
Myeloproliferative Neoplasms: Diseases Mediated by Chronic Activation of Signal Transducer and Activator of Transcription (STAT) Proteins



Abstract
:Simple Summary
Abstract
1. Introduction
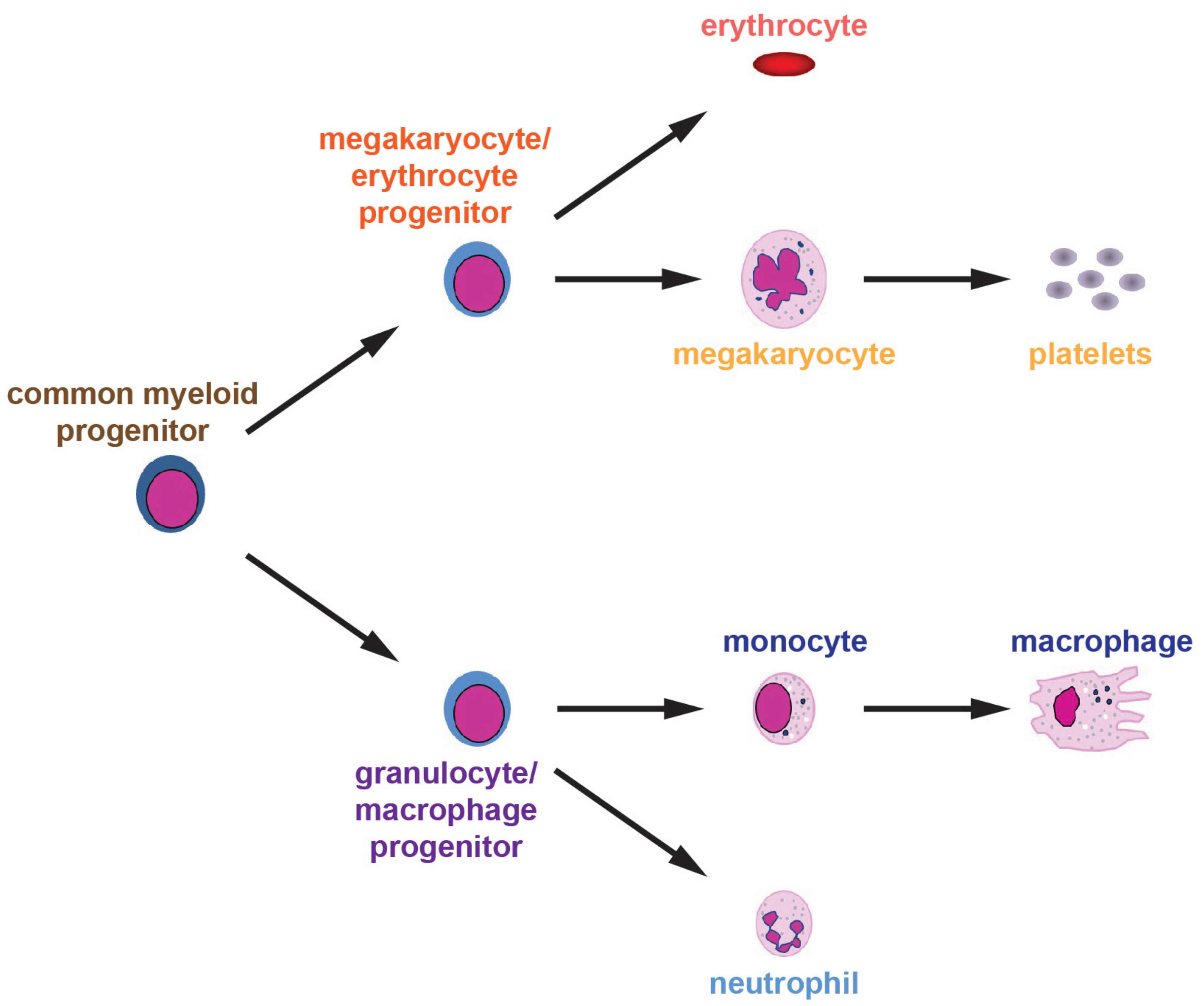
2. Normal Hematopoiesis and Its Control
3. Underlying Genetic Causes of MPNs
3.1. BCR::ABL1
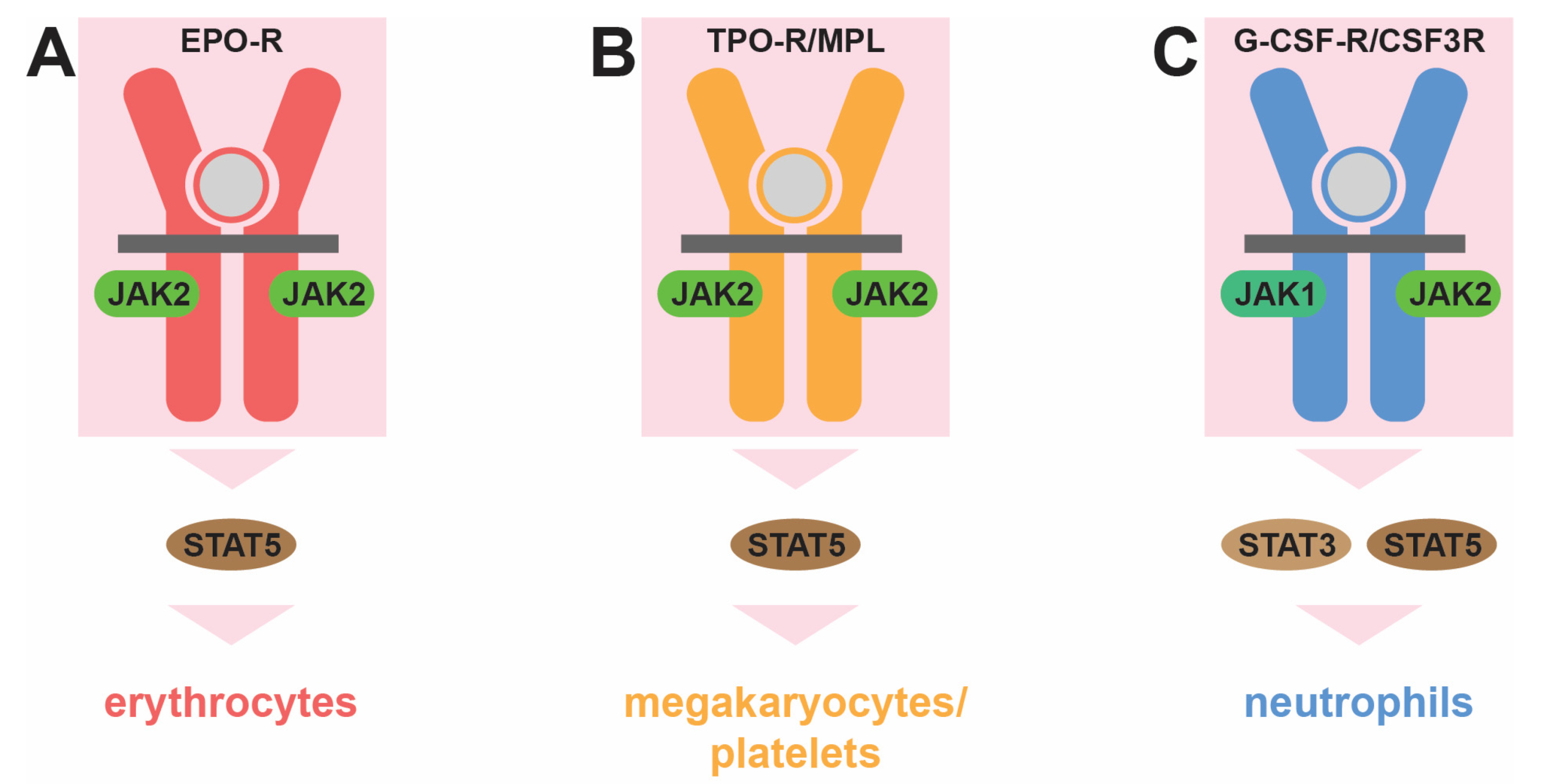
3.2. JAK2
3.3. MPL
3.4. CALR
3.5. CSF3R
3.6. Other Mutations/Variants
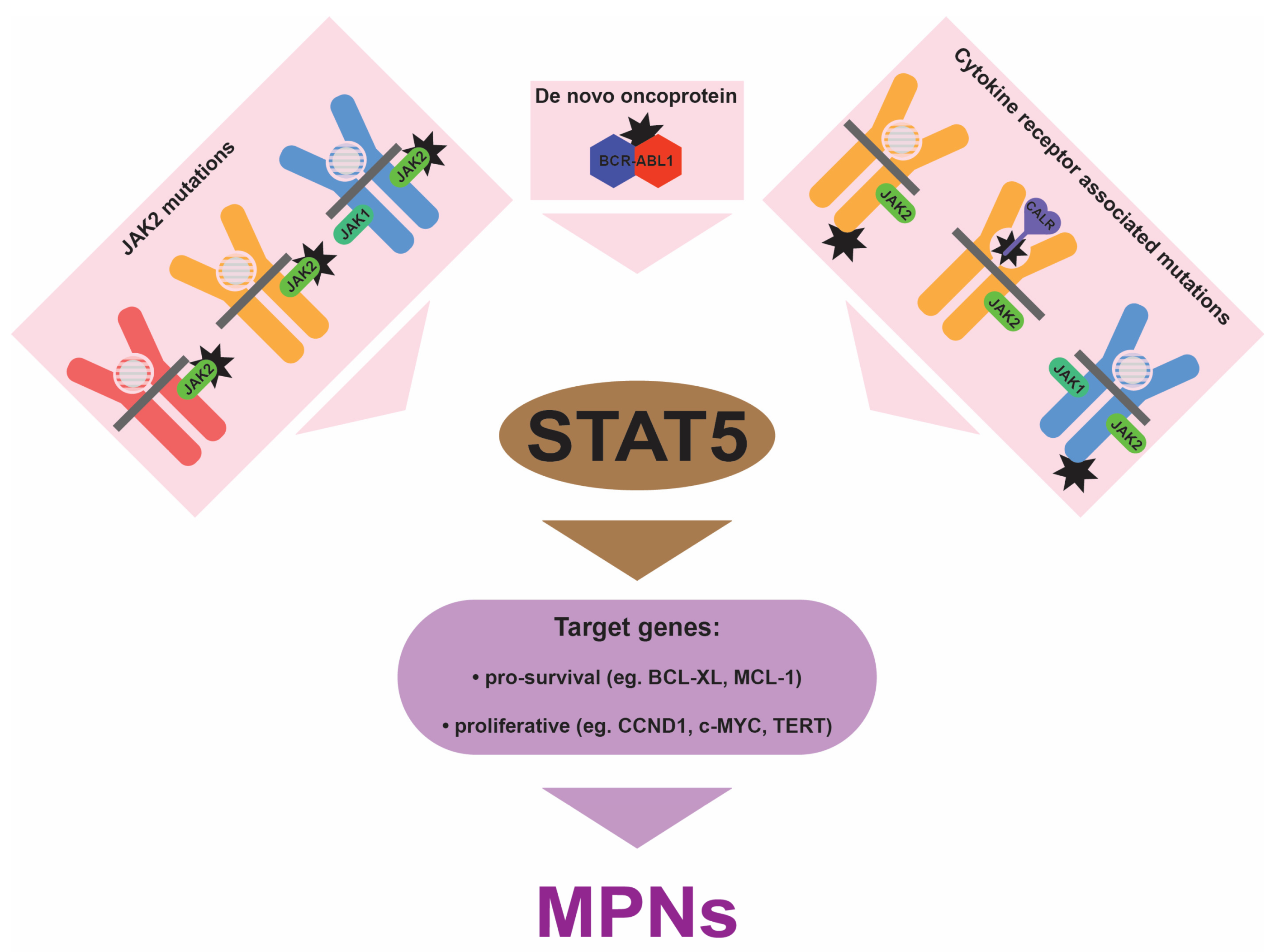
4. Central Role for STAT5
5. Implications for the Clinic
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Mughal, T.I.; Barbui, T.; Abdel-Wahab, O.; Kralovics, R.; Jamieson, C.; Kvasnicka, H.M.; Mullaly, A.; Rampal, R.; Mesa, R.; Kiladjian, J.J.; et al. Novel insights into the biology and treatment of chronic myeloproliferative neoplasms. Leuk. Lymphoma 2015, 56, 1938–1948. [Google Scholar] [CrossRef]
- Osman, A.E.G.; Deininger, M.W. Chronic myeloid leukemia: Modern therapies, current challenges and future directions. Blood Rev. 2021, 49, 100825. [Google Scholar] [CrossRef]
- Lin, Q.; Mao, L.; Shao, L.; Zhu, L.; Han, Q.; Zhu, H.; Jin, J.; You, L. Global, regional, and national burden of chronic myeloid leukemia, 1990-2017: A systematic analysis for the Global Burden of Disease study 2017. Front. Oncol. 2020, 10, 580759. [Google Scholar] [CrossRef]
- McMullin, M.F.; Anderson, L.A. Aetiology of myeloproliferative meoplasms. Cancers 2020, 12, 1810. [Google Scholar] [CrossRef]
- Baumeister, J.; Chatain, N.; Sofias, A.M.; Lammers, T.; Koschmieder, S. Progression of myeloproliferative neoplasms (MPN): Diagnostic and therapeutic perspectives. Cells 2021, 10, 3551. [Google Scholar] [CrossRef]
- Marchetti, M.; Ghirardi, A.; Masciulli, A.; Carobbio, A.; Palandri, F.; Vianelli, N.; Rossi, E.; Betti, S.; Di Veroli, A.; Iurlo, A.; et al. Second cancers in MPN: Survival analysis from an international study. Am. J. Hematol. 2020, 95, 295–301. [Google Scholar] [CrossRef]
- Edginton-White, B.; Bonifer, C. The transcriptional regulation of normal and malignant blood cell development. FEBS J. 2022, 289, 1240–1255. [Google Scholar] [CrossRef]
- Barbarani, G.; Fugazza, C.; Strouboulis, J.; Ronchi, A.E. The pleiotropic effects of GATA1 and KLF1 in physiological erythropoiesis and in dyserythropoietic disorders. Front. Physiol. 2019, 10, 91. [Google Scholar] [CrossRef] [PubMed]
- Friedman, A.D. C/EBPalpha in normal and malignant myelopoiesis. Int. J. Hematol. 2015, 101, 330–341. [Google Scholar] [CrossRef]
- Li, G.; Hao, W.; Hu, W. Transcription factor PU.1 and immune cell differentiation. Int. J. Mol. Med. 2020, 46, 1943–1950. [Google Scholar] [CrossRef]
- Stockley, J.; Morgan, N.V.; Bem, D.; Lowe, G.C.; Lordkipanidze, M.; Dawood, B.; Simpson, M.A.; Macfarlane, K.; Horner, K.; Leo, V.C.; et al. Enrichment of FLI1 and RUNX1 mutations in families with excessive bleeding and platelet dense granule secretion defects. Blood 2013, 122, 4090–4093. [Google Scholar] [CrossRef]
- Marin-Quilez, A.; Garcia-Tunon, I.; Fernandez-Infante, C.; Hernandez-Cano, L.; Palma-Barqueros, V.; Vuelta, E.; Sanchez-Martin, M.; Gonzalez-Porras, J.R.; Guerrero, C.; Benito, R.; et al. Characterization of the platelet phenotype caused by a germline RUNX1 variant in a CRISPR/Cas9-generated murine model. Thromb. Haemost. 2021, 121, 1193–1205. [Google Scholar] [CrossRef]
