
Next-Generation CEA-CAR-NK-92 Cells against Solid Tumors: Overcoming Tumor Microenvironment Challenges in Colorectal Cancer
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Multicellular Tumor Spheroid Culture (3D Cell Culture)
2.3. Lentivirus Vector Production and Transduction
2.4. Cytotoxicity Assays
2.5. Migration Assay
2.6. Flow Cytometry
2.7. Immunocytochemistry and Confocal Microscopy
2.8. Trogocytosis Assays
2.9. Data Analysis
3. Results
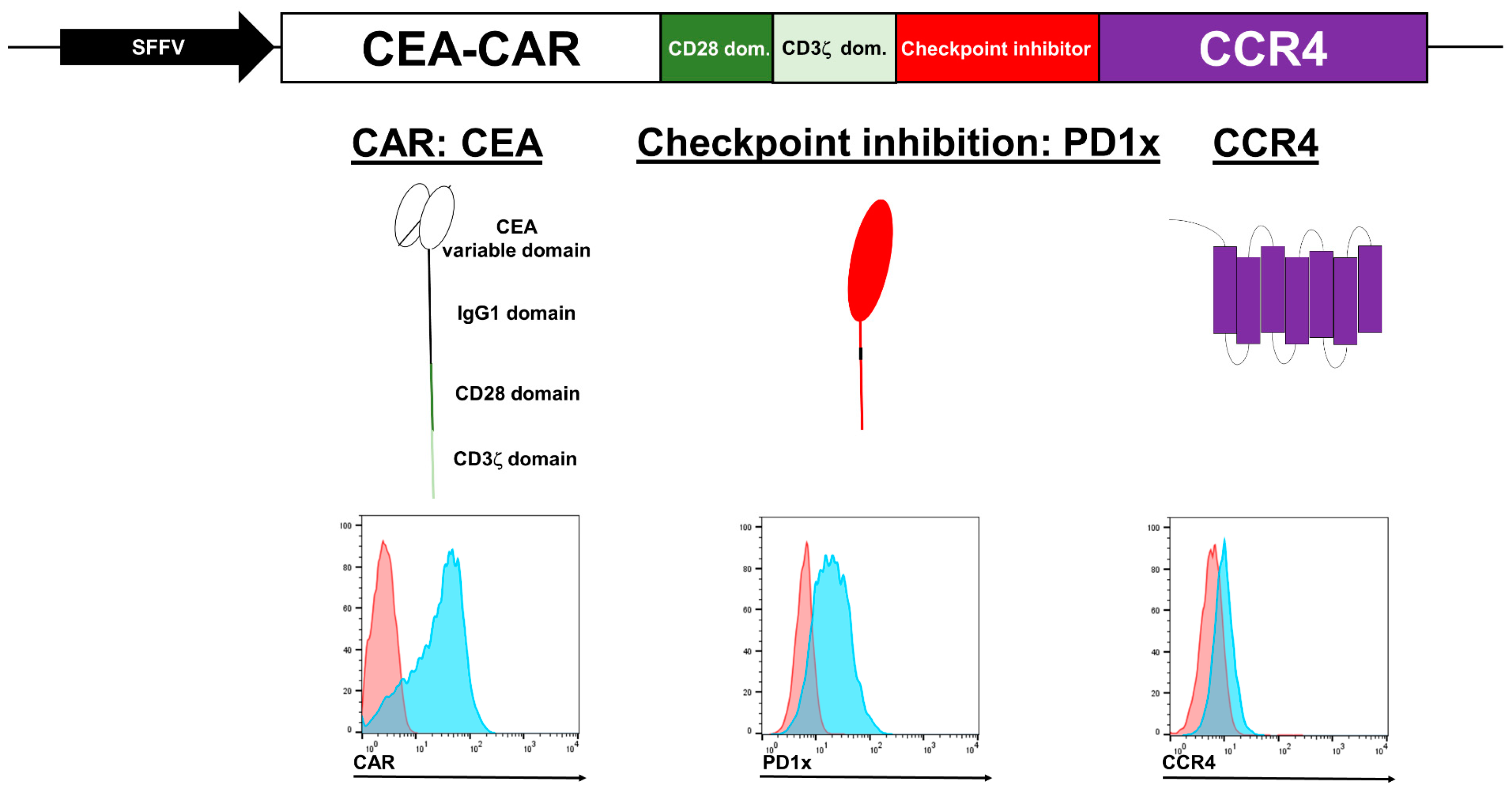
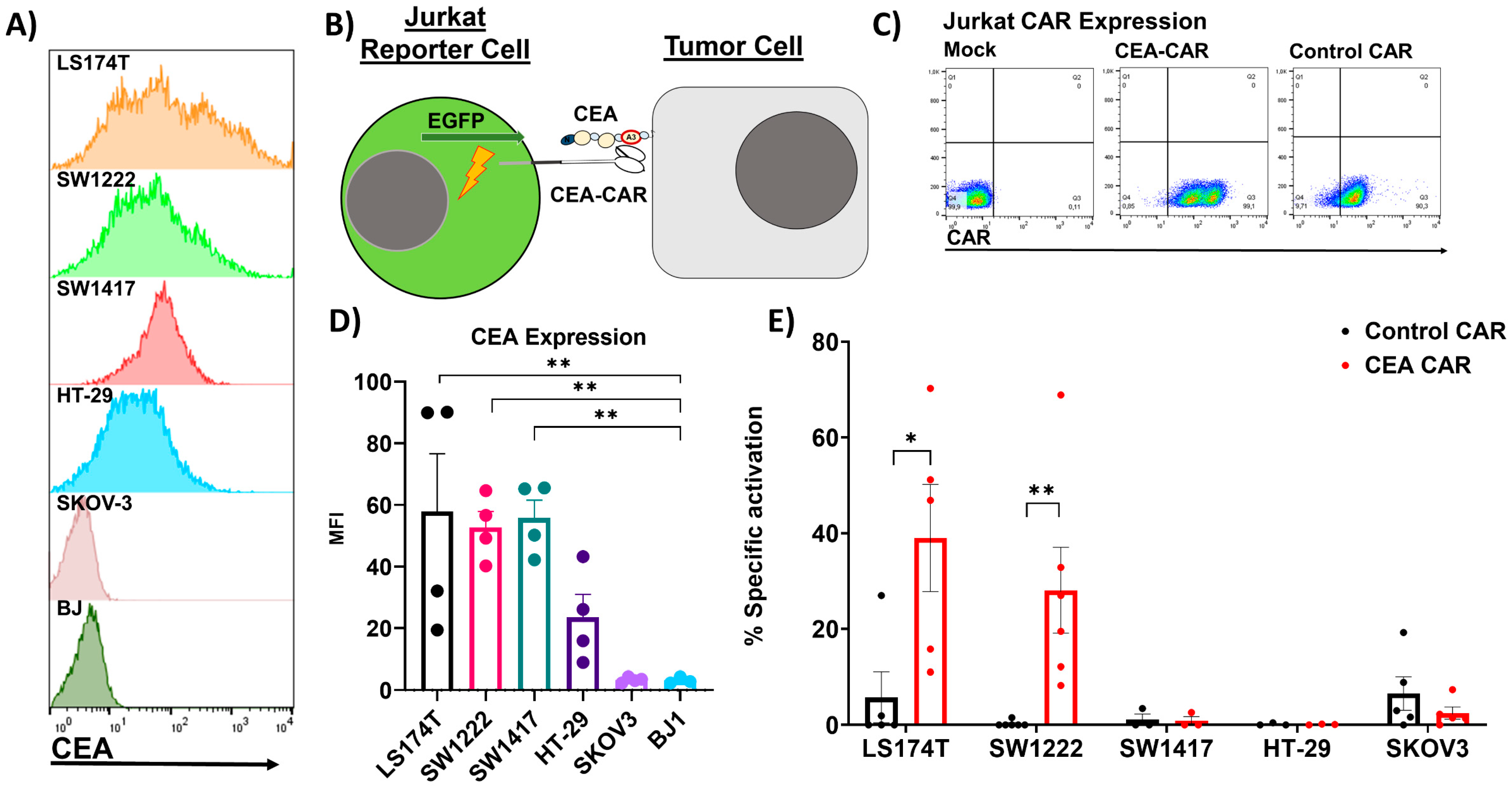
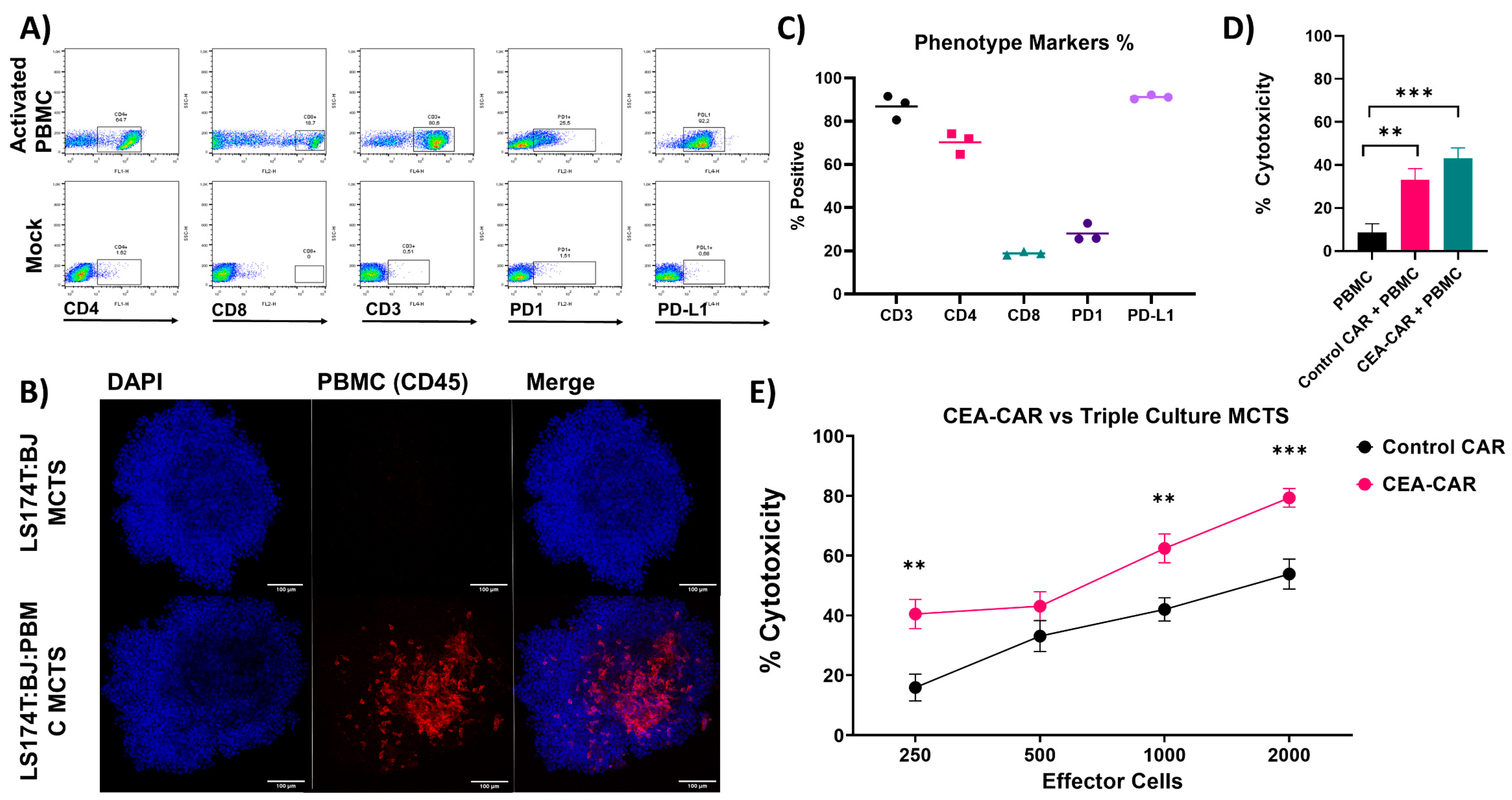
3.1. NK Cell CEA-CAR Validation
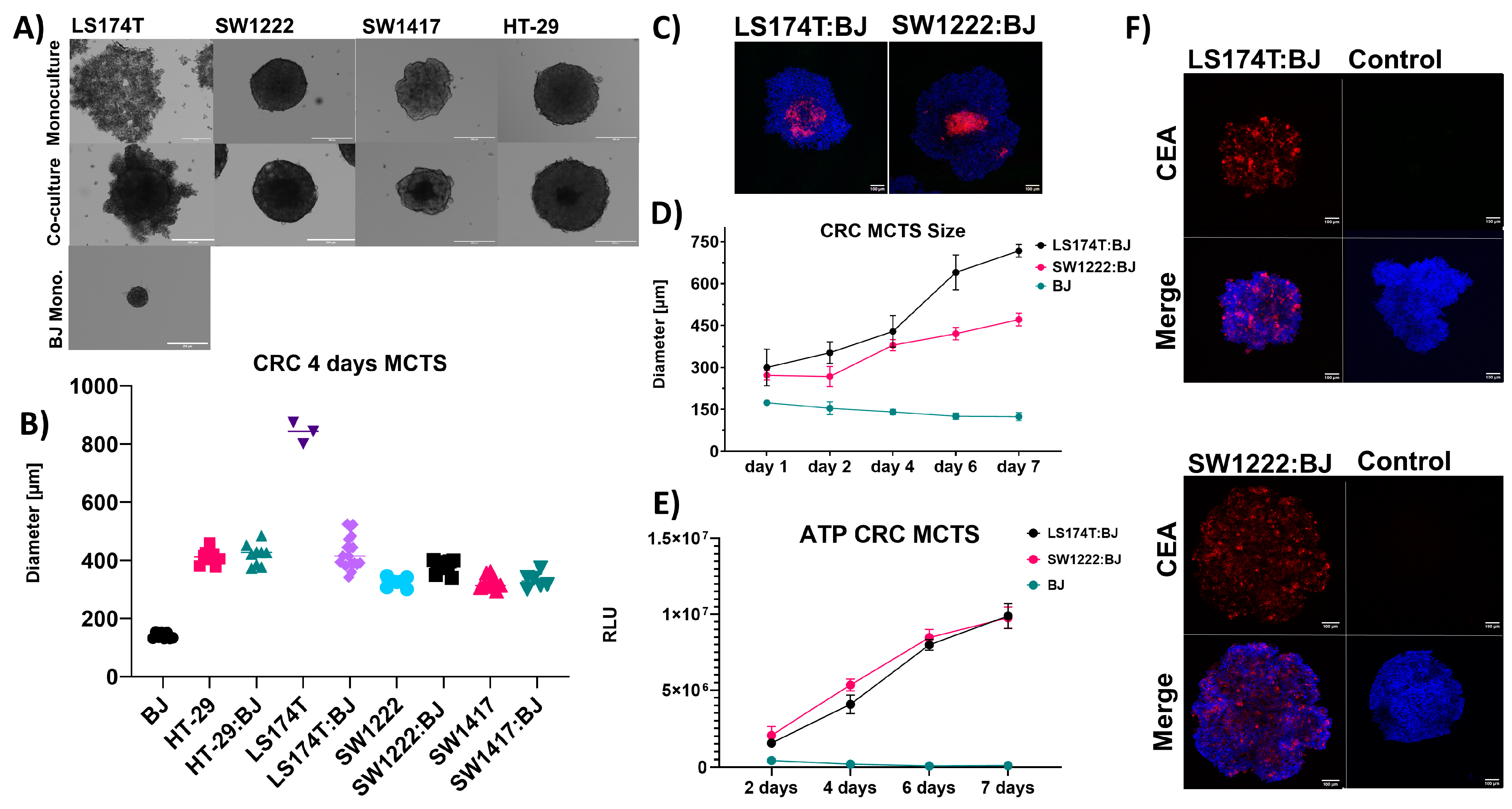
3.2. Solid Tumor Modeling
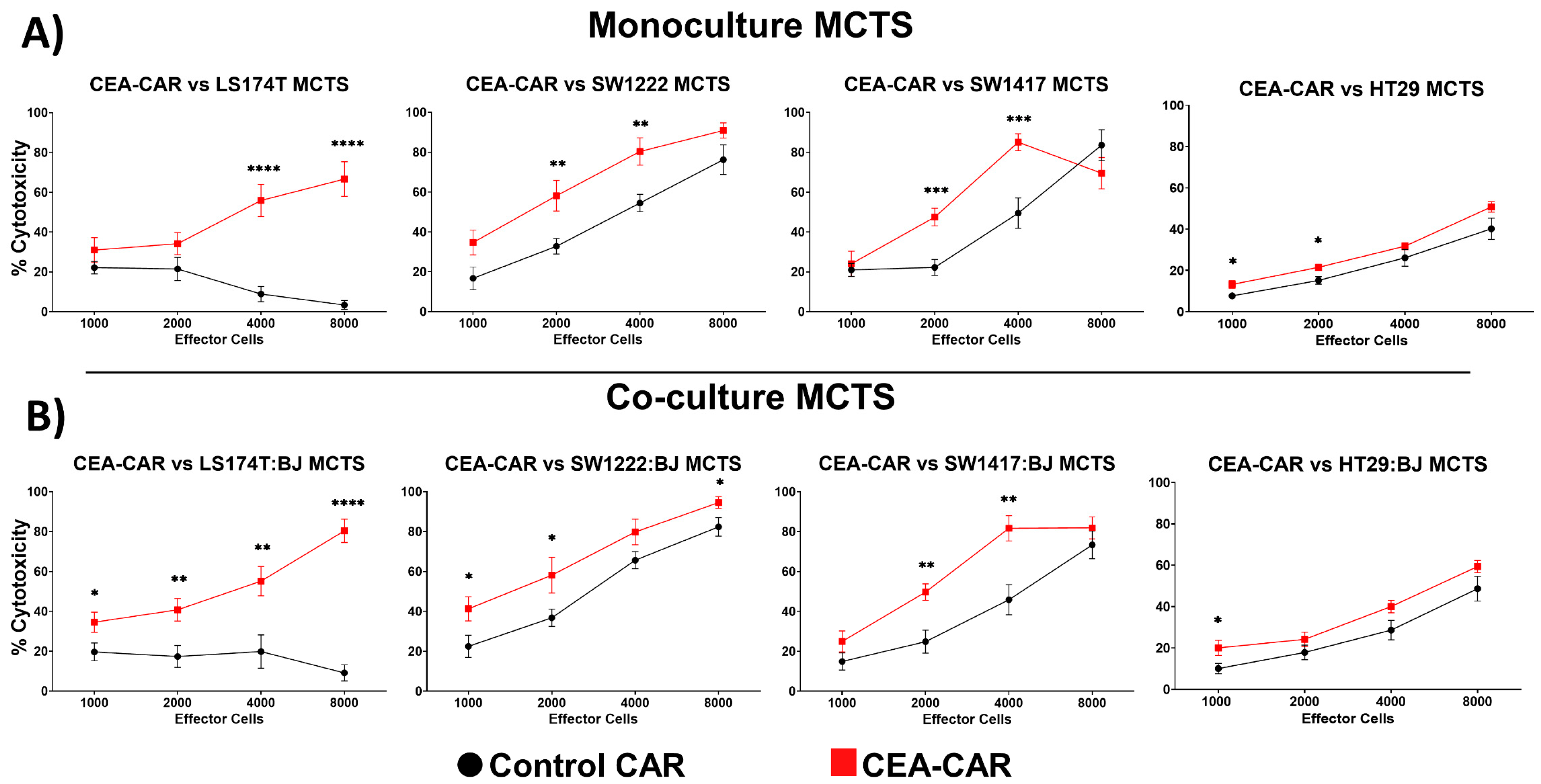
3.3. CEA-CAR NK Cells vs. Solid Tumor Models
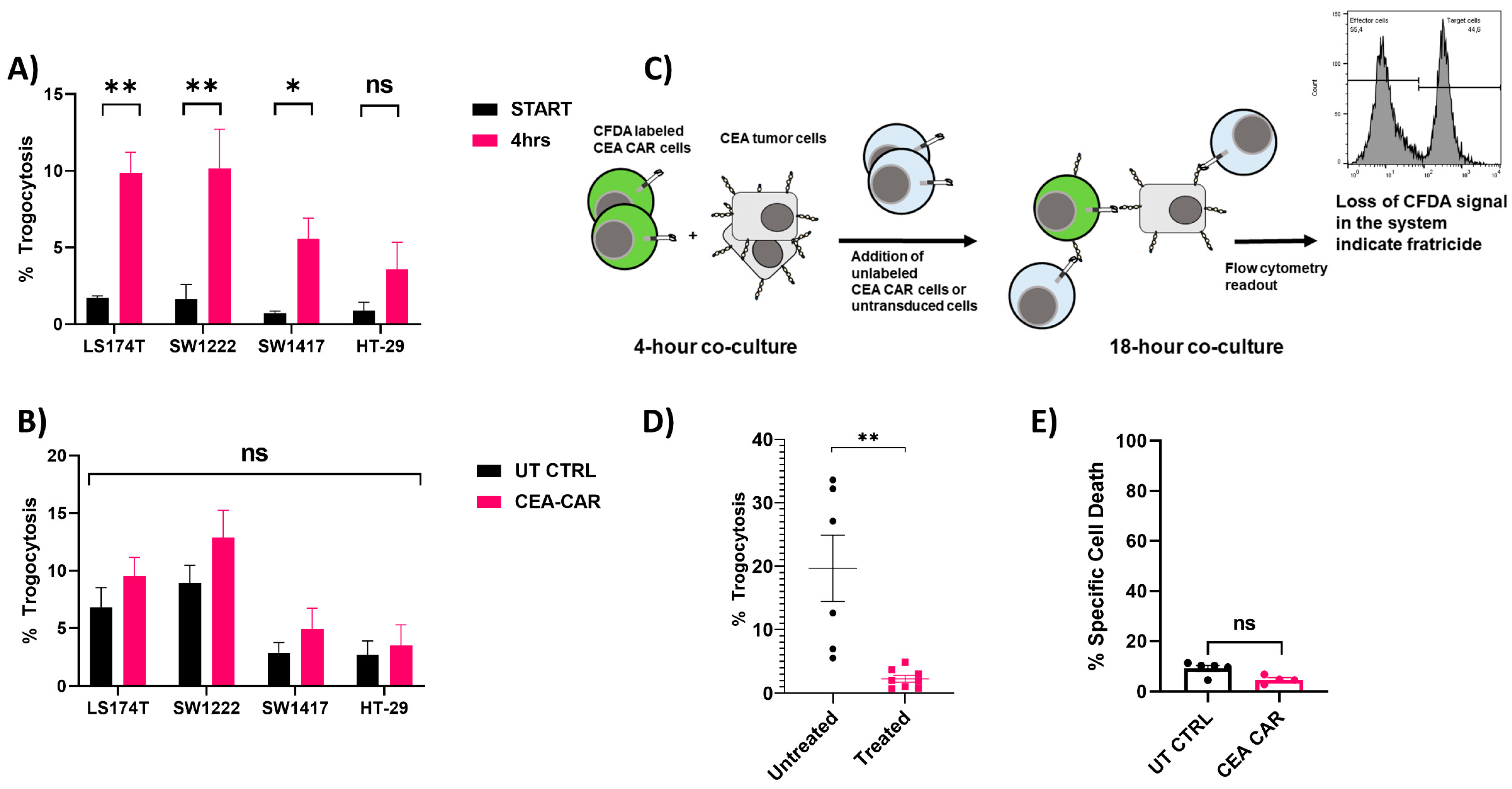
3.4. Trogocytosis and Trogocytosis-Induced Fratricide
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marofi, F.; Abdul-Rasheed, O.F.; Rahman, H.S.; Budi, H.S.; Jalil, A.T.; Yumashev, A.V.; Hassanzadeh, A.; Yazdanifar, M.; Motavalli, R.; Chartrand, M.S.; et al. CAR-NK Cell in Cancer Immunotherapy; A Promising Frontier. Cancer Sci. 2021, 112, 3427–3436. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, K.; Rouce, R.; Liu, E.; Shpall, E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol. Ther. 2017, 25, 1769–1781. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; Büning, H.; Sauer, M.; Schambach, A. Use of Cell and Genome Modification Technologies to Generate Improved “Off-the-Shelf” CAR T and CAR NK Cells. Front. Immunol. 2020, 11, 1965. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Rubnitz, J.E.; Inaba, H.; Ribeiro, R.C.; Pounds, S.; Rooney, B.; Bell, T.; Pui, C.-H.; Leung, W. NKAML: A Pilot Study to Determine the Safety and Feasibility of Haploidentical Natural Killer Cell Transplantation in Childhood Acute Myeloid Leukemia. J. Clin. Oncol. 2010, 28, 955–959. [Google Scholar] [CrossRef]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful Adoptive Transfer and in Vivo Expansion of Human Haploidentical NK Cells in Patients with Cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef]
- Woan, K.V.; Kim, H.; Bjordahl, R.; Davis, Z.B.; Gaidarova, S.; Goulding, J.; Hancock, B.; Mahmood, S.; Abujarour, R.; Wang, H.; et al. Harnessing Features of Adaptive NK Cells to Generate iPSC-Derived NK Cells for Enhanced Immunotherapy. Cell Stem Cell 2021, 28, 2062–2075.e5. [Google Scholar] [CrossRef] [PubMed]
