moretti, U.; et al. LargeScale Postmarketing Surveillance of Biological Drugs for ImmuneMediated Infammatory Diseases through an Italian Distributed MultiDatabase Healthcare Network: The VALORE Project. BioDrugs 2021, 35, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Italian Medicines Agency. 648/96 List. Available online: https://www.aifa.gov.it/en/legge-648-96 (accessed on 9 November 2023).
- Harris, P.A.; Taylor, R.; Thielke, R.; Payne, J.; Gonzalez, N.; Conde, J.G. Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inform. 2009, 42, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Halimi, V.; Daci, A.; Ancevska Netkovska, K.; Suturkova, L.; Babar, Z.U.; Grozdanova, A. Clinical and Regulatory Concerns of Biosimilars: A Review of Literature. Int. J. Environ. Res. Public Health 2020, 17, 5800. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.P.; Blauvelt, A.; Rifkin, R.M.; Danese, S.; Gokhale, S.B.; Woollett, G. Switching Reference Medicines to Biosimilars: A Systematic Literature Review of Clinical Outcomes. Drugs 2018, 78, 463–478. [Google Scholar] [CrossRef] [PubMed]
- Otremba, B.; Borchardt, J.; Kuske, A.; Hollnagel-Schmitz, M.; Losch, F.O. Real-world use and acceptance of rituximab biosimilars in non-hodgkin lymphoma in an oncologist network in Germany. Future Oncol. 2020, 16, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zheng, Z.; Li, N.; Zheng, B.; Liu, M.; Cai, H. Efficacy and safety of rituximab biosimilars or reference product as first-line treatment in patients with low-tumour-burden follicular lymphoma: A systematic review and meta-analysis. J. Clin. Pharm. Ther. 2022, 47, 1923–1931. [Google Scholar] [CrossRef] [PubMed]
- Poddubnaya, I.V.; Alekseev, S.M.; Kaplanov, K.D.; Lukavetskyy, L.M.; Rekhtman, G.B.; Dolai, T.K.; Attili, V.S.S.; Bermúdez, C.D.; Isaev, A.A.; Chernyaeva, E.V.; et al. Proposed rituximab biosimilar BCD-020 versus reference rituximab for treatment of patients with indolent non-Hodgkin lymphomas: An international multicenter randomized trial. Hematol. Oncol. 2020, 38, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Moots, R.; Azevedo, V.; Coindreau, J.L.; Dörner, T.; Mahgoub, E.; Mysler, E.; Scheinberg, M.; Marshall, L. Switching between reference biologics and biosimilars for the treatment of rheumatology, gastroenterology, and dermatology inflammatory conditions: Considerations for the clinician. Curr. Rheumatol. Rep. 2017, 19, 37. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, K.K.; Olsen, I.C.; Goll, G.L.; Lorentzen, M.; Bolstad, N.; Haavardsholm, E.A.; Lundin, K.E.A.; Mørk, C.; Jahnsen, J.; Kvien, T.K. Switching from originator infliximab to biosimilar CT-P13 compared with maintained treatment with originator infliximab (NOR-SWITCH): A 52-week, randomised, double-blind, non-inferiority trial. Lancet 2017, 389, 2304–2316. [Google Scholar] [CrossRef] [PubMed]
