
Targeting PARP-1 and DNA Damage Response Defects in Colorectal Cancer Chemotherapy with Established and Novel PARP Inhibitors
 , ,
, ,  , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Test Compounds
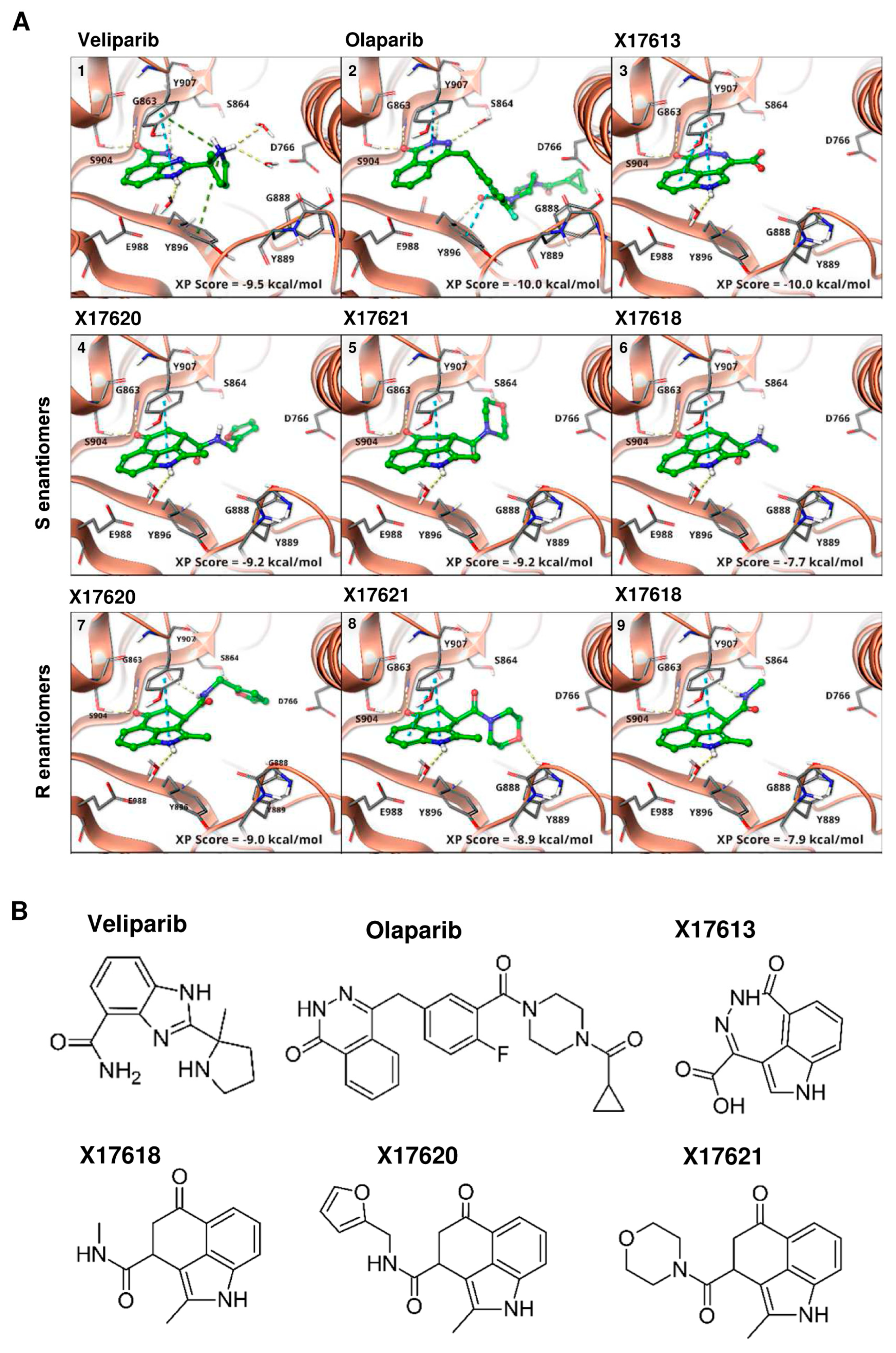
2.2. Molecular Docking
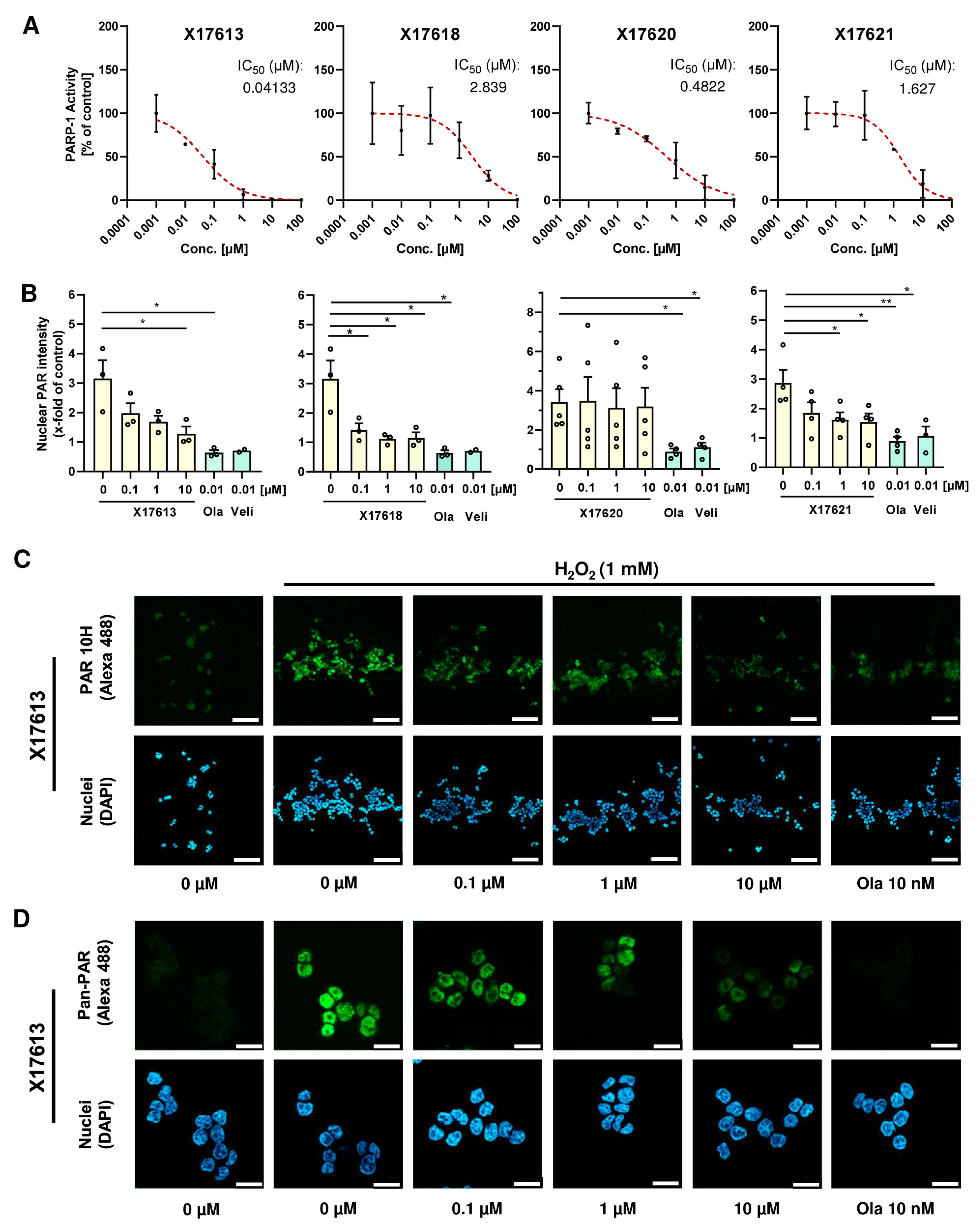