- Jochems, C.; Hodge, J.W.; Fantini, M.; Fujii, R.; Maurice, Y.M.; Greiner, J.W.; Padget, M.R.; Tritsch, S.R.; Tsang, K.Y.; Campbell, K.S.; et al. An NK Cell Line (haNK) Expressing High Levels of Granzyme and Engineered to Express the High Affinity CD16 Allele. Oncotarget 2016, 7, 86359–86373. [Google Scholar] [CrossRef]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord Blood NK Cells Engineered to Express IL-15 and a CD19-Targeted CAR Show Long-Term Persistence and Potent Antitumor Activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef]
- Daher, M.; Basar, R.; Gokdemir, E.; Baran, N.; Uprety, N.; Nunez Cortes, A.K.; Mendt, M.; Kerbauy, L.N.; Banerjee, P.P.; Shanley, M.; et al. Targeting a Cytokine Checkpoint Enhances the Fitness of Armored Cord Blood CAR-NK Cells. Blood 2021, 137, 624–636. [Google Scholar] [CrossRef]
- Beauchemin, N.; Arabzadeh, A. Carcinoembryonic Antigen-Related Cell Adhesion Molecules (CEACAMs) in Cancer Progression and Metastasis. Cancer Metastasis Rev. 2013, 32, 643–671. [Google Scholar] [CrossRef] [PubMed]
- Albinger, N.; Pfeifer, R.; Nitsche, M.; Mertlitz, S.; Campe, J.; Stein, K.; Kreyenberg, H.; Schubert, R.; Quadflieg, M.; Schneider, D.; et al. Primary CD33-Targeting CAR-NK Cells for the Treatment of Acute Myeloid Leukemia. Blood Cancer J. 2022, 12, 61. [Google Scholar] [CrossRef]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural Killer Cells for Immunotherapy—Advantages of the NK-92 Cell Line over Blood NK Cells. Front. Immunol. 2016, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Klöß, S.; Oberschmidt, O.; Morgan, M.; Dahlke, J.; Arseniev, L.; Huppert, V.; Granzin, M.; Gardlowski, T.; Matthies, N.; Soltenborn, S.; et al. Optimization of Human NK Cell Manufacturing: Fully Automated Separation, Improved Ex Vivo Expansion Using IL-21 with Autologous Feeder Cells, and Generation of Anti-CD123-CAR-Expressing Effector Cells. Hum. Gene Ther. 2017, 28, 897–913. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Meng, Y.; Feng, X.; Han, Z. CAR-NK Cells for Cancer Immunotherapy: From Bench to Bedside. Biomark. Res. 2022, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Lamers-Kok, N.; Panella, D.; Georgoudaki, A.-M.; Liu, H.; Özkazanc, D.; Kučerová, L.; Duru, A.D.; Spanholtz, J.; Raimo, M. Natural Killer Cells in Clinical Development as Non-Engineered, Engineered, and Combination Therapies. J. Hematol. Oncol. 2022, 15, 164. [Google Scholar] [CrossRef] [PubMed]
