
Beyond PD(L)-1 Blockade in Microsatellite-Instable Cancers: Current Landscape of Immune Co-Inhibitory Receptor Targeting
 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
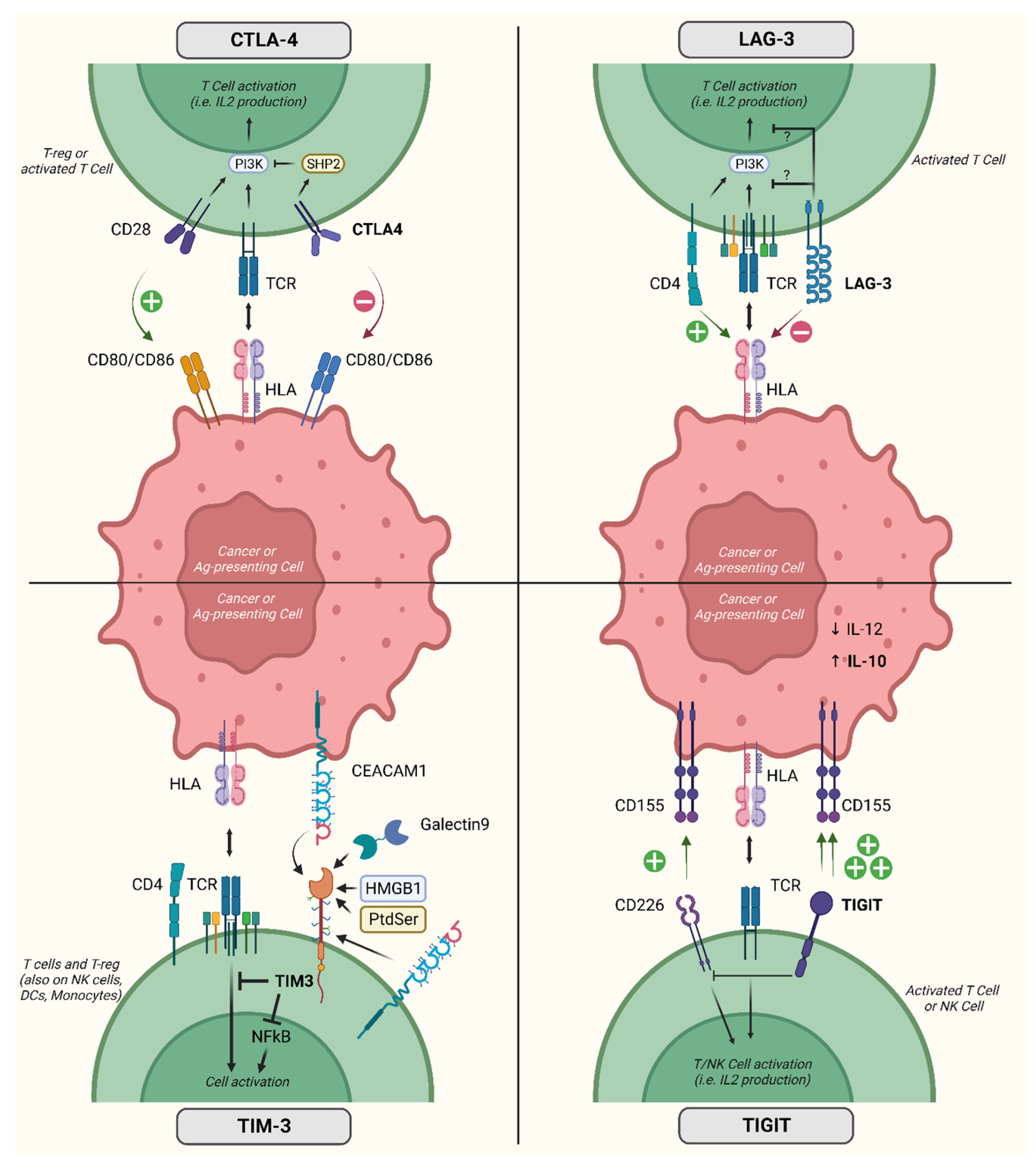
2. Immune Checkpoint Molecules
3. Clinical Evidence on Co-Inhibitory Receptor Targeting in MSI-H Cancers
3.1. CTLA4
3.2. TIM-3
3.3. LAG
3.4. TIGIT
4. Ongoing Trials Enrolling Patients with MSI-H Cancers
5. Other Immunotherapeutic Strategies for MSI-H Cancers
5.1. Tumor Microenvironment
5.2. Cytokines and Cytokine Superagonists
5.3. Bispecific T-Cell Engagers
5.4. Other T Cell Immunotherapies
5.5. Oncolytic Viruses
5.6. Vaccines
6. Discussion and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Subramanian, S.; Mishra, R.K.; Singh, L. Genome-wide analysis of microsatellite repeats in humans: Their abundance and density in specific genomic regions. Genome Biol. 2003, 4, R13. [Google Scholar] [CrossRef] [PubMed]
- Eckert, K.A.; Hile, S.E. Every microsatellite is different: Intrinsic DNA features dictate mutagenesis of common microsatellites present in the human genome. Mol. Carcinog. 2009, 48, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Olave, M.C.; Graham, R.P. Mismatch repair deficiency: The what, how and why it is important. Genes Chromosom. Cancer 2022, 61, 314–321. [Google Scholar] [CrossRef]
- A Loeb, L. Microsatellite instability: Marker of a mutator phenotype in cancer. Cancer Res. 1994, 54, 5059–5063. [Google Scholar] [PubMed]
- Amato, M.; Franco, R.; Facchini, G.; Addeo, R.; Ciardiello, F.; Berretta, M.; Vita, G.; Sgambato, A.; Pignata, S.; Caraglia, M.; et al. Microsatellite Instability: From the Implementation of the Detection to a Prognostic and Predictive Role in Cancers. Int. J. Mol. Sci. 2022, 23, 8726. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Bartley, A.N.; Mills, A.M.; Konnick, E.; Overman, M.; Ventura, C.B.; Souter, L.; Colasacco, C.; Stadler, Z.K.; Kerr, S.; E Howitt, B.; et al. Mismatch Repair and Microsatellite Instability Testing for Immune Checkpoint Inhibitor Therapy: Guideline from the College of American Pathologists in Collaboration with the Association for Molecular Pathology and Fight Colorectal Cancer. Arch. Pathol. Lab. Med. 2022, 146, 1194–1210. [Google Scholar] [CrossRef]
- Marcus, V.A.; Madlensky, L.; Gryfe, R.; Kim, H.; So, K.; Millar, A.; Temple, L.K.; Hsieh, E.; Hiruki, T.; Narod, S.; et al. Immunohistochemistry for hMLH1 and hMSH2: A practical test for DNA mismatch repair-deficient tumors. Am. J. Surg. Pathol. 1999, 23, 1248–1255. [Google Scholar] [CrossRef]
- Wong, H.-L.; Christie, M.; Gately, L.; Tie, J.; Lee, B.; Semira, C.; Lok, S.W.; Wong, R.; Gibbs, P. Mismatch repair deficiency assessment by immunohistochemistry: For Lynch syndrome screening and beyond. Futur. Oncol. 2018, 14, 2725–2739. [Google Scholar] [CrossRef]
- Svrcek, M.; Lascols, O.; Cohen, R.; Collura, A.; Jonchère, V.; Fléjou, J.-F.; Buhard, O.; Duval, A. MSI/MMR-deficient tumor diagnosis: Which standard for screening and for diagnosis? Diagnostic modalities for the colon and other sites: Differences between tumors. Bull. Du Cancer 2019, 106, 119–128. [Google Scholar] [CrossRef]
- Vikas, P.; Messersmith, H.; Compton, C.; Sholl, L.; Broaddus, R.R.; Davis, A.; Estevez-Diz, M.; Garje, R.; Konstantinopoulos, P.A.; Leiser, A.; et al. Mismatch Repair and Microsatellite Instability Testing for Immune Checkpoint Inhibitor Therapy: ASCO Endorsement of College of American Pathologists Guideline. J. Clin. Oncol. 2023, 41, 1943–1948. [Google Scholar] [CrossRef] [PubMed]
- Hause, R.J.; Pritchard, C.C.; Shendure, J.; Salipante, S.J. Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 2016, 22, 1342–1350. [Google Scholar] [CrossRef]