- Palii, C.G.; Cheng, Q.; Gillespie, M.A.; Shannon, P.; Mazurczyk, M.; Napolitani, G.; Price, N.D.; Ranish, J.A.; Morrissey, E.; Higgs, D.R.; et al. Single-cell proteomics reveal that quantitative changes in co-expressed lineage-specific transcription factors determine cell fate. Cell Stem Cell 2019, 24, 812–820.e815. [Google Scholar] [CrossRef]
- Caulier, A.L.; Sankaran, V.G. Molecular and cellular mechanisms that regulate human erythropoiesis. Blood 2022, 139, 2450–2459. [Google Scholar] [CrossRef]
- Hitchcock, I.S.; Hafer, M.; Sangkhae, V.; Tucker, J.A. The thrombopoietin receptor: Revisiting the master regulator of platelet production. Platelets 2021, 32, 770–778. [Google Scholar] [CrossRef]
- Liongue, C.; Wright, C.; Russell, A.P.; Ward, A.C. Granulocyte colony-stimulating factor receptor: Stimulating granulopoiesis and much more. Int. J. Biochem. Cell. Biol. 2009, 41, 2372–2375. [Google Scholar] [CrossRef]
- Awasthi, N.; Liongue, C.; Ward, A.C. STAT proteins: A kaleidoscope of canonical and non-canonical functions in immunity and cancer. J. Hematol. Oncol. 2021, 14, 198. [Google Scholar] [CrossRef]
- Kautz, L.; Nemeth, E. Molecular liaisons between erythropoiesis and iron metabolism. Blood 2014, 124, 479–482. [Google Scholar] [CrossRef]
- Shih, H.M.; Wu, C.J.; Lin, S.L. Physiology and pathophysiology of renal erythropoietin-producing cells. J. Formos. Med. Assoc. 2018, 117, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Grozovsky, R.; Begonja, A.J.; Liu, K.; Visner, G.; Hartwig, J.H.; Falet, H.; Hoffmeister, K.M. The Ashwell-Morell receptor regulates hepatic thrombopoietin production via JAK2-STAT3 signaling. Nat. Med. 2015, 21, 47–54. [Google Scholar] [CrossRef]
- Pedersen, C.C.; Borup, R.; Fischer-Nielsen, A.; Mora-Jensen, H.; Fossum, A.; Cowland, J.B.; Borregaard, N. Changes in gene expression during G-CSF-induced emergency granulopoiesis in humans. J. Immunol. 2016, 197, 1989–1999. [Google Scholar] [CrossRef]
- Sobah, M.L.; Liongue, C.; Ward, A.C. SOCS proteins in immunity, inflammatory diseases and immune-related cancer. Front. Med. 2021, 8, 727987. [Google Scholar] [CrossRef]
- Cross, N.C.; Daley, G.Q.; Green, A.R.; Hughes, T.P.; Jamieson, C.; Manley, P.; Mughal, T.; Perrotti, D.; Radich, J.; Skoda, R.; et al. BCR-ABL1-positive CML and BCR-ABL1-negative chronic myeloproliferative disorders: Some common and contrasting features. Leukemia 2008, 22, 1975–1989. [Google Scholar] [CrossRef]
- Maxson, J.E.; Tyner, J.W. Genomics of chronic neutrophilic leukemia. Blood 2017, 129, 715–722. [Google Scholar] [CrossRef]
- Kjaer, L. Clonal hematopoiesis and mutations of myeloproliferative neoplasms. Cancers 2020, 12, 2100. [Google Scholar] [CrossRef]
- Bellanne-Chantelot, C.; Rabadan Moraes, G.; Schmaltz-Panneau, B.; Marty, C.; Vainchenker, W.; Plo, I. Germline genetic factors in the pathogenesis of myeloproliferative neoplasms. Blood Rev. 2020, 42, 100710. [Google Scholar] [CrossRef]
- Luque Paz, D.; Kralovics, R.; Skoda, R.C. Genetic basis and molecular profiling in myeloproliferative neoplasms. Blood 2023, 141, 1909–1921. [Google Scholar] [CrossRef]