- Fabian, K.P.; Padget, M.R.; Donahue, R.N.; Solocinski, K.; Robbins, Y.; Allen, C.T.; Lee, J.H.; Rabizadeh, S.; Soon-Shiong, P.; Schlom, J.; et al. PD-L1 Targeting High-Affinity NK (t-haNK) Cells Induce Direct Antitumor Effects and Target Suppressive MDSC Populations. J. Immunother. Cancer 2020, 8, e000450. [Google Scholar] [CrossRef] [PubMed]
- Nowakowska, P.; Romanski, A.; Miller, N.; Odendahl, M.; Bonig, H.; Zhang, C.; Seifried, E.; Wels, W.S.; Tonn, T. Clinical Grade Manufacturing of Genetically Modified, CAR-Expressing NK-92 Cells for the Treatment of ErbB2-Positive Malignancies. Cancer Immunol. Immunother. 2018, 67, 25–38. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, W.; Yang, J.; Yang, J.; Wang, W. Chimeric Antigen Receptor Engineered Natural Killer Cells for Cancer Therapy. Exp. Hematol. Oncol. 2023, 12, 70. [Google Scholar] [CrossRef]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-Man Clinical Trial of CAR NK-92 Cells: Safety Test of CD33-CAR NK-92 Cells in Patients with Relapsed and Refractory Acute Myeloid Leukemia. Am. J. Cancer Res. 2018, 8, 1083–1089. [Google Scholar]
- Weeber, F.; Ooft, S.N.; Dijkstra, K.K.; Voest, E.E. Tumor Organoids as a Pre-Clinical Cancer Model for Drug Discovery. Cell Chem. Biol. 2017, 24, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Herter, S.; Morra, L.; Schlenker, R.; Sulcova, J.; Fahrni, L.; Waldhauer, I.; Lehmann, S.; Reisländer, T.; Agarkova, I.; Kelm, J.M.; et al. A Novel Three-Dimensional Heterotypic Spheroid Model for the Assessment of the Activity of Cancer Immunotherapy Agents. Cancer Immunol. Immunother. 2017, 66, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Goudar, V.S.; Koduri, M.P.; Ta, Y.-N.N.; Chen, Y.; Chu, L.-A.; Lu, L.-S.; Tseng, F.-G. Impact of a Desmoplastic Tumor Microenvironment for Colon Cancer Drug Sensitivity: A Study with 3D Chimeric Tumor Spheroids. ACS Appl. Mater. Interfaces 2021, 13, 48478–48491. [Google Scholar] [CrossRef]
- Ho, W.Y.; Yeap, S.K.; Ho, C.L.; Rahim, R.A.; Alitheen, N.B. Development of Multicellular Tumor Spheroid (MCTS) Culture from Breast Cancer Cell and a High Throughput Screening Method Using the MTT Assay. PLoS ONE 2012, 7, e44640. [Google Scholar] [CrossRef] [PubMed]
- Ong, L.J.Y.; Chia, S.; Wong, S.Q.R.; Zhang, X.; Chua, H.; Loo, J.M.; Chua, W.Y.; Chua, C.; Tan, E.; Hentze, H.; et al. A Comparative Study of Tumour-on-Chip Models with Patient-Derived Xenografts for Predicting Chemotherapy Efficacy in Colorectal Cancer Patients. Front. Bioeng. Biotechnol. 2022, 10, 952726. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J. Applications of Patient-Derived Tumor Xenograft Models and Tumor Organoids. J. Hematol. Oncol. 2020, 13, 4. [Google Scholar] [CrossRef]