- Le, D.T.; Kim, T.W.; Van Cutsem, E.; Geva, R.; Jäger, D.; Hara, H.; Burge, M.; O’neil, B.; Kavan, P.; Yoshino, T.; et al. Phase II open-label study of pembrolizumab in treatment-refractory, microsatellite instability–high/mismatch repair–deficient metastatic colorectal cancer: KEYNOTE-164. J. Clin. Oncol. 2020, 38, 11–19. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.-J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Oaknin, A.; Gilbert, L.; Tinker, A.V.; Brown, J.; Mathews, C.; Press, J.; Sabatier, R.; O’malley, D.M.; Samouelian, V.; Boni, V.; et al. Safety and antitumor activity of dostarlimab in patients with advanced or recurrent DNA mismatch repair deficient/microsatellite instability-high (dMMR/MSI-H) or proficient/stable (MMRp/MSS) endometrial cancer: Interim results from GARNET—A phase I, single-arm study. J. Immunother. Cancer 2022, 10, e003777. [Google Scholar] [CrossRef]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site—When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair–Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Andre, T.; Berton, D.; Curigliano, G.; Ellard, S.; Pérez, J.M.T.; Arkenau, H.-T.; Abdeddaim, C.; Moreno, V.; Guo, W.; Im, E.; et al. Safety and efficacy of anti–PD-1 antibody dostarlimab in patients (pts) with mismatch repair-deficient (dMMR) solid cancers: Results from GARNET study. J. Clin. Oncol. 2021, 39 (Suppl. S3), 9. [Google Scholar] [CrossRef]
- Linsley, P.S.; Bradshaw, J.; Greene, J.; Peach, R.; Bennett, K.L.; Mittler, R.S. Intracellular Trafficking of CTLA-4 and Focal Localization Towards Sites of TCR Engagement. Immunity 1996, 4, 535–543. [Google Scholar] [CrossRef]
- Walker, L.S.K.; Sansom, D.M. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat. Rev. Immunol. 2011, 11, 852–863. [Google Scholar] [CrossRef]
- Oyewole-Said, D.; Konduri, V.; Vazquez-Perez, J.; Weldon, S.A.; Levitt, J.M.; Decker, W.K. Beyond T-Cells: Functional Characterization of CTLA-4 Expression in Immune and Non-Immune Cell Types. Front. Immunol. 2020, 11, 608024. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Kane, L.P. T Cell Ig and Mucin Domain Proteins and Immunity. J. Immunol. 2010, 184, 2743–2749. [Google Scholar] [CrossRef]
- Kobayashi, N.; Karisola, P.; Peña-Cruz, V.; Dorfman, D.M.; Jinushi, M.; Umetsu, S.E.; Butte, M.J.; Nagumo, H.; Chernova, I.; Zhu, B.; et al. TIM-1 and TIM-4 Glycoproteins Bind Phosphatidylserine and Mediate Uptake of Apoptotic Cells. Immunity 2007, 27, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, S.; Sabatos, C.A.; Xiao, S.; Illes, Z.; Cha, E.K.; Sobel, R.A.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. Tim-2 regulates T helper type 2 responses and autoimmunity. J. Exp. Med. 2005, 202, 437–444. [Google Scholar] [CrossRef]
- Han, G.; Chen, G.; Shen, B.; Li, Y. Tim-3: An Activation Marker and Activation Limiter of Innate Immune Cells. Front. Immunol. 2013, 4, 449. [Google Scholar] [CrossRef] [PubMed]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Banerjee, H.; Kane, L.P. Immune regulation by Tim-3. F1000Research 2018, 7, 316. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Shayan, G.; Srivastava, R.; Li, J.; Schmitt, N.; Kane, L.P.; Ferris, R.L. Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. OncoImmunology 2017, 6, e1261779. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.-M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef] [PubMed]
- Blake, S.J.; Dougall, W.C.; Miles, J.J.; Teng, M.W.; Smyth, M.J. Molecular Pathways: Targeting CD96 and TIGIT for Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 5183–5188. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.M.; Nelson, C.A.; Šedý, J.R. Balancing co-stimulation and inhibition with BTLA and HVEM. Nat. Rev. Immunol. 2006, 6, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Ning, Z.; Liu, K.; Xiong, H. Roles of BTLA in Immunity and Immune Disorders. Front. Immunol. 2021, 12, 654960. [Google Scholar] [CrossRef] [PubMed]
- Vlad, G.; Chang, C.-C.; Colovai, A.I.; Berloco, P.; Cortesini, R.; Suciu-Foca, N. Immunoglobulin-like transcript 3: A crucial regulator of dendritic cell function. Hum. Immunol. 2009, 70, 340–344. [Google Scholar] [CrossRef]
- Chang, C.C.; Ciubotariu, R.; Manavalan, J.S.; Yuan, J.; Colovai, A.I.; Piazza, F.; Lederman, S.; Colonna, M.; Cortesini, R.; Dallafavera, R.; et al. Tolerization of dendritic cells by TS cells: The crucial role of inhibitory receptors ILT3 and ILT4. Nat. Immunol. 2002, 3, 237–243. [Google Scholar] [CrossRef]