- Nangalia, J.; Green, A.R. Myeloproliferative neoplasms: From origins to outcomes. Blood 2017, 130, 2475–2483. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, G.; McMullin, M.F.; Mills, K. Molecular pathogenesis of the myeloproliferative neoplasms. J. Hematol. Oncol. 2021, 14, 103. [Google Scholar] [CrossRef]
- Quintas-Cardama, A.; Cortes, J. Molecular biology of BCR-ABL1-positive chronic myeloid leukemia. Blood 2009, 113, 1619–1630. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2016 update on diagnosis, therapy, and monitoring. Am. J. Hematol. 2016, 91, 252–265. [Google Scholar] [CrossRef]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.P.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef]
- Passamonti, F.; Elena, C.; Schnittger, S.; Skoda, R.C.; Green, A.R.; Girodon, F.; Kiladjian, J.J.; McMullin, M.F.; Ruggeri, M.; Besses, C.; et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood 2011, 117, 2813–2816. [Google Scholar] [CrossRef]
- Wilmes, S.; Hafer, M.; Vuorio, J.; Tucker, J.A.; Winkelmann, H.; Lochte, S.; Stanly, T.A.; Pulgar Prieto, K.D.; Poojari, C.; Sharma, V.; et al. Mechanism of homodimeric cytokine receptor activation and dysregulation by oncogenic mutations. Science 2020, 367, 643–652. [Google Scholar] [CrossRef]
- Bose, P.; Verstovsek, S. Updates in the management of polycythemia vera and essential thrombocythemia. Ther. Adv. Hematol. 2019, 10, 2040620719870052. [Google Scholar] [CrossRef]
- Ding, J.; Komatsu, H.; Wakita, A.; Kato-Uranishi, M.; Ito, M.; Satoh, A.; Tsuboi, K.; Nitta, M.; Miyazaki, H.; Iida, S.; et al. Familial essential thrombocythemia associated with a dominant-positive activating mutation of the c-MPL gene, which encodes for the receptor for thrombopoietin. Blood 2004, 103, 4198–4200. [Google Scholar] [CrossRef]
- Plo, I.; Bellanne-Chantelot, C.; Mosca, M.; Mazzi, S.; Marty, C.; Vainchenker, W. Genetic alterations of the thrombopoietin/MPL/JAK2 axis impacting megakaryopoiesis. Front. Endocrinol. 2017, 8, 234. [Google Scholar] [CrossRef]
- Pecquet, C.; Chachoua, I.; Roy, A.; Balligand, T.; Vertenoeil, G.; Leroy, E.; Albu, R.I.; Defour, J.P.; Nivarthi, H.; Hug, E.; et al. Calreticulin mutants as oncogenic rogue chaperones for TpoR and traffic-defective pathogenic TpoR mutants. Blood 2019, 133, 2669–2681. [Google Scholar] [CrossRef]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhang, C.; Ma, X.; Guan, M. Clinical relevance between CALR mutation and myeloproliferative neoplasms. Stem Cell Investig. 2015, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Liongue, C.; Ward, A.C. Granulocyte colony-stimulating factor receptor mutations in myeloid malignancy. Front. Oncol. 2014, 4, 93. [Google Scholar] [CrossRef] [PubMed]
- Maxson, J.E.; Luty, S.B.; MacManiman, J.D.; Abel, M.L.; Druker, B.J.; Tyner, J.W. Ligand independence of the T618I mutation in the colony-stimulating factor 3 receptor (CSF3R) protein results from loss of O-linked glycosylation and increased receptor dimerization. J. Biol. Chem. 2014, 289, 5820–5827. [Google Scholar] [CrossRef]