- Nath, S.; Devi, G.R. Three-Dimensional Culture Systems in Cancer Research: Focus on Tumor Spheroid Model. Pharmacol. Ther. 2016, 163, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Bauleth-Ramos, T.; Feijão, T.; Gonçalves, A.; Shahbazi, M.-A.; Liu, Z.; Barrias, C.; Oliveira, M.J.; Granja, P.; Santos, H.A.; Sarmento, B. Colorectal Cancer Triple Co-Culture Spheroid Model to Assess the Biocompatibility and Anticancer Properties of Polymeric Nanoparticles. J. Control. Release 2020, 323, 398–411. [Google Scholar] [CrossRef]
- Zanoni, M.; Piccinini, F.; Arienti, C.; Zamagni, A.; Santi, S.; Polico, R.; Bevilacqua, A.; Tesei, A. 3D Tumor Spheroid Models for in Vitro Therapeutic Screening: A Systematic Approach to Enhance the Biological Relevance of Data Obtained. Sci. Rep. 2016, 6, 19103. [Google Scholar] [CrossRef]
- Däster, S.; Amatruda, N.; Calabrese, D.; Ivanek, R.; Turrini, E.; Droeser, R.A.; Zajac, P.; Fimognari, C.; Spagnoli, G.C.; Iezzi, G.; et al. Induction of Hypoxia and Necrosis in Multicellular Tumor Spheroids Is Associated with Resistance to Chemotherapy Treatment. Oncotarget 2016, 8, 1725–1736. [Google Scholar] [CrossRef]
- Vinci, M.; Gowan, S.; Boxall, F.; Patterson, L.; Zimmermann, M.; Court, W.; Lomas, C.; Mendiola, M.; Hardisson, D.; Eccles, S.A. Advances in Establishment and Analysis of Three-Dimensional Tumor Spheroid-Based Functional Assays for Target Validation and Drug Evaluation. BMC Biol. 2012, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Lin, C.; Cheng, J.; Su, J.; Zhao, H.; Liu, T.; Wen, X.; Zhao, P. Generation of Multicellular Tumor Spheroids with Microwell-Based Agarose Scaffolds for Drug Testing. PLoS ONE 2015, 10, e0130348. [Google Scholar] [CrossRef] [PubMed]
- Weydert, Z.; Lal-Nag, M.; Mathews-Greiner, L.; Thiel, C.; Cordes, H.; Küpfer, L.; Guye, P.; Kelm, J.M.; Ferrer, M. A 3D Heterotypic Multicellular Tumor Spheroid Assay Platform to Discriminate Drug Effects on Stroma versus Cancer Cells. SLAS Discov. Adv. Sci. Drug Discov. 2020, 25, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Gonzalez, A.; Huang, Y.-W.; Porpiglia, E.; Donoso, K.; Gonzalez, V.D.; Fantl, W.J. Measuring Trogocytosis between Ovarian Tumor and Natural Killer Cells. STAR Protoc. 2022, 3, 101425. [Google Scholar] [CrossRef] [PubMed]