- Deng, M.; Chen, H.; Liu, X.; Huang, R.; He, Y.; Yoo, B.; Xie, J.; John, S.; Zhang, N.; An, Z.; et al. Leukocyte immunoglobulin-like receptor subfamily B: Therapeutic targets in cancer. Antib. Ther. 2021, 4, 16–33. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, N.; Xue, Y.; Zhang, M.; Li, Y.; Si, Y.; Bian, X.; Jia, Y.; Wang, Y. Expression of immunoglobulin-like transcript (ILT)2 and ILT3 in human gastric cancer and its clinical significance. Mol. Med. Rep. 2012, 5, 910–916. [Google Scholar] [CrossRef]
- de Goeje, P.L.; Bezemer, K.; E Heuvers, M.; Dingemans, A.-M.C.; Groen, H.J.; Smit, E.F.; Hoogsteden, H.C.; Hendriks, R.W.; Aerts, J.G.; Hegmans, J.P. Immunoglobulin-like transcript 3 is expressed by myeloid-derived suppressor cells and correlates with survival in patients with non-small cell lung cancer. OncoImmunology 2015, 4, e1014242. [Google Scholar] [CrossRef]
- Maruhashi, T.; Sugiura, D.; Okazaki, I.-M.; Okazaki, T. LAG-3: From molecular functions to clinical applications. J. Immunother. Cancer 2020, 8, e001014. [Google Scholar] [CrossRef] [PubMed]
- Graydon, C.G.; Mohideen, S.; Fowke, K.R. LAG3′s Enigmatic Mechanism of Action. Front. Immunol. 2021, 11, 3444. [Google Scholar] [CrossRef] [PubMed]
- Huo, J.-L.; Wang, Y.-T.; Fu, W.-J.; Lu, N.; Liu, Z.-S. The promising immune checkpoint LAG-3 in cancer immunotherapy: From basic research to clinical application. Front. Immunol. 2022, 13, 956090. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.; Maddineni, S.; Mathews, I.I.; Sperberg, R.A.P.; Huang, P.-S.; Cochran, J.R. Structure and Functional Binding Epitope of V-domain Ig Suppressor of T Cell Activation. Cell Rep. 2019, 28, 2509–2516.e5. [Google Scholar] [CrossRef] [PubMed]
- Hosseinkhani, N.; Derakhshani, A.; Shadbad, M.A.; Argentiero, A.; Racanelli, V.; Kazemi, T.; Mokhtarzadeh, A.; Brunetti, O.; Silvestris, N.; Baradaran, B. The Role of V-Domain Ig Suppressor of T Cell Activation (VISTA) in Cancer Therapy: Lessons Learned and the Road Ahead. Front. Immunol. 2021, 12, 676181. [Google Scholar] [CrossRef]
- Prasad, D.V.R.; Nguyen, T.; Li, Z.; Yang, Y.; Duong, J.; Wang, Y.; Dong, C. Murine B7-H3 Is a Negative Regulator of T Cells. J. Immunol. 2004, 173, 2500–2506. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.-T.; Jin, W.-L. B7-H3/CD276: An Emerging Cancer Immunotherapy. Front. Immunol. 2021, 12, 701006. [Google Scholar] [CrossRef]
- Cai, D.; Li, J.; Liu, D.; Hong, S.; Qiao, Q.; Sun, Q.; Li, P.; Lyu, N.; Sun, T.; Xie, S.; et al. Tumor-expressed B7-H3 mediates the inhibition of antitumor T-cell functions in ovarian cancer insensitive to PD-1 blockade therapy. Cell. Mol. Immunol. 2020, 17, 227–236. [Google Scholar] [CrossRef]
- Yang, S.; Wei, W.; Zhao, Q. B7-H3, a checkpoint molecule, as a target for cancer immunotherapy. Int. J. Biol. Sci. 2020, 16, 1767–1773. [Google Scholar] [CrossRef]
- André, T.; Lonardi, S.; Wong, K.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Nivolumab plus low-dose ipilimumab in previously treated patients with microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: 4-year follow-up from CheckMate 142. Ann. Oncol. 2022, 33, 1052–1060. [Google Scholar] [CrossRef]
- Lenz, H.-J.; Van Cutsem, E.; Limon, M.L.; Wong, K.Y.M.; Hendlisz, A.; Aglietta, M.; García-Alfonso, P.; Neyns, B.; Luppi, G.; Cardin, D.B.; et al. First-Line Nivolumab Plus Low-Dose Ipilimumab for Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: The Phase II CheckMate 142 Study. J. Clin. Oncol. 2022, 40, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Verschoor, Y.L.; Berg, J.v.D.; Beets, G.; Sikorska, K.; Aalbers, A.; van Lent, A.; Grootscholten, C.; Huibregtse, I.; Marsman, H.; Oosterling, S.; et al. Neoadjuvant nivolumab, ipilimumab, and celecoxib in MMR-proficient and MMR-deficient colon cancers: Final clinical analysis of the NICHE study. J. Clin. Oncol. 2022, 40, 3511. [Google Scholar] [CrossRef]
- Neoadjuvant Immune Checkpoint Inhibition in Locally Advanced MMR-Deficient Colon Cancer: The NICHE-2 Study|OncologyPRO. Available online: https://www.annalsofoncology.org/article/S0923-7534(22)03894-7/fulltext (accessed on 22 February 2023).