- Glintborg, B.; Sørensen, I.J.; Loft, A.G.; Lindegaard, H.; Linauskas, A.; Hendricks, O.; Hansen, I.M.J.; Jensen, D.V.; Manilo, N.; Espesen, J.; et al. A nationwide non-medical switch from originator infliximab to biosimilar CT-P13 in 802 patients with inflammatory arthritis: 1-year clinical outcomes from the DANBIO registry. Ann. Rheum. Dis. 2017, 76, 1426–1431. [Google Scholar] [CrossRef] [PubMed]


| Characteristics | All Patients | Switch to MabSC | Switch to IV (OR/BIO) | No Switchers | p |
|---|---|---|---|---|---|
| Number of patients | 505 | 78 | 35 | 392 | |
| Female [n. (%)] | 212 (42.0%) | 29 (37.2%) | 12 (34.3%) | 171 (43.6%) | 0.36 |
| BMI (kg/m2) [median (IQR)] | 25.0 (22.5–27.6) | 25.8 (22.8–28.3) | 24.4 (21.9–28.1) | 25.0 (22.5–27.3) | 0.43 |
| Age at diagnosis (years) [median (IQR)] | 65.8 (57.0–73.3) | 67.1 (56.1–73.4) | 68.8 (59.4–73.8) | 65.3 (57.1–73.2) | 0.50 |
| Age at baseline (years) [median (IQR)] | 66.8 (57.5–73.9) | 67.6 (57.1–73.8) | 69.2 (59.8–74.1) | 66.6 (57.4–74.0) | 0.59 |
| Duration of disease (days) [median (IQR)] | 390.0 (276.0–637.0) | 534.0 (388.0–787.0) | 732.0 (358.0–965.5) | 361.0 (265.0–544.0) | <0.001 |
| Diagnosis | <0.001 | ||||
| Indolent non-Hodgkin lymphoma | 96 (19.0%) | 28 (35.9%) | 5 (14.3%) | 63 (16.1%) | |
| Aggressive non-Hodgkin lymphoma | 244 (48.3%) | 49 (62.8%) | 10 (28.6%) | 185 (47.2%) | |
| Unspecified non-Hodgkin lymphoma | 113 (22.4%) | 1 (1.3%) | 17 (48.6%) | 95 (24.2%) | |
| Chronic lymphocytic leukemia | 33 (6.5%) | 0 (0.0%) | 2 (5.7%) | 31 (7.9%) | |
| Law 648-96 | 19 (3.8%) | 0 (0.0%) | 1 (2.9%) | 18 (4.6%) | |
| Time of follow-up (days) [median (IQR)] | 317 (217–461) | 445 (335–812) | 498 (255–823) | 295 (210–404) | <0.001 |
| Number of concomitant medications | 0.96 | ||||
| 0 | 114 (22.6%) | 20 (25.6%) | 7 (20.0%) | 87 (22.2%) | |
| 1–3 | 246 (48.7%) | 37 (47.4%) | 17 (48.6%) | 192 (49.0%) | |
| ≥4 | 145 (28.7%) | 21 (26.9%) | 11 (31.4%) | 113 (28.8%) | |
| Number of comorbidities | 0.55 | ||||
| 0 | 161 (31.9%) | 25 (32.1%) | 10 (28.6%) | 126 (32.1%) | |
| 1–2 | 179 (35.4%) | 27 (34.6%) | 17 (48.6%) | 135 (34.4%) | |
| ≥3 | 165 (32.7%) | 26 (33.3%) | 8 (22.9%) | 131 (33.4%) | |
| Performance status [n (%)] | 0.035 | ||||
| 0 | 210 (41.6%) | 39 (50.0%) | 6 (17.1%) | 165 (42.1%) | |
| 1 | 156 (30.9%) | 23 (29.5%) | 15 (42.9%) | 118 (30.1%) | |
| 2 | 25 (5.0%) | 1 (1.3%) | 2 (5.7%) | 22 (5.6%) | |
| ≥3 | 14 (2.8%) | 0 (0.0%) | 1 (2.9%) | 13 (3.3%) | |
| NA | 100 (19.8%) | 15 (19.2%) | 11 (31.4%) | 74 (18.9%) | |
| Number of infusions [mean] | 7.3 ± 3.8 | 10.8 ± 5.3 | 9.7 ± 5.3 | 6.4 ± 2.7 | <0.001 |
| All Patients | n. At Least 1 ADR | % At Least 1 ADR | |
|---|---|---|---|
| Diagnosis | |||
| Aggressive non-Hodgkin lymphoma | 244 | 38 | 15.6 |