- Schirrmann, T.; Pecher, G. Human Natural Killer Cell Line Modified with a Chimeric Immunoglobulin T-Cell Receptor Gene Leads to Tumor Growth Inhibition In Vivo. Cancer Gene Ther. 2002, 9, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.; Koch, D.; Sircar, R.; Heuser, C.; Diehl, V.; Kruis, W.; Pohl, C.; Abken, H. A Chimeric Receptor That Selectively Targets Membrane-Bound Carcinoembryonic Antigen (mCEA) in the Presence of Soluble CEA. Gene Ther. 1999, 6, 300–304. [Google Scholar] [CrossRef]
- Bosslet, K.; Steinsträsser, A.; Schwarz, A.; Harthus, H.P.; Lüben, G.; Kuhlmann, L.; Sedlacek, H.H. Quantitative Considerations Supporting the Irrelevance of Circulating Serum CEA for the Immunoscintigraphic Visualization of CEA Expressing Carcinomas. Eur. J. Nucl. Med. 1988, 14, 523–528. [Google Scholar] [CrossRef]
- Kohli, K.; Pillarisetty, V.G.; Kim, T.S. Key Chemokines Direct Migration of Immune Cells in Solid Tumors. Cancer Gene Ther. 2022, 29, 10–21. [Google Scholar] [CrossRef]
- Raftery, M.J.; Franzén, A.S.; Radecke, C.; Boulifa, A.; Schönrich, G.; Stintzing, S.; Blohmer, J.-U.; Pecher, G. Next Generation CD44v6-Specific CAR-NK Cells Effective against Triple Negative Breast Cancer. Int. J. Mol. Sci. 2023, 24, 9038. [Google Scholar] [CrossRef]
- Hsu, J.; Hodgins, J.J.; Marathe, M.; Nicolai, C.J.; Bourgeois-Daigneault, M.-C.; Trevino, T.N.; Azimi, C.S.; Scheer, A.K.; Randolph, H.E.; Thompson, T.W.; et al. Contribution of NK Cells to Immunotherapy Mediated by PD-1/PD-L1 Blockade. J. Clin. Investig. 2018, 128, 4654–4668. [Google Scholar] [CrossRef]
- Klingemann, H. The NK-92 Cell Line—30 Years Later: Its Impact on Natural Killer Cell Research and Treatment of Cancer. Cytotherapy 2023, 25, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, O.; Wall, J.B.J.; Zheng, M.; Zhou, Y.; Wang, L.; Ruth Vaseghi, H.; Qian, L.; Liu, J. Systematic Comparison of 2A Peptides for Cloning Multi-Genes in a Polycistronic Vector. Sci. Rep. 2017, 7, 2193. [Google Scholar] [CrossRef] [PubMed]
- Krokhotin, A.; Du, H.; Hirabayashi, K.; Popov, K.; Kurokawa, T.; Wan, X.; Ferrone, S.; Dotti, G.; Dokholyan, N.V. Computationally Guided Design of Single-Chain Variable Fragment Improves Specificity of Chimeric Antigen Receptors. Mol. Ther.-Oncolytics 2019, 15, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.A.; Schildgen, V.; Heuser, C.; Finnern, R.; Gilham, D.E.; Abken, H. T Cell Activation by Antibody-Like Immunoreceptors: The Position of the Binding Epitope within the Target Molecule Determines the Efficiency of Activation of Redirected T Cells1. J. Immunol. 2007, 178, 4650–4657. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wei, Q.; Brzostek, J.; Gascoigne, N.R.J. Signaling from T Cell Receptors (TCRs) and Chimeric Antigen Receptors (CARs) on T Cells. Cell Mol. Immunol. 2020, 17, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Feigl, F.F.; Stahringer, A.; Peindl, M.; Dandekar, G.; Koehl, U.; Fricke, S.; Schmiedel, D. Efficient Redirection of NK Cells by Genetic Modification with Chemokine Receptors CCR4 and CCR2B. Int. J. Mol. Sci. 2023, 24, 3129. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The Biology and Function of Fibroblasts in Cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Yakavets, I.; Francois, A.; Benoit, A.; Merlin, J.-L.; Bezdetnaya, L.; Vogin, G. Advanced Co-Culture 3D Breast Cancer Model for Investigation of Fibrosis Induced by External Stimuli: Optimization Study. Sci. Rep. 2020, 10, 21273. [Google Scholar] [CrossRef]
- Imamura, Y.; Mukohara, T.; Shimono, Y.; Funakoshi, Y.; Chayahara, N.; Toyoda, M.; Kiyota, N.; Takao, S.; Kono, S.; Nakatsura, T.; et al. Comparison of 2D- and 3D-Culture Models as Drug-Testing Platforms in Breast Cancer. Oncol. Rep. 2015, 33, 1837–1843. [Google Scholar] [CrossRef]
- Ayuso, J.M.; Truttschel, R.; Gong, M.M.; Humayun, M.; Virumbrales-Munoz, M.; Vitek, R.; Felder, M.; Gillies, S.D.; Sondel, P.; Wisinski, K.B.; et al. Evaluating Natural Killer Cell Cytotoxicity against Solid Tumors Using a Microfluidic Model. OncoImmunology 2019, 8, 1553477. [Google Scholar] [CrossRef]
- Keller, F.; Rudolf, R.; Hafner, M. Towards Optimized Breast Cancer 3D Spheroid Mono- and Co-Culture Models for Pharmacological Research and Screening. J. Cell. Biotechnol. 2019, 5, 89–101. [Google Scholar] [CrossRef]
- Li, Y.; Basar, R.; Wang, G.; Liu, E.; Moyes, J.S.; Li, L.; Kerbauy, L.N.; Uprety, N.; Fathi, M.; Rezvan, A.; et al. KIR-Based Inhibitory CARs Overcome CAR-NK Cell Trogocytosis-Mediated Fratricide and Tumor Escape. Nat. Med. 2022, 28, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Schoutrop, E.; Renken, S.; Micallef Nilsson, I.; Hahn, P.; Poiret, T.; Kiessling, R.; Wickström, S.L.; Mattsson, J.; Magalhaes, I. Trogocytosis and Fratricide Killing Impede MSLN-Directed CAR T Cell Functionality. Oncoimmunology 2022, 11, 2093426. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Kuroki, M.; Arakawa, F.; Haruno, M.; Kuwahara, M.; Ozaki, H.; Senba, T.; Matsuoka, Y. Binding Reactivity of Monoclonal Anti-Carcinoembryonic Antigen (CEA) Antibodies with Cell Membrane-Bound CEA and with Free CEA in Solution. Immunol. Investig. 1996, 25, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Peterson, C.Y.; Sriram, D.; Mahipal, A. Early Stage Colon Cancer: Current Treatment Standards, Evolving Paradigms, and Future Directions. World J. Gastrointest. Oncol. 2020, 12, 808–832. [Google Scholar] [CrossRef]