- Pietrantonio, F.; Raimondi, A.; Lonardi, S.; Murgioni, S.; Cardellino, G.G.; Tamberi, S.; Strippoli, A.; Palermo, F.; Prisciandaro, M.; Randon, G.; et al. INFINITY: A multicentre, single-arm, multi-cohort, phase II trial of tremelimumab and durvalumab as neoadjuvant treatment of patients with microsatellite instability-high (MSI) resectable gastric or gastroesophageal junction adenocarcinoma (GAC/GEJAC). J. Clin. Oncol. 2023, 41 (Suppl. S4), 358. [Google Scholar] [CrossRef]
- Hollebecque, A.; Chung, H.C.; de Miguel, M.J.; Italiano, A.; Machiels, J.-P.; Lin, C.-C.; Dhani, N.C.; Peeters, M.; Moreno, V.; Su, W.-C.; et al. Safety and Antitumor Activity of α-PD-L1 Antibody as Monotherapy or in Combination with α-TIM-3 Antibody in Patients with Microsatellite Instability–High/Mismatch Repair–Deficient Tumors. Clin. Cancer Res. 2021, 27, 6393–6404. [Google Scholar] [CrossRef] [PubMed]
- Bever, K.; Wang, H.; Durham, J.; Apostol, C.; Azad, N.; Browner, I.; Gaillard, S.; Laheru, D.; Lee, V.; Sharfman, W.; et al. 711 Interim results of a phase 2 study of nivolumab and relatlimab in advanced mismatch repair deficient (dMMR) cancers resistant to prior PD-(L)1 inhibition. J. Immunother Cancer 2022, 10 (Suppl. S2), A743. [Google Scholar] [CrossRef]
- Kim, T.W.; Bedard, P.L.; LoRusso, P.; Gordon, M.S.; Bendell, J.; Oh, D.-Y.; Ahn, M.-J.; Garralda, E.; D’angelo, S.P.; Desai, J.; et al. Anti-TIGIT Antibody Tiragolumab Alone or with Atezolizumab in Patients with Advanced Solid Tumors: A Phase 1a/1b Nonrandomized Controlled Trial. JAMA Oncol. 2023, 9, 1574–1582. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Pietrantonio, F.; Avallone, A.; Gumus, M.; Wyrwicz, L.; Kim, J.G.; Yalcin, S.; Kwiatkowski, M.; Lonardi, S.; Zolnierek, J.; et al. KEYSTEP-008: Phase II trial of pembrolizumab-based combination in MSI-H/dMMR metastatic colorectal cancer. Future Oncol. 2023, 37, 2445–2452. [Google Scholar] [CrossRef]
- Belli, C.; Antonarelli, G.; Repetto, M.; Bielo, L.B.; Crimini, E.; Curigliano, G. Targeting Cellular Components of the Tumor Microenvironment in Solid Malignancies. Cancers 2022, 14, 4278. [Google Scholar] [CrossRef]
- Woo, S.-R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.K.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef]
- Ng, K.W.; Marshall, E.A.; Bell, J.C.; Lam, W.L. cGAS–STING and Cancer: Dichotomous Roles in Tumor Immunity and Development. Trends Immunol. 2018, 39, 44–54. [Google Scholar] [CrossRef]
- Corrales, L.; McWhirter, S.M.; Dubensky, T.W.; Gajewski, T.F. The host STING pathway at the interface of cancer and immunity. J. Clin. Investig. 2016, 126, 2404–2411. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Guan, W. STING signaling promotes apoptosis, necrosis, and cell death: An overview and update. Mediat. Inflamm. 2018, 2018, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Kunac, N.; Degoricija, M.; Viculin, J.; Omerović, J.; Terzić, J.; Vilović, K.; Korac-Prlic, J. Activation of cGAS-STING Pathway Is Associated with MSI-H Stage IV Colorectal Cancer. Cancers 2023, 15, 221. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Kanne, D.B.; Leong, M.; Glickman, L.H.; McWhirter, S.M.; Lemmens, E.; Mechette, K.; Leong, J.J.; Lauer, P.; Liu, W.; et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci. Transl. Med. 2015, 7, 283ra52. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, X.; Qin, N.; Qiao, Y.; Xing, S.; Liu, W.; Feng, F.; Liu, Z.; Sun, H. STING, a promising target for small molecular immune modulator: A review. Eur. J. Med. Chem. 2021, 211, 113113. [Google Scholar] [CrossRef] [PubMed]
- Pu, F.; Chen, F.; Liu, J.; Zhang, Z.; Shao, Z. Immune Regulation of the cGAS-STING Signaling Pathway in the Tumor Microenvironment and Its Clinical Application. OncoTargets Ther. 2021, 14, 1501–1516. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.C.; Hung, W.C. Pyruvate kinase M2 fuels multiple aspects of cancer cells: From cellular metabolism, transcriptional regulation to extracellular signaling. Mol. Cancer 2018, 17, 1–9. [Google Scholar] [CrossRef]
- Warner, S.L.; Carpenter, K.J.; Bearss, D.J. Activators of PKM2 in cancer metabolism. Futur. Med. Chem. 2014, 6, 1167–1178. [Google Scholar] [CrossRef]
- Chhipa, A.S.; Patel, S. Targeting pyruvate kinase muscle isoform 2 (PKM2) in cancer: What do we know so far? Life Sci. 2021, 280, 119694. [Google Scholar] [CrossRef]
- Chen, M.; Liu, H.; Li, Z.; Ming, A.L.; Chen, H. Mechanism of PKM2 affecting cancer immunity and metabolism in Tumor Microenvironment. J. Cancer 2021, 12, 3566–3574. [Google Scholar] [CrossRef]
- Sommakia, S.; Matsumura, Y.; Allred, C.; Pathi, S.; Tyagi, E.; Foulks, J.; Siddiqui, A.; Warner, S. The PKM2 activator and molecular glue TP-1454 modulates tumor-immune responses by destabilizing T-regulatory cells. Eur. J. Cancer 2022, 174, S47. [Google Scholar] [CrossRef]
- Horn, L.A.; Chariou, P.L.; Gameiro, S.R.; Qin, H.; Iida, M.; Fousek, K.; Meyer, T.J.; Cam, M.; Flies, D.; Langermann, S.; et al. Remodeling the tumor microenvironment via blockade of LAIR-1 and TGF-β signaling enables PD-L1–mediated tumor eradication. J. Clin. Investig. 2022, 132, e155148. [Google Scholar] [CrossRef] [PubMed]