| Unspecified non-Hodgkin lymphoma | 113 | 21 | 18.6 |
| Indolent non-Hodgkin lymphoma | 96 | 18 | 18.8 |
| Chronic lymphocytic leukemia | 33 | 5 | 15.2 |
| Law 648-96 | 19 | 3 | 15.8 |
| No switch during the study period | 392 | 81 | 20.7 |
| Originator—Mabthera IV | 4 | 3 | 75.0 |
| Originator—Mabthera SC | 1 | 0 | 0.0 |
| Biosimilar–Rixathon | 130 | 32 | 24.6 |
| Biosimilar—Truxima | 257 | 46 | 17.9 |
| Switch during the study period | 113 | 4 | 3.5 |
| sw protocol | 57 | 1 | 1.8 |
| sw or | 2 | 0 | 0.0 |
| sw ob | 21 | 1 | 4.8 |
| sw bb | 25 | 2 | 8.0 |
| sw bo | 8 | 0 | 0.0 |
| Total | 505 | 85 | 16.8 |
| Type of Events (Cases, n = 85) | ADRs Related to Rituximab | ||||
|---|---|---|---|---|---|
| n. Any Grade | n. Grade 1–2 | n. Grade 3–5 | % Grade 1–2 | % Grade 3–5 | |
| General disorders and administration site conditions | 50 | 46 | 4 | 92.0 | 8.0 |
| Respiratory, thoracic, and mediastinal disorders | 16 | 15 | 1 | 93.8 | 6.3 |
| Gastrointestinal disorders | 12 | 10 | 2 | 83.3 | 16.7 |
| Infections and infestations | 11 | 9 | 2 | 81.8 | 18.2 |
| Blood and lymphatic system disorders | 8 | 2 | 6 | 25.0 | 75.0 |
| Skin and subcutaneous tissue disorders | 8 | 8 | 0 | 100.0 | 0.0 |
| Nervous system disorders | 6 | 4 | 2 | 66.7 | 33.3 |
| Musculoskeletal and connective tissue disorders | 5 | 4 | 1 | 80.0 | 20.0 |
| Cardiac disorders | 4 | 3 | 1 | 75.0 | 25.0 |
| Eye disorders | 1 | 1 | 0 | 100.0 | 0.0 |
| Psychiatric disorders | 1 | 1 | 0 | 100.0 | 0.0 |
| Secondary cancer | 1 | 0 | 1 | 0.0 | 100.0 |
| Vascular disorders | 1 | 1 | 0 | 100.0 | 0.0 |
| Total | 124 | 104 | 20 | 83.9 | 16.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urru, S.A.M.; Mayer, F.; Spila Alegiani, S.; Paoloni, F.; Guella, A.; Murru, R.; Bucaneve, G.; Formoso, G.; Racanelli, V.; Ferrarini, I.; et al. The Importance of Real-World Data in Evaluating the Safety of Biosimilars: A Descriptive Study of Clinical Practice in an Oncohematological Italian Population. Cancers 2024, 16, 3419. https://doi.org/10.3390/cancers16193419
Urru SAM, Mayer F, Spila Alegiani S, Paoloni F, Guella A, Murru R, Bucaneve G, Formoso G, Racanelli V, Ferrarini I, et al. The Importance of Real-World Data in Evaluating the Safety of Biosimilars: A Descriptive Study of Clinical Practice in an Oncohematological Italian Population. Cancers. 2024; 16(19):3419. https://doi.org/10.3390/cancers16193419
Chicago/Turabian StyleUrru, Silvana A. M., Flavia Mayer, Stefania Spila Alegiani, Francesca Paoloni, Anna Guella, Roberta Murru, Giampaolo Bucaneve, Giulio Formoso, Vito Racanelli, Isacco Ferrarini, and et al. 2024. "The Importance of Real-World Data in Evaluating the Safety of Biosimilars: A Descriptive Study of Clinical Practice in an Oncohematological Italian Population" Cancers 16, no. 19: 3419. https://doi.org/10.3390/cancers16193419