- Xi, Y.; Xu, P. Global Colorectal Cancer Burden in 2020 and Projections to 2040. Transl. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef]
- Hall, C.; Clarke, L.; Pal, A.; Buchwald, P.; Eglinton, T.; Wakeman, C.; Frizelle, F. A Review of the Role of Carcinoembryonic Antigen in Clinical Practice. Ann. Coloproctol. 2019, 35, 294–305. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Z.; Yang, Z.; Wang, M.; Li, S.; Li, Y.; Zhang, R.; Xiong, Z.; Wei, Z.; Shen, J.; et al. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA+ Metastatic Colorectal Cancers. Mol. Ther. 2017, 25, 1248–1258. [Google Scholar] [CrossRef]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J.-Y. Comprehensive Review of Targeted Therapy for Colorectal Cancer. Sig. Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Ou, B.; Zhao, J.; Guan, S.; Feng, H.; Wangpu, X.; Zhu, C.; Zong, Y.; Ma, J.; Sun, J.; Shen, X.; et al. CCR4 Promotes Metastasis via ERK/NF-κB/MMP13 Pathway and Acts Downstream of TNF-α in Colorectal Cancer. Oncotarget 2016, 7, 47637–47649. [Google Scholar] [CrossRef]
- Priwitaningrum, D.L.; Blondé, J.-B.G.; Sridhar, A.; van Baarlen, J.; Hennink, W.E.; Storm, G.; Le Gac, S.; Prakash, J. Tumor Stroma-Containing 3D Spheroid Arrays: A Tool to Study Nanoparticle Penetration. J. Control. Release 2016, 244, 257–268. [Google Scholar] [CrossRef]
- Chiavarina, B.; Costanza, B.; Ronca, R.; Blomme, A.; Rezzola, S.; Chiodelli, P.; Giguelay, A.; Belthier, G.; Doumont, G.; Van Simaeys, G.; et al. Metastatic Colorectal Cancer Cells Maintain the TGFβ Program and Use TGFBI to Fuel Angiogenesis. Theranostics 2021, 11, 1626–1640. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- Roelands, J.; Kuppen, P.J.K.; Vermeulen, L.; Maccalli, C.; Decock, J.; Wang, E.; Marincola, F.M.; Bedognetti, D.; Hendrickx, W. Immunogenomic Classification of Colorectal Cancer and Therapeutic Implications. Int. J. Mol. Sci. 2017, 18, 2229. [Google Scholar] [CrossRef]
- Bazzi, Z.A.; Sneddon, S.; Zhang, P.G.Y.; Tai, I.T. Characterization of the Immune Cell Landscape in CRC: Clinical Implications of Tumour-Infiltrating Leukocytes in Early- and Late-Stage CRC. Front. Immunol. 2023, 13, 978862. [Google Scholar] [CrossRef]