- Lebbink, R.J.; Berg, M.C.W.v.D.; de Ruiter, T.; Raynal, N.; van Roon, J.A.G.; Lenting, P.J.; Jin, B.; Meyaard, L. The Soluble Leukocyte-Associated Ig-Like Receptor (LAIR)-2 Antagonizes the Collagen/LAIR-1 Inhibitory Immune Interaction. J. Immunol. 2008, 180, 1662–1669. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, S.; Li, J.; Li, J.; Li, B. Cancer immunotherapy based on blocking immune suppression mediated by an immune modulator LAIR-1. OncoImmunology 2020, 9, 1740477. [Google Scholar] [CrossRef]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef]
- Wolfson, B.; Franks, S.E.; Hodge, J.W. Stay on Target: Reengaging Cancer Vaccines in Combination Immunotherapy. Vaccines 2021, 9, 509. [Google Scholar] [CrossRef]
- Waldmann, T.A. The shared and contrasting roles of IL2 and IL15 in the life and death of normal and neoplastic lymphocytes: Implications for cancer therapy. Cancer Immunol. Res. 2015, 3, 219–227. [Google Scholar] [CrossRef]
- Yang, Y.; Lundqvist, A. Immunomodulatory Effects of IL-2 and IL-15; Implications for Cancer Immunotherapy. Cancers 2020, 12, 3586. [Google Scholar] [CrossRef]
- Knudson, K.M.; Hodge, J.W.; Schlom, J.; Gameiro, S.R. Rationale for IL-15 superagonists in cancer immunotherapy. Expert Opin. Biol. Ther. 2020, 20, 705–709. [Google Scholar] [CrossRef]
- Wrangle, J.M.; Awad, M.M.; Badin, F.B.; Rubinstein, M.P.; Bhar, P.; Garner, C.; Reddy, S.K.; Soon-Shiong, P. Preliminary data from QUILT 3.055: A phase 2 multi-cohort study of N803 (IL-15 superagonist) in combination with checkpoint inhibitors (CPI). J. Clin. Oncol. 2021, 39 (Suppl. S15), 2596. [Google Scholar] [CrossRef]
- Fousek, K.; Horn, L.A.; Qin, H.; Dahut, M.; Iida, M.; Yacubovich, D.; Hamilton, D.H.; Thomas, A.; Schlom, J.; Palena, C. An Interleukin-15 Superagonist Enables Antitumor Efficacy of Natural Killer Cells Against All Molecular Variants of SCLC. J. Thorac. Oncol. 2023, 18, 350–368. [Google Scholar] [CrossRef] [PubMed]
- Van der Meer, J.M.R.; Maas, R.J.A.; Guldevall, K.; Klarenaar, K.; de Jonge, P.K.J.D.; Evert, J.S.H.-V.; van der Waart, A.B.; Cany, J.; Safrit, J.T.; Lee, J.H.; et al. IL-15 superagonist N-803 improves IFNγ production and killing of leukemia and ovarian cancer cells by CD34+ progenitor-derived NK cells. Cancer Immunol. Immunother. 2021, 70, 1305–1321. [Google Scholar] [CrossRef] [PubMed]
- Chamie, K.; Chang, S.S.; Gonzalgo, M.; Kramolowsky, E.V.; Sexton, W.J.; Bhar, P.; Reddy, S.K.; Soon-Shiong, P. Final clinical results of pivotal trial of IL-15RαFc superagonist N-803 with BCG in BCG-unresponsive CIS and papillary nonmuscle-invasive bladder cancer (NMIBC). J. Clin. Oncol. 2022, 40 (Suppl. S16), 4508. [Google Scholar] [CrossRef]
- Rosser, C.J.; Nix, J.; Ferguson, L.; Hernandez, L.; Wong, H.C. Phase Ib trial of ALT-803, an IL-15 superagonist, plus BCG for the treatment of BCG-naïve patients with non-muscle-invasive bladder cancer. NMIBC 2018, 36 (Suppl. S16), 510. [Google Scholar] [CrossRef]
- Antosova, Z.; Podzimkova, N.; Tomala, J.; Augustynkova, K.; Sajnerova, K.; Nedvedova, E.; Sirova, M.; de Martynoff, G.; Bechard, D.; Moebius, U.; et al. SOT101 induces NK cell cytotoxicity and potentiates antibody-dependent cell cytotoxicity and anti-tumor activity. Front. Immunol. 2022, 13, 989895. [Google Scholar] [CrossRef]
- Champiat, S.; Marabelle, A.; Galvao, V.; LoRusso, P.; Grell, P.; Cassier, P.; Gomez-Roca, C.; Korakis, I.; Naing, A.; Jelinkova, L.P.; et al. Abstract CT040: SOT101, an IL-2/IL-15 Rβγ superagonist, in combination with pembrolizumab in patients with advanced solid tumors: Interim safety and efficacy results from the AURELIO-03 dose escalation trial. Cancer Res. 2022, 82 (Suppl. S12), CT040. [Google Scholar] [CrossRef]
- Garralda, E.; Naing, A.; Galvao, V.; LoRusso, P.; Grell, P.; Cassier, P.A.; Gomez-Roca, C.A.; Korakis, I.; Bechard, D.; Jelinkova, L.P.; et al. Interim safety and efficacy results from AURELIO-03: A phase 1 dose escalation study of the IL-2/IL-15 receptor βγ superagonist SOT101 as a single agent and in combination with pembrolizumab in patients with advanced solid tumors. J. Clin. Oncol. 2022, 40 (Suppl. S16), 2502. [Google Scholar] [CrossRef]
- Champiat, S.; Marabelle, A.; Davar, D.; Grell, P.; Ouali, K.; Patrikidou, A.; Schoenenberger, A.; Jelinkova, L.P.; Adkins, I.; Tillmanns, S.; et al. 716 AURELIO-04: A phase 2, open-label, single-arm, multicenter study to determine the efficacy and safety of SOT101 in combination with pembrolizumab in patients with selected advanced solid tumors. J. Immunother Cancer 2022, 40 (Suppl. S2), A749. [Google Scholar]
- Azadi, A.; Golchini, A.; Delazar, S.; Kahaki, F.A.; Dehnavi, S.M.; Payandeh, Z.; Eyvazi, S. Recent Advances on Immune Targeted Therapy of Colorectal Cancer Using bi-Specific Antibodies and Therapeutic Vaccines. Biol. Proced. Online 2021, 23, 1–13. [Google Scholar] [CrossRef]
- Klein, C.; Schaefer, W.; Regula, J.T. The use of CrossMAb technology for the generation of bi- and multispecific antibodies. mAbs 2016, 8, 1010–1020. [Google Scholar] [CrossRef]