- Yin, B.; Chen, X.; Gao, F.; Li, J.; Wang, H.W. Analysis of gene mutation characteristics in patients with chronic neutrophilic leukaemia. Hematology 2019, 24, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Plo, I.; Zhang, Y.; Le Couedic, J.P.; Nakatake, M.; Boulet, J.M.; Itaya, M.; Smith, S.O.; Debili, N.; Constantinescu, S.N.; Vainchenker, W.; et al. An activating mutation in the CSF3R gene induces a hereditary chronic neutrophilia. J. Exp. Med. 2009, 206, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Druhan, L.J.; McMahon, D.P.; Steuerwald, N.; Price, A.E.; Lance, A.; Gerber, J.M.; Avalos, B.R. Chronic neutrophilic leukemia in a child with a CSF3R T618I germ line mutation. Blood 2016, 128, 2097–2099. [Google Scholar] [CrossRef]
- Lee, J.; Godfrey, A.L.; Nangalia, J. Genomic heterogeneity in myeloproliferative neoplasms and applications to clinical practice. Blood Rev. 2020, 42, 100708. [Google Scholar] [CrossRef]
- Vaughan, A.M.; Kappe, S.H.; Ploss, A.; Mikolajczak, S.A. Development of humanized mouse models to study human malaria parasite infection. Future Microbiol. 2012, 7, 657–665. [Google Scholar] [CrossRef]
- Pasquier, F.; Marty, C.; Balligand, T.; Verdier, F.; Grosjean, S.; Gryshkova, V.; Raslova, H.; Constantinescu, S.N.; Casadevall, N.; Vainchenker, W.; et al. New pathogenic mechanisms induced by germline erythropoietin receptor mutations in primary erythrocytosis. Haematologica 2018, 103, 575–586. [Google Scholar] [CrossRef]
- Zmajkovic, J.; Lundberg, P.; Nienhold, R.; Torgersen, M.L.; Sundan, A.; Waage, A.; Skoda, R.C. A gain-of-function mutation in EPO in familial erythrocytosis. N. Engl. J. Med. 2018, 378, 924–930. [Google Scholar] [CrossRef]
- Maurer, B.; Kollmann, S.; Pickem, J.; Hoelbl-Kovacic, A.; Sexl, V. STAT5A and STAT5B-twins with different personalities in hematopoiesis and leukemia. Cancers 2019, 11, 1726. [Google Scholar] [CrossRef]
- Cross, N.C.P.; Hoade, Y.; Tapper, W.J.; Carreno-Tarragona, G.; Fanelli, T.; Jawhar, M.; Naumann, N.; Pieniak, I.; Lubke, J.; Ali, S.; et al. Recurrent activating STAT5B N642H mutation in myeloid neoplasms with eosinophilia. Leukemia 2019, 33, 415–425. [Google Scholar] [CrossRef]
- Luo, Q.; Shen, J.; Yang, Y.; Tang, H.; Shi, M.; Liu, J.; Liu, Z.; Shi, X.; Yi, Y. CSF3R T618I, ASXL1 G942 fs and STAT5B N642H trimutation co-contribute to a rare chronic neutrophilic leukaemia manifested by rapidly progressive leucocytosis, severe infections, persistent fever and deep venous thrombosis. Br. J. Haematol. 2018, 180, 892–894. [Google Scholar] [CrossRef]
- Pardanani, A.; Fridley, B.L.; Lasho, T.L.; Gilliland, D.G.; Tefferi, A. Host genetic variation contributes to phenotypic diversity in myeloproliferative disorders. Blood 2008, 111, 2785–2789. [Google Scholar] [CrossRef]
- Jones, A.V.; Campbell, P.J.; Beer, P.A.; Schnittger, S.; Vannucchi, A.M.; Zoi, K.; Percy, M.J.; McMullin, M.F.; Scott, L.M.; Tapper, W.; et al. The JAK2 46/1 haplotype predisposes to MPL-mutated myeloproliferative neoplasms. Blood 2010, 115, 4517–4523. [Google Scholar] [CrossRef]
- Lesteven, E.; Picque, M.; Conejero Tonetti, C.; Giraudier, S.; Varin-Blank, N.; Velazquez, L.; Kiladjian, J.J.; Cassinat, B.; Baran-Marszak, F. Association of a single-nucleotide polymorphism in the SH2B3 gene with JAK2V617F-positive myeloproliferative neoplasms. Blood 2014, 123, 794–796. [Google Scholar] [CrossRef]