- Ko, Y.S.; Pyo, J.-S. Clinicopathological Significance and Prognostic Role of Tumor-Infiltrating Lymphocytes in Colorectal Cancer. Int. J. Biol. Markers 2019, 34, 132–138. [Google Scholar] [CrossRef]
- Idos, G.E.; Kwok, J.; Bonthala, N.; Kysh, L.; Gruber, S.B.; Qu, C. The Prognostic Implications of Tumor Infiltrating Lymphocytes in Colorectal Cancer: A Systematic Review and Meta-Analysis. Sci. Rep. 2020, 10, 3360. [Google Scholar] [CrossRef]
- Becht, E.; de Reyniès, A.; Giraldo, N.A.; Pilati, C.; Buttard, B.; Lacroix, L.; Selves, J.; Sautès-Fridman, C.; Laurent-Puig, P.; Fridman, W.H. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy. Clin. Cancer Res. 2016, 22, 4057–4066. [Google Scholar] [CrossRef]
- Giraldo, N.A.; Becht, E.; Pagès, F.; Skliris, G.; Verkarre, V.; Vano, Y.; Mejean, A.; Saint-Aubert, N.; Lacroix, L.; Natario, I.; et al. Orchestration and Prognostic Significance of Immune Checkpoints in the Microenvironment of Primary and Metastatic Renal Cell Cancer. Clin. Cancer Res. 2015, 21, 3031–3040. [Google Scholar] [CrossRef]
- Daubeuf, S.; Lindorfer, M.A.; Taylor, R.P.; Joly, E.; Hudrisier, D. The Direction of Plasma Membrane Exchange between Lymphocytes and Accessory Cells by Trogocytosis Is Influenced by the Nature of the Accessory Cell. J. Immunol. 2010, 184, 1897–1908. [Google Scholar] [CrossRef]
- Gonzalez, V.D.; Huang, Y.-W.; Delgado-Gonzalez, A.; Chen, S.-Y.; Donoso, K.; Sachs, K.; Gentles, A.J.; Allard, G.M.; Kolahi, K.S.; Howitt, B.E.; et al. High-Grade Serous Ovarian Tumor Cells Modulate NK Cell Function to Create an Immune-Tolerant Microenvironment. Cell Rep. 2021, 36, 109632. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franzén, A.S.; Boulifa, A.; Radecke, C.; Stintzing, S.; Raftery, M.J.; Pecher, G. Next-Generation CEA-CAR-NK-92 Cells against Solid Tumors: Overcoming Tumor Microenvironment Challenges in Colorectal Cancer. Cancers 2024, 16, 388. https://doi.org/10.3390/cancers16020388
Franzén AS, Boulifa A, Radecke C, Stintzing S, Raftery MJ, Pecher G. Next-Generation CEA-CAR-NK-92 Cells against Solid Tumors: Overcoming Tumor Microenvironment Challenges in Colorectal Cancer. Cancers. 2024; 16(2):388. https://doi.org/10.3390/cancers16020388
Chicago/Turabian StyleFranzén, Alexander Sebastian, Abdelhadi Boulifa, Clarissa Radecke, Sebastian Stintzing, Martin J. Raftery, and Gabriele Pecher. 2024. "Next-Generation CEA-CAR-NK-92 Cells against Solid Tumors: Overcoming Tumor Microenvironment Challenges in Colorectal Cancer" Cancers 16, no. 2: 388. https://doi.org/10.3390/cancers16020388