- Dorigo, O.; Fiset, S.; MacDonald, L.D.; Bramhecha, Y.; Hrytsenko, O.; Dirk, B.; Rosu, G.N.; Stanford, M. DPX-Survivac, a novel T-cell immunotherapy, to induce robust T-cell responses in advanced ovarian cancer. J. Clin. Oncol. 2020, 38 (Suppl. S5), 6. [Google Scholar] [CrossRef]
- Jafari, M.; Kadkhodazadeh, M.; Shapourabadi, M.B.; Goradel, N.H.; Shokrgozar, M.A.; Arashkia, A.; Abdoli, S.; Sharifzadeh, Z. Immunovirotherapy: The role of antibody based therapeutics combination with oncolytic viruses. Front. Immunol. 2022, 13, 1012806. [Google Scholar] [CrossRef] [PubMed]
- de Gruijl, T.D.; Janssen, A.B.; van Beusechem, V.W. Arming oncolytic viruses to leverage antitumor immunity. Expert Opin. Biol. Ther. 2015, 15, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e10. [Google Scholar] [CrossRef] [PubMed]
- Lan, Q.; Xia, S.; Wang, Q.; Xu, W.; Huang, H.; Jiang, S.; Lu, L. Development of oncolytic virotherapy: From genetic modification to combination therapy. Front. Med. 2020, 14, 160–184. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef] [PubMed]
- Malogolovkin, A.; Gasanov, N.; Egorov, A.; Weener, M.; Ivanov, R.; Karabelsky, A. Combinatorial approaches for cancer treatment using oncolytic viruses: Projecting the perspectives through clinical trials outcomes. Viruses 2021, 13, 1271. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, O.; dos Santos, J.M.; Hemminki, A. Oncolytic viruses for cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 1–15. [Google Scholar] [CrossRef]
- Ren, Y.; Miao, J.-M.; Wang, Y.-Y.; Fan, Z.; Kong, X.-B.; Yang, L.; Cheng, G. Oncolytic viruses combined with immune checkpoint therapy for colorectal cancer is a promising treatment option. Front. Immunol. 2022, 13, 961796. [Google Scholar] [CrossRef]
- Kloor, M.; Von Knebel Doeberitz, M. The Immune Biology of Microsatellite-Unstable Cancer. Trends Cancer 2016, 2, 121–133. [Google Scholar] [CrossRef]
- Ballhausen, A.; Przybilla, M.J.; Jendrusch, M.; Haupt, S.; Pfaffendorf, E.; Seidler, F.; Witt, J.; Sanchez, A.H.; Urban, K.; Draxlbauer, M.; et al. The shared frameshift mutation landscape of microsatellite-unstable cancers suggests immunoediting during tumor evolution. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Sanchez, A.; Grossman, M.; Yeung, K.; Sei, S.S.; Lipkin, S.; Kloor, M. Vaccines for immunoprevention of DNA mismatch repair deficient cancers. J. Immunother. Cancer 2022, 10, e004416. [Google Scholar] [CrossRef] [PubMed]
- Roudko, V.; Bozkus, C.C.; Greenbaum, B.; Lucas, A.; Samstein, R.; Bhardwaj, N. Lynch Syndrome and MSI-H Cancers: From Mechanisms to ‘Off-The-Shelf’ Cancer Vaccines. Front. Immunol. 2021, 12, 757804. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Yang, M.; Cao, Y. Tumor antigens and vaccines in colorectal cancer. Med. Drug Discov. 2022, 16, 100144. [Google Scholar] [CrossRef]
- Kloor, M.; Reuschenbach, M.; Pauligk, C.; Karbach, J.; Rafiyan, M.-R.; Al-Batran, S.-E.; Tariverdian, M.; Jäger, E.; von Knebel Doeberitz, M. A frameshift peptide neoantigen-based vaccine for mismatch repair-deficient cancers: A phase I/IIa clinical trial. Clin. Cancer Res. 2020, 26, 4503–4510. [Google Scholar] [CrossRef]
- Fakih, M.; Le, D.T.; Pedersen, K.S.; Shields, A.F.; Shah, M.A.; Mukherjee, S.; Delaite, P.; Faivre, T.; Morena, D.A.; Leoni, G.; et al. First clinical and immunogenicity results including all subjects enrolled in a phase I study of Nous-209, an off-the-shelf immunotherapy, with pembrolizumab, for the treatment of tumors with a deficiency in mismatch repair/microsatellite instability (dMMR/MSI). J. Clin. Oncol. 2022, 40 (Suppl. S16), 2515. [Google Scholar] [CrossRef]
- Andre, T.; Berton, D.; Curigliano, G.; Jimenez-Rodriguez, B.; Ellard, S.; Gravina, A.; Miller, R.; Tinker, A.; Jewell, A.; Pikiel, J.; et al. Efficacy and safety of dostarlimab in patients (pts) with mismatch repair deficient (dMMR) solid tumors: Analysis of 2 cohorts in the GARNET study. J. Clin. Oncol. 2022, 40, 2587. [Google Scholar] [CrossRef]
- Xiang, Z.; Li, J.; Zhang, Z.; Cen, C.; Chen, W.; Jiang, B.; Meng, Y.; Wang, Y.; Berglund, B.; Zhai, G.; et al. Comprehensive Evaluation of Anti-PD-1, Anti-PD-L1, Anti-CTLA-4 and Their Combined Immunotherapy in Clinical Trials: A Systematic Review and Meta-analysis. Front. Pharmacol. 2022, 13, 883655. [Google Scholar] [CrossRef]
- Yang, F.; Wang, J.F.; Wang, Y.; Liu, B.; Molina, J.R. Comparative Analysis of Predictive Biomarkers for PD-1/PD-L1 Inhibitors in Cancers: Developments and Challenges. Cancers 2022, 14, 109. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Agent | Target | Trial | Ph. | No. | Tumor Type | Setting | Study Treatment | Endpoint |
|---|---|---|---|---|---|---|---|---|
| Ipilimumab | CTLA4 | NCT04969887 (MOST-CIRCUIT) * | 2 | 240 | Cohort 4: dMMR/MSI-H AST | M+ pretreated | Ipilimumab + Nivolumab | ORR; 6-month PFS |
| NCT04730544 * | 2 | 96 | dMRR/MSI-H CCR | M+ 1-2L | Ipilimumab + Nivolumab | AEs; PFS | ||
| NCT04008030 (CheckMate 8HW) * | 3 | 831 | dMRR/MSI-H CCR | Part 1: M+ pretreated Part 2: M+ 1L | A: Nivolumab B: Nivolumab + Ipilimumab C: ICC | PFS | ||