- Rabadan Moraes, G.; Pasquier, F.; Marzac, C.; Deconinck, E.; Damanti, C.C.; Leroy, G.; El-Khoury, M.; El Nemer, W.; Kiladjian, J.J.; Raslova, H.; et al. An inherited gain-of-function risk allele in EPOR predisposes to familial JAK2(V617F) myeloproliferative neoplasms. Br. J. Haematol. 2022, 198, 131–136. [Google Scholar] [CrossRef]
- Turakhia, S.K.; Murugesan, G.; Cotta, C.V.; Theil, K.S. Thrombocytosis and STAT5 activation in chronic myelogenous leukaemia are not associated with JAK2 V617F or calreticulin mutations. J. Clin. Pathol. 2016, 69, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Wisniewska-Chudy, E.; Szylberg, L.; Dworacki, G.; Mizera-Nyczak, E.; Marszalek, A. pSTAT5 and ERK exhibit different expression in myeloproliferative neoplasms. Oncol. Rep. 2017, 37, 2295–2307. [Google Scholar] [CrossRef] [PubMed]
- Beer, P.A.; Campbell, P.J.; Scott, L.M.; Bench, A.J.; Erber, W.N.; Bareford, D.; Wilkins, B.S.; Reilly, J.T.; Hasselbalch, H.C.; Bowman, R.; et al. MPL mutations in myeloproliferative disorders: Analysis of the PT-1 cohort. Blood 2008, 112, 141–149. [Google Scholar] [CrossRef]
- Zhang, H.; Coblentz, C.; Watanabe-Smith, K.; Means, S.; Means, J.; Maxson, J.E.; Tyner, J.W. Gain-of-function mutations in granulocyte colony-stimulating factor receptor (CSF3R) reveal distinct mechanisms of CSF3R activation. J. Biol. Chem. 2018, 293, 7387–7396. [Google Scholar] [CrossRef] [PubMed]
- Sakr, H.; Clark Schneider, K.; Murugesan, G.; Bodo, J.; Hsi, E.D.; Cook, J.R. pSTAT3/pSTAT5 signaling patterns in molecularly defined subsets of myeloproliferative neoplasms. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 147–152. [Google Scholar] [CrossRef]
- Weber, A.; Borghouts, C.; Brendel, C.; Moriggl, R.; Delis, N.; Brill, B.; Vafaizadeh, V.; Groner, B. Stat5 exerts distinct, vital functions in the cytoplasm and nucleus of Bcr-Abl+ K562 and Jak2(V617F)+ HEL leukemia Cells. Cancers 2015, 7, 503–537. [Google Scholar] [CrossRef] [PubMed]
- Walz, C.; Ahmed, W.; Lazarides, K.; Betancur, M.; Patel, N.; Hennighausen, L.; Zaleskas, V.M.; Van Etten, R.A. Essential role for Stat5a/b in myeloproliferative neoplasms induced by BCR-ABL1 and JAK2(V617F) in mice. Blood 2012, 119, 3550–3560. [Google Scholar] [CrossRef]
- Yan, D.; Hutchison, R.E.; Mohi, G. Critical requirement for Stat5 in a mouse model of polycythemia vera. Blood 2012, 119, 3539–3549. [Google Scholar] [CrossRef]
- Grisouard, J.; Shimizu, T.; Duek, A.; Kubovcakova, L.; Hao-Shen, H.; Dirnhofer, S.; Skoda, R.C. Deletion of Stat3 in hematopoietic cells enhances thrombocytosis and shortens survival in a JAK2-V617F mouse model of MPN. Blood 2015, 125, 2131–2140. [Google Scholar] [CrossRef] [PubMed]
- Duek, A.; Lundberg, P.; Shimizu, T.; Grisouard, J.; Karow, A.; Kubovcakova, L.; Hao-Shen, H.; Dirnhofer, S.; Skoda, R.C. Loss of Stat1 decreases megakaryopoiesis and favors erythropoiesis in a JAK2-V617F-driven mouse model of MPNs. Blood 2014, 123, 3943–3950. [Google Scholar] [CrossRef]
- Kollmann, S.; Grundschober, E.; Maurer, B.; Warsch, W.; Grausenburger, R.; Edlinger, L.; Huuhtanen, J.; Lagger, S.; Hennighausen, L.; Valent, P.; et al. Twins with different personalities: STAT5B-but not STAT5A-has a key role in BCR/ABL-induced leukemia. Leukemia 2019, 33, 1583–1597. [Google Scholar] [CrossRef]
- Belizaire, R.; Koochaki, S.H.J.; Udeshi, N.D.; Vedder, A.; Sun, L.; Svinkina, T.; Hartigan, C.; McConkey, M.; Kovalcik, V.; Bizuayehu, A.; et al. CBL mutations drive PI3K/AKT signaling via increased interaction with LYN and PIK3R1. Blood 2021, 137, 2209–2220. [Google Scholar] [CrossRef]
- Grand, F.H.; Hidalgo-Curtis, C.E.; Ernst, T.; Zoi, K.; Zoi, C.; McGuire, C.; Kreil, S.; Jones, A.; Score, J.; Metzgeroth, G.; et al. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood 2009, 113, 6182–6192. [Google Scholar] [CrossRef]
- Maslah, N.; Cassinat, B.; Verger, E.; Kiladjian, J.J.; Velazquez, L. The role of LNK/SH2B3 genetic alterations in myeloproliferative neoplasms and other hematological disorders. Leukemia 2017, 31, 1661–1670. [Google Scholar] [CrossRef]
- Mandal, M.; Powers, S.E.; Maienschein-Cline, M.; Bartom, E.T.; Hamel, K.M.; Kee, B.L.; Dinner, A.R.; Clark, M.R. Epigenetic repression of the Igk locus by STAT5-mediated recruitment of the histone methyltransferase Ezh2. Nat. Immunol. 2011, 12, 1212–1220. [Google Scholar] [CrossRef]
- Yoo, K.H.; Oh, S.; Kang, K.; Hensel, T.; Robinson, G.W.; Hennighausen, L. Loss of EZH2 results in precocious mammary gland development and activation of STAT5-dependent genes. Nucleic Acids Res. 2015, 43, 8774–8789. [Google Scholar] [CrossRef]
- Goyal, H.; Chachoua, I.; Pecquet, C.; Vainchenker, W.; Constantinescu, S.N. A p53-JAK-STAT connection involved in myeloproliferative neoplasm pathogenesis and progression to secondary acute myeloid leukemia. Blood Rev. 2020, 42, 100712. [Google Scholar] [CrossRef] [PubMed]
- Ma, A.C.; Fan, A.; Ward, A.C.; Liongue, C.; Lewis, R.S.; Cheng, S.H.; Chan, P.K.; Yip, S.F.; Liang, R.; Leung, A.Y. A novel zebrafish jak2a(V581F) model shared features of human JAK2(V617F) polycythemia vera. Exp. Hematol. 2009, 37, 1379–1386. [Google Scholar] [CrossRef]
- Lewis, R.S.; Stephenson, S.E.M.; Ward, A.C. Constitutive activation of zebrafish Stat5 expands hematopoietic cell populations in vivo. Exp. Hematol. 2006, 34, 179–187. [Google Scholar] [CrossRef]
- Harrison, D.A.; Binari, R.; Nahreini, T.S.; Gilman, M.; Perrimon, N. Activation of a Drosophila Janus kinase (JAK) causes hematopoietic neoplasia and developmental defects. EMBO J. 1995, 14, 2857–2865. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.S.; Melnick, M.B.; Perrimon, N. Marelle acts downstream of the Drosophila HOP/JAK kinase and encodes a protein similar to the mammalian STATs. Cell 1996, 84, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, G.O.L.; Cramer, S.D.; Winer, H.Y.; Hixon, J.A.; Li, W.; Yunes, J.A.; Durum, S.K. Mutations that collaborate with IL-7Ra signaling pathways to drive ALL. Adv. Biol. Regul. 2021, 80, 100788. [Google Scholar] [CrossRef]
- Teramo, A.; Barila, G.; Calabretto, G.; Vicenzetto, C.; Gasparini, V.R.; Semenzato, G.; Zambello, R. Insights into genetic landscape of large granular lymphocyte leukemia. Front. Oncol. 2020, 10, 152. [Google Scholar] [CrossRef]
- Andersson, E.I.; Bruck, O.; Braun, T.; Mannisto, S.; Saikko, L.; Lagstrom, S.; Ellonen, P.; Leppa, S.; Herling, M.; Kovanen, P.E.; et al. STAT3 mutation is associated with STAT3 activation in CD30+ ALK− ALCL. Cancers 2020, 12, 702. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Vannucchi, A.M. Current management strategies for polycythemia vera and essential thrombocythemia. Blood Rev. 2020, 42, 100714. [Google Scholar] [CrossRef]