| Quavonlimab | CTLA4 | NCT04895722 (KEYSTEP-008) * | 2 | 320 | MSI-H CRC | M+ pretreated (cohort A); M+ untreated (cohort B) | Cohort A Pembrolizumab vs. Quavonlimab/pembrolizuma; Cohort B: Pembrolizumab vs. Quavonlimab/Pembrolizumab vs. Favezelimab/Pembrolizumab vs. Vibostolimab/pembrolizumab vs. MK-4830 + pembrolizumab | ORR |
| Cadonlimab | bsAb PD-1/CTLA-4 | NCT05426005 | 2 | 25 | dMRR/MSI-H CCR | M+ pretreated | Cadonlimab | ORR |
| NCT04556253 | 2 | 29 | dMRR/MSI-H CCR and dMRR/MSI-H gastric cancer | Perioperative | Cadonlimab | pCR | ||
| NCT05815290 | 2 | 50 | dMRR/MSI-H CCR | Neoadj | Cadonlimab | pCR | ||
| Ociperlimab | TIGIT | NCT04746924 | 3 | 660 | NSCLC | M+ pretreated. PD-L1 ≥ 50% | A: Ociperlimab + Tislezumab B: Pembrolizumab C: Tislezumab | PFS; OS |
| NCT04866017 | 3 | 700 | NSCLC | Stage III following CRT | A: Ociperlimab + Tislelizumab B: Tislelizumab C: Durvalumab | PFS | ||
| NCT04732494 | 2 | 120 | ESCC | M+ 2L PD-L1 ≥ 10% | A: Ociperlimab + Tislelizumab B: Tislelizumab | ORR | ||
| NCT05023109 | 2 | 45 | BTC | M+ 1L | Ociperlimab + Tislelizumab + Gemcitabine + Cisplatin | ORR | ||
| NCT05014815 | 2 | 270 | NSCLC | M+ 1L | Ociperlimab + Tislelizumab + chemotherapy | PFS | ||
| Tiragolumab | TIGIT | NCT04543617 (SKYSCRAPER-07) | 3 | 750 | ESCC | Following CRT | A: Tiragolumab + Atezolizumab B: Atezolizumab C: Placebo | PFS; OS |
| NCT04294810 (SKYSCRAPER-01) | 3 | 660 | NSCLC | M+ 1L PD-L1 ≥ 50% | A: Tiragolumab + Atezolizumab B: Atezolizumab | PFS; OS; AEs | ||
| NCT05805501 | 2 | 210 | RCC | M+ 1L | Tiragolumab + RO7247669 (bsAb PD1-LAG3) + Axitinib | PFS | ||
| NCT03708224 | 2 | 55 | HNSCC | Neoadj | Tiragolumab + Atezolizumab + Tocilizumab | ≥40% increase in infiltrating CD3; R0 resection rate | ||
| NCT05009069 | 2 | 76 | Rectal cancer | Following Neoadj CRT | A: Adj Tiragolumab + Atezolizumab B: Adj Atezolizumab | pCR | ||
| NCT05483400 * (TIRACAN) | 2 | 97 | Cohort A: HNSCC Cohort B: AST MSI-H Cohort C: melanoma | Cohort A: neoadj Cohort B: M+ pretreated Cohort C: M+ pretreated | Tiragolumab + Atezolizumab | Cohort A: Pcr Cohort B and C: ORR | ||
| Domvanalimab (AB154) | TIGIT | NCT05568095 (STAR-221) | 3 | 970 | Gastric, GEJ or esophageal adenocarcinoma | M+ 1L | A: Domvanalimab + Zimberelimab + Oxaliplatin + 5-FU + Capecitabine B: Nivolumab + Oxaliplatin + 5-FU + Capecitabine | OS |
| NCT04736173 (ARC-10) | 3 | 750 | NSCLC | M+ 1L PD-L1 ≥ 50% | A: Carboplatin + Pemetrexed + Paclitaxel B: Zimberelimab C: Domvanalimab + Zimberelimab D: Pembrolizumab | OS | ||
| NCT04791839 | 2 | 30 | NSCLC | Cohort A: M+ 1L PD-L1 1–49% Cohort B: M+ 1L PD-L1 ≥ 50% | Domvanalimab + Zimberelimab + Etrumadenant (A2R antagonist) | ORR | ||
| NCT05130177 | 2 | 26 | Melanoma | M+ ≥ 1L progressed on PD-1 | Domvanalimab + Zimberelimab | ORR | ||
| Vibostolimab | TIGIT | NCT05665595 (MK-7684A-010/KEYVIBE-010) | 1560 | Melanoma | Adj | A: Vibostolimab + Pembrolizumab B: Pembrolizumab | RFS | |
| NCT04895722 (KEYSTEP-008) * | 2 | 320 | MSI-H CRC | M+ untreated (cohort B) | Cohort B: Pembrolizumab vs. Quavonlimab/Pembrolizumab vs. Favezelimab/Pembrolizumab vs. Vibostolimab/pembrolizumab vs. MK-4830 + pembrolizumab | ORR | ||
| HLX301 | bsAb TIGIT-PD-L1 | NCT05102214 | 1/2 | 150 | AST | M+ pretreated | HLX301 | AEs; DLTs, RP2D; ORR; DCR |
| NCT05390528 | 1/2 | 30 | AST or Lymphoma | M+ pretreated | HLX301 | AEs | ||
| M6223 | TIGIT | NCT05327530 | 2 | 252 | Bladder | M+ pretreated | Cohort C: Avelumab + M6223 | PFS |
| Fianlimab | LAG3 | NCT05352672 | 3 | 1590 | Melanoma | M+ 1L | A: Fianlimab + Cemiplimab B: Pembrolizumab C: Cemiplimab | PFS |
| NCT05608291 | 3 | 1530 | Melanoma | Adj | A: Fianlimab + Cemiplimab B: Pembrolizumab | RFS | ||
| INCAGN02385 | LAG3 | NCT05287113 | 2 | 162 | HNSCC | M+ PD-L1 ≥ 1% | A: Retifanlimab B: Retifanlimab + INCAGN02385 C: Retifanlimab + INCAGN02385 + INCAGN02390 | PFS |
| NCT04586244 | 2 | 45 | Bladder | Neoadj cisplatin inelegible | Cohort D: INCAGN02385 + Retifanlimab | CD8 increase | ||
| Eftilagimod Alpha | MHC II agonist | NCT05747794 (AIPAC-003) | 2/3 | 849 | Breast | TNBC 1L; HR+/HER2- ≥ 2L | A: Eftilagimod Alpha + Paclitaxel B: Paclitaxel | DLTs; AEs; OS |
| NCT04811027 | 2 | 154 | HNSCC | Cohort A and B: M+ 1L PD-L1 CPS ≥ 1% Cohort C: M+ 1L PD-L1 CPS < 1% | A: Eftilagimod alpha + Pembrolizumab B: Pembrolizumab C: Eftilagimod alpha + Pembrolizumab | ORR | ||
| Relatlimab | LAG3 | NCT04080804 | 2 | 60 | HNSCC | Neoadj | A: Relatlimab + Nivolumab B: Nivolumab + Ipilimumab C: Nivolumab | AEs |
| NCT05134948 | 1/2 | 24 | AST | M+ pretreated | Relatlimab + Nivolumab | AEs | ||
| NCT05704647 | 2 | 30 | Melanoma | M+ pretreated with active brain metastases | Relatlimab + Nivolumab | AEs | ||
| NCT04095208 | 2 | 67 | Soft-tissue sarcoma | M+ pretreated | Relatlimab + Nivolumab | ORR | ||
| NCT04095208 | 2 | 61 | Skin squamous cell carcinoma | 1L | Relatlimab + Nivolumab | ORR | ||
| NCT03642067 | 2 | 96 | CRR | ≥2L | Relatlimab + Nivolumab | ORR | ||
| NCT03607890 * | 2 | 42 | AST | M+ MSI-H resistant to PD1/PD-L1 therapy | Relatlimab + Nivolumab | ORR | ||
| NCT04205552 (NEOpredict) | 2 | 90 | NSCLC | Neoadj | A: Nivolumab B: Nivoluamb + Relatlimab | AEs | ||
| NCT03026140 * (NICHE-3) | 2 | 268 | CRC | Neoadj Cohort 5: pMRR/MSS; Cohort 6: dMRR/MSI-H | Cohort 5: Relatlimab + Nivolumab Cohort 6: Relatlimab + Nivolumab | AEs, DFS | ||
| Favezelimab | LAG3 | NCT04895722 (KEYSTEP-008) * | 2 | 320 | MSI-H CRC | M+ untreated (cohort B) | Cohort B: Pembrolizumab vs. Quavonlimab/Pembrolizumab vs. Favezelimab/Pembrolizumab vs. Vibostolimab/pembrolizumab vs. MK-4830 + pembrolizumab | ORR |
| RO7247669 | bsAb PD1-LAG3 | NCT05805501 | 2 | 210 | RCC | M+ 1L | RO7247669 (bsAb PD1-LAG3) + Tiragolumab + Axitinib | PFS |
| NCT04140500 | 1/2 | 320 | AST | M+ pretreated | RO7247669 | AEs; DLT; ORR; DCR | ||
| NCT05805501 | 2 | 210 | RCC | M+ pretreated | Cohort A: RO7247669 + Axitinib Cohort B: RO7247669 + Tiragolumab + Axitinib | PFS | ||
| HLX26 | LAG3 | NCT05787613 | 2 | 60 | NSCLC | M+ 1L | HLX26 + Serplulimab + Pemetrexed + Nab-paclitaxel + Carboplatin | DLT; MTD; ORR |
| TSR-022 | TIM-3 | NCT03680508 | 2 | 42 | HCC | M+ 1L | A: TSR-022 + TSR-042 | ORR |
| NCT04139902 | 2 | 56 | Melanoma | Neoadj | A: TSR-022 + Dostalimab B: Dostarlimab | MPR | ||
| AZD7789 | bsAb PD1-TIM3 | NCT04931654 | 1/2 | 81 | NSCLC | M+ ≥ 2L; part B2 IO-naive | A: AZD7789 | AEs, DLT, ORR |
| Lomvastomig | bsAb PD1-TIM3 | NCT04785820 | 2 | 210 | Esophageal SCC | M+ ≥ 2L | A: Lomvastomig B: Tobemstomig (bsAb PD1-LAG3) C: Nivolumab | OS |
| INCAGN02390 | TIM-3 | NCT05287113 | 2 | 162 | HNSCC | M+ PD-L1 ≥ 1% | A: Retifanlimab B: Retifanlimab + INCAGN02385 C: Retifanlimab + INCAGN02385 + INCAGN02390 | PFS |
| NCT04586244 | 2 | 45 | Bladder | Neoadj | Cohort E: INCAGN02385 + Retifanlimab + INCAGN02390 | CD8 increase | ||
| TQB2618 | TIM-3 | NCT05645315 | 1/2 | 127 | Cohort 1: solid tumors Cohort 2: NSCLC | Cohort 1: pretreated MC Cohort 2: 1L NSCLC PD-L1 ≥ 1% | A: TQB2618 + TQB2450 | DLT; ORR |
| NCT05563480 | 2 | 90 | Nasopharyngeal carcinoma | Part 1: M+ ≥ 2L Part 2: M+ 1L | A: TQB2618 + Pempulimab + Chemotherapy B: Penpulimab + Chemotherapy C: TQB2618 + Pempulimab | MTD; ORR; PFS | ||
| JS004 | BTLA | NCT05664971 | 1/2 | 240 | NSCLC; SCLC | Cohort 1-3: NSCLC M+ 1-3L; Cohort 4: SCLC M+ 1L | Cohort 1: JS004 + Toripalimab Cohort 2: JS004 + Toripalimab + Docetaxel Cohort 3: JS004 + Toripalimab + Pemetrexed + Cisplatin or Carboplatin Cohort 4: JS004 + Toripalimab + Carboplatin + Etoposide | AEs; SAEs; ORR |
| NCT04929080 | 1/2 | 149 | Nasopharyngeal carcinoma, HNSCC | M+ ≥ 2L | JS004 alone or + Toripalimab | AEs; ORR | ||
| KVA12123 | VISTA | NCT05708950 | 1/2 | 314 | AST | M+ pretreated | KVA12123 alone or + Pembrolizumab | AEs; RP2D |
| MK-4830 | ILT-4 | NCT04895722 (KEYSTEP-008) * | 2 | 320 | MSI-H CRC | M+ untreated (cohort B) | Cohort B: Pembrolizumab vs. Quavonlimab/Pembrolizumab vs. Favezelimab/Pembrolizumab vs. Vibostolimab/pembrolizumab vs. MK-4830 + pembrolizumab | ORR |
| Class | Target/Mechanism of Action | Example Drugs | Tumor Types | Trials |
|---|---|---|---|---|
| Tumor microenvironment | STING agonists | E7766 | Solid tumors and lymphomas | NCT04144140 |
| Cyclic dinucleotide analogues | Solid tumors | - | ||
| PKM2 inhibitors | TP-1454 | Solid tumors, MSI-H CRC | NCT04328740 * | |
| LAIR-1 inhibitors | NC410 | Solid tumors | NCT04408599 NCT05572684 | |
| Cytokine superagonists | IL15 superagonist | N-803 | Solid tumors, various combination therapies with chemotherapy, anti-VEGF, antibody-drug conjugates, anti-PD1 | NCT03228667 NCT03022825 NCT02138734 NCT04390399 NCT03520686 NCT05096663 NCT04927884 |
| SOT-101 Monotherapy or + anti-PD1 SOT101 + anti-PD1 | Solid tumors | AURELIO-03 (NCT04234113) AURELIO-04 (NCT05256381) * | ||
| Bispecific T-cell engagers | CEA + CD3 | Cibisatamab | MSS-CRC | NCT03866239 |
| Oncolytic viruses | Cancer cells with release of antigens | RP1 +/− anti-PD1 | Solid tumors | NCT03767348 * |
| Vaccines | Various cancer antigens | MSI-induced FSPs combined with MONTANIDE ISA-51 | dMMR cancers | NCT01461148 * |
| Nous-209 | dMMR/MSI-H cancers | NCT04041310 * | ||
| Other T cell immunotherapies | Induction of survivin-specific T cell reactions | DPX-Survivac | Solid tumors, MSI-H tumors | NCT03836352 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crimini, E.; Boscolo Bielo, L.; Berton Giachetti, P.P.M.; Pellizzari, G.; Antonarelli, G.; Taurelli Salimbeni, B.; Repetto, M.; Belli, C.; Curigliano, G. Beyond PD(L)-1 Blockade in Microsatellite-Instable Cancers: Current Landscape of Immune Co-Inhibitory Receptor Targeting. Cancers 2024, 16, 281. https://doi.org/10.3390/cancers16020281
Crimini E, Boscolo Bielo L, Berton Giachetti PPM, Pellizzari G, Antonarelli G, Taurelli Salimbeni B, Repetto M, Belli C, Curigliano G. Beyond PD(L)-1 Blockade in Microsatellite-Instable Cancers: Current Landscape of Immune Co-Inhibitory Receptor Targeting. Cancers. 2024; 16(2):281. https://doi.org/10.3390/cancers16020281
Chicago/Turabian StyleCrimini, Edoardo, Luca Boscolo Bielo, Pier Paolo Maria Berton Giachetti, Gloria Pellizzari, Gabriele Antonarelli, Beatrice Taurelli Salimbeni, Matteo Repetto, Carmen Belli, and Giuseppe Curigliano. 2024. "Beyond PD(L)-1 Blockade in Microsatellite-Instable Cancers: Current Landscape of Immune Co-Inhibitory Receptor Targeting" Cancers 16, no. 2: 281. https://doi.org/10.3390/cancers16020281