- Ge, H.; Wang, C.; Tian, C.; Diao, Y.; Wang, W.; Ma, X.; Zhang, J.; Li, H.; Zhao, Z.; Zhu, L. Efficacy of WWQ-131, a highly selective JAK2 inhibitor, in mouse models of myeloproliferative neoplasms. Biomed. Pharmacother. 2022, 156, 113884. [Google Scholar] [CrossRef]
- Wang, X.; Haylock, D.; Hu, C.S.; Kowalczyk, W.; Jiang, T.; Qiu, J.; Mosoyan, G.; He, W.; Marshall, N.; Mascarenhas, J.; et al. A thrombopoietin receptor antagonist is capable of depleting myelofibrosis hematopoietic stem and progenitor cells. Blood 2016, 127, 3398–3409. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Wingelhofer, B.; Maurer, B.; Heyes, E.C.; Cumaraswamy, A.A.; Berger-Becvar, A.; de Araujo, E.D.; Orlova, A.; Freund, P.; Ruge, F.; Park, J.; et al. Pharmacologic inhibition of STAT5 in acute myeloid leukemia. Leukemia 2018, 32, 1135–1146. [Google Scholar] [CrossRef]
- Hadzijusufovic, E.; Keller, A.; Berger, D.; Greiner, G.; Wingelhofer, B.; Witzeneder, N.; Ivanov, D.; Pecnard, E.; Nivarthi, H.; Schur, F.K.M.; et al. STAT5 is expressed in CD34+/CD38− stem cells and serves as a potential molecular target in Ph-negative myeloproliferative neoplasms. Cancers 2020, 12, 1021. [Google Scholar] [CrossRef]
- Juen, L.; Brachet-Botineau, M.; Parmenon, C.; Bourgeais, J.; Herault, O.; Gouilleux, F.; Viaud-Massuard, M.C.; Prie, G. New inhibitor targeting signal transducer and activator of transcription 5 (STAT5) signaling in myeloid leukemias. J. Med. Chem. 2017, 60, 6119–6136. [Google Scholar] [CrossRef]
- Liao, Z.; Gu, L.; Vergalli, J.; Mariani, S.A.; De Dominici, M.; Lokareddy, R.K.; Dagvadorj, A.; Purushottamachar, P.; McCue, P.A.; Trabulsi, E.; et al. Structure-based screen identifies a potent small molecule inhibitor of Stat5a/b with therapeutic potential for prostate cancer and chronic myeloid leukemia. Mol. Cancer Ther. 2015, 14, 1777–1793. [Google Scholar] [CrossRef]
- Elumalai, N.; Berg, A.; Rubner, S.; Blechschmidt, L.; Song, C.; Natarajan, K.; Matysik, J.; Berg, T. Rational development of Stafib-2: A selective, nanomolar inhibitor of the transcription factor STAT5b. Sci. Rep. 2017, 7, 819. [Google Scholar] [CrossRef] [PubMed]
- Mencalha, A.L.; Du Rocher, B.; Salles, D.; Binato, R.; Abdelhay, E. LLL-3, a STAT3 inhibitor, represses BCR-ABL-positive cell proliferation, activates apoptosis and improves the effects of Imatinib mesylate. Cancer Chemother. Pharmacol. 2010, 65, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zeng, J.; Shi, M.; Zhao, S.; Bai, W.; Cao, W.; Tu, Z.; Huang, Z.; Feng, W. Targeted blockage of signal transducer and activator of transcription 5 signaling pathway with decoy oligodeoxynucleotides suppresses leukemic K562 cell growth. DNA Cell Biol. 2011, 30, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Turkson, J.; Zhang, S.; Mora, L.B.; Burns, A.; Sebti, S.; Jove, R. A novel platinum compound inhibits constitutive Stat3 signaling and induces cell cycle arrest and apoptosis of malignant cells. J. Biol. Chem. 2005, 280, 32979–32988. [Google Scholar] [CrossRef] [PubMed]
- Siddiquee, K.; Zhang, S.; Guida, W.C.; Blaskovich, M.A.; Greedy, B.; Lawrence, H.R.; Yip, M.L.; Jove, R.; McLaughlin, M.M.; Lawrence, N.J.; et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7391–7396. [Google Scholar] [CrossRef] [PubMed]
- Bartalucci, N.; Calabresi, L.; Balliu, M.; Martinelli, S.; Rossi, M.C.; Villeval, J.L.; Annunziato, F.; Guglielmelli, P.; Vannucchi, A.M. Inhibitors of the PI3K/mTOR pathway prevent STAT5 phosphorylation in JAK2V617F mutated cells through PP2A/CIP2A axis. Oncotarget 2017, 8, 96710–96724. [Google Scholar] [CrossRef]
- Fenerich, B.A.; Fernandes, J.C.; Rodrigues Alves, A.P.N.; Coelho-Silva, J.L.; Scopim-Ribeiro, R.; Scheucher, P.S.; Eide, C.A.; Tognon, C.E.; Druker, B.J.; Rego, E.M.; et al. NT157 has antineoplastic effects and inhibits IRS1/2 and STAT3/5 in JAK2(V617F)-positive myeloproliferative neoplasm cells. Signal Transduct. Target. Ther. 2020, 5, 5. [Google Scholar] [CrossRef]
- Austin, R.J.; Straube, J.; Bruedigam, C.; Pali, G.; Jacquelin, S.; Vu, T.; Green, J.; Grasel, J.; Lansink, L.; Cooper, L.; et al. Distinct effects of ruxolitinib and interferon-alpha on murine JAK2V617F myeloproliferative neoplasm hematopoietic stem cell populations. Leukemia 2020, 34, 1075–1089. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| MPN Category | Genetic Change | Frequency |
|---|---|---|
| CML | BCR::ABL1 | >95% |
| PV | JAK2 GOF | ~98% |
| ET | JAK2 GOF | 55% |
| MPL GOF | 5–7% | |
| CALR GOF | 25–30% | |
| PMF | JAK2 GOF | 60% |
| MPL GOF | 7–10% | |
| CALR GOF | 20–30% | |
| Hereditary thrombocytosis | THPO GOF | nd |
| JAK2 GOF | nd | |
| MPL GOF | nd | |
| Hereditary erythrocytosis | EPOR GOF | nd |
| EPO GOF | nd | |
| VHL LOF | nd | |
| CNL | CSF3R GOF | >80% |
| Hypereosinophilic syndrome | STAT5B GOF | nd |
| Disease | Treatment | Effect | Application |
|---|---|---|---|
| CML | imatinib, dasatinib, nilotinib, bosutinib, ponatinib | ABL1-selective inhibitors | 1st line therapy |
| PV | hydroxyurea, phlebotomy | cytoreductive | 1st line therapy |
| aspirin (low dose) | antithrombotic | 1st line therapy | |
| ruxolitinib | JAK2-selective inhibitors | 2nd line therapy | |
| MF | ruxolitinib, fedratinib, pacritinib | JAK2-selective inhibitors | 1st line therapy |
| Various MPNs | interferon α | antiproliferative, pro-apoptotic | alternative therapy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liongue, C.; Ward, A.C. Myeloproliferative Neoplasms: Diseases Mediated by Chronic Activation of Signal Transducer and Activator of Transcription (STAT) Proteins. Cancers 2024, 16, 313. https://doi.org/10.3390/cancers16020313
Liongue C, Ward AC. Myeloproliferative Neoplasms: Diseases Mediated by Chronic Activation of Signal Transducer and Activator of Transcription (STAT) Proteins. Cancers. 2024; 16(2):313. https://doi.org/10.3390/cancers16020313
Chicago/Turabian StyleLiongue, Clifford, and Alister C. Ward. 2024. "Myeloproliferative Neoplasms: Diseases Mediated by Chronic Activation of Signal Transducer and Activator of Transcription (STAT) Proteins" Cancers 16, no. 2: 313. https://doi.org/10.3390/cancers16020313
APA StyleLiongue, C., & Ward, A. C. (2024). Myeloproliferative Neoplasms: Diseases Mediated by Chronic Activation of Signal Transducer and Activator of Transcription (STAT) Proteins. Cancers, 16(2), 313. https://doi.org/10.3390/cancers16020313