2.3. PARP Inhibitor Assay
2.4. Cell Culture and Treatments
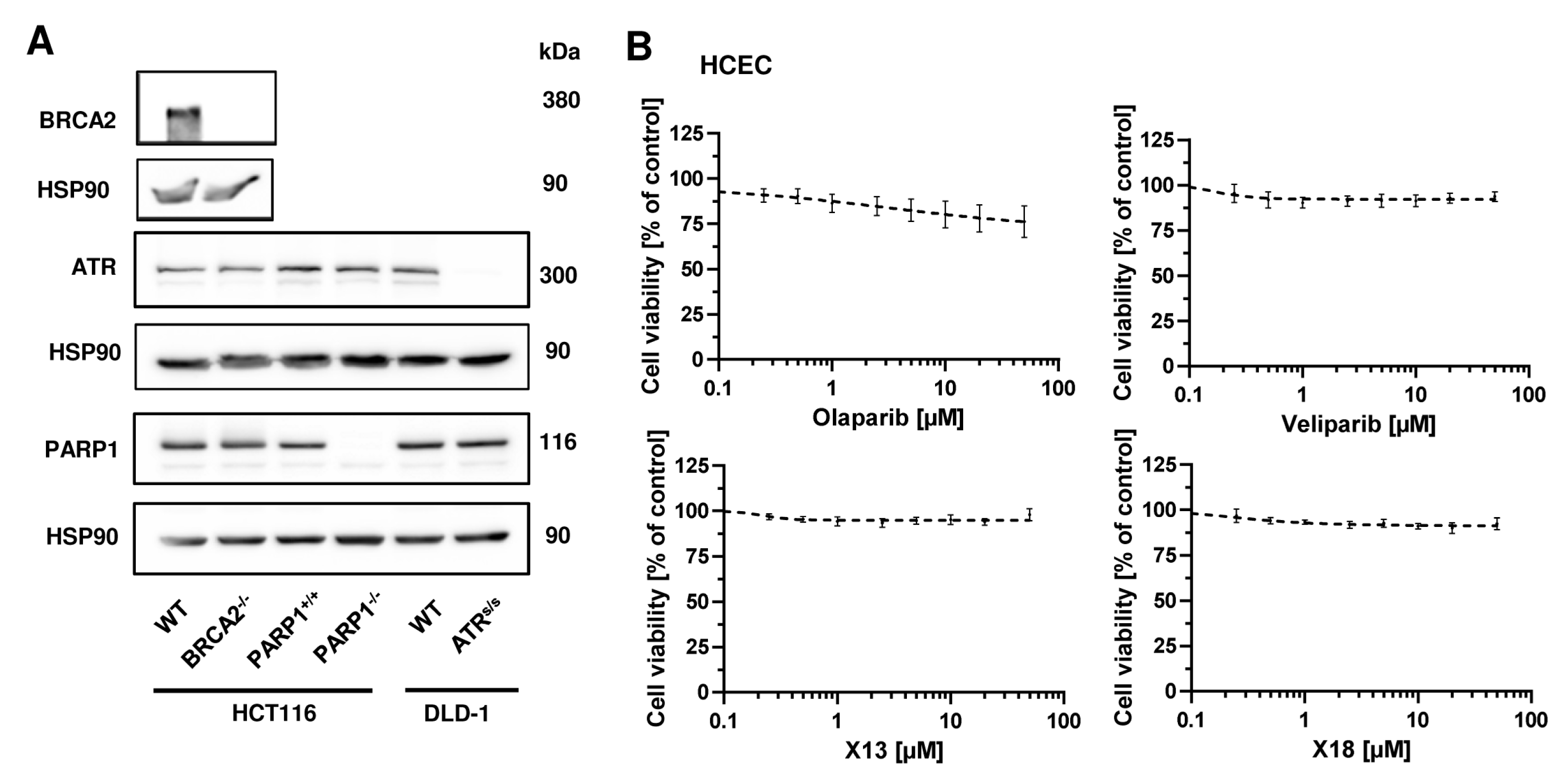
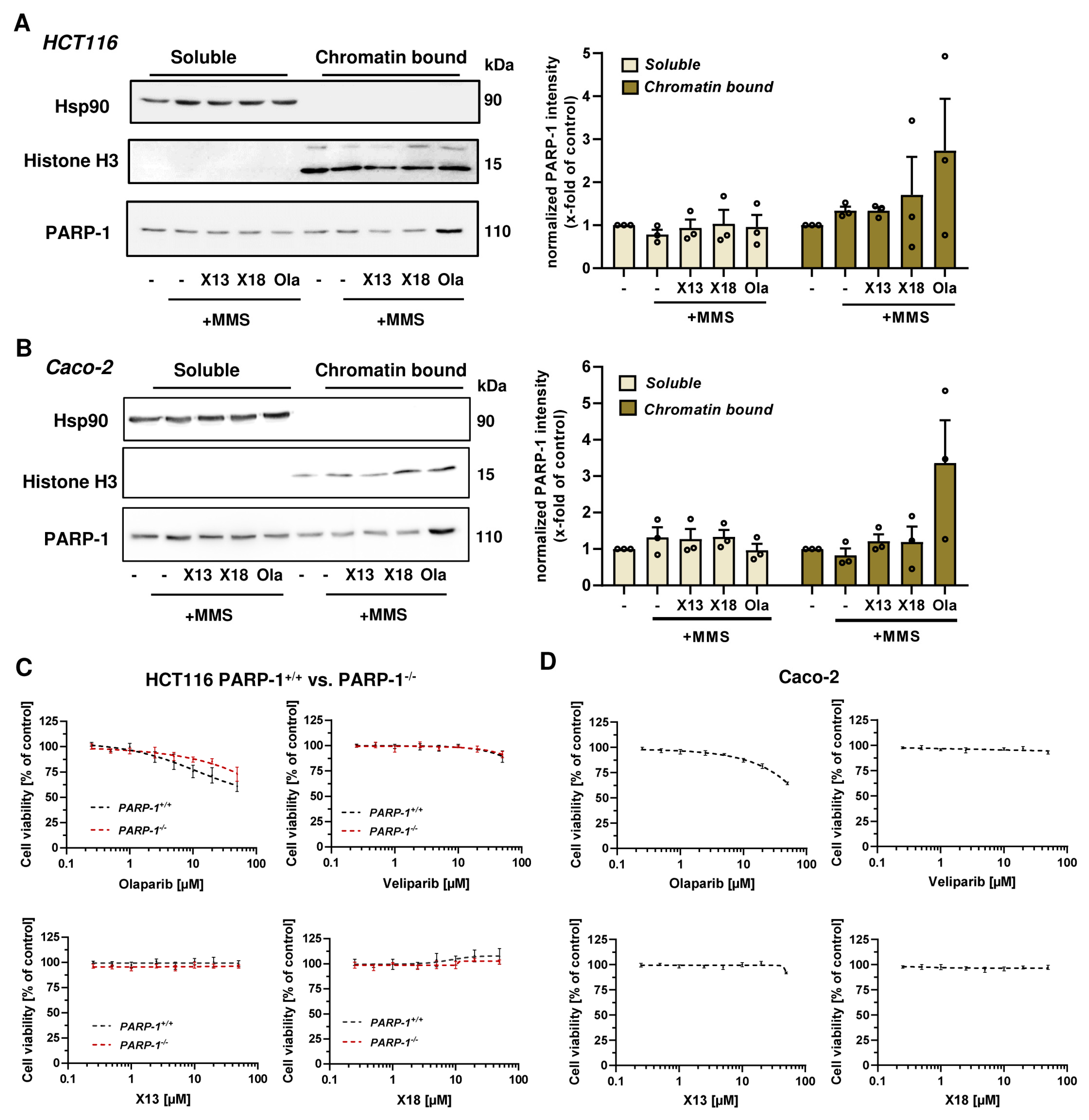
2.5. Cytotoxicity Testing of PARP Inhibitors
2.6. PAR Immunofluorescence Analysis
2.7. Chromatin Retention Assay and Western Blot Analysis
2.8. Statistics
3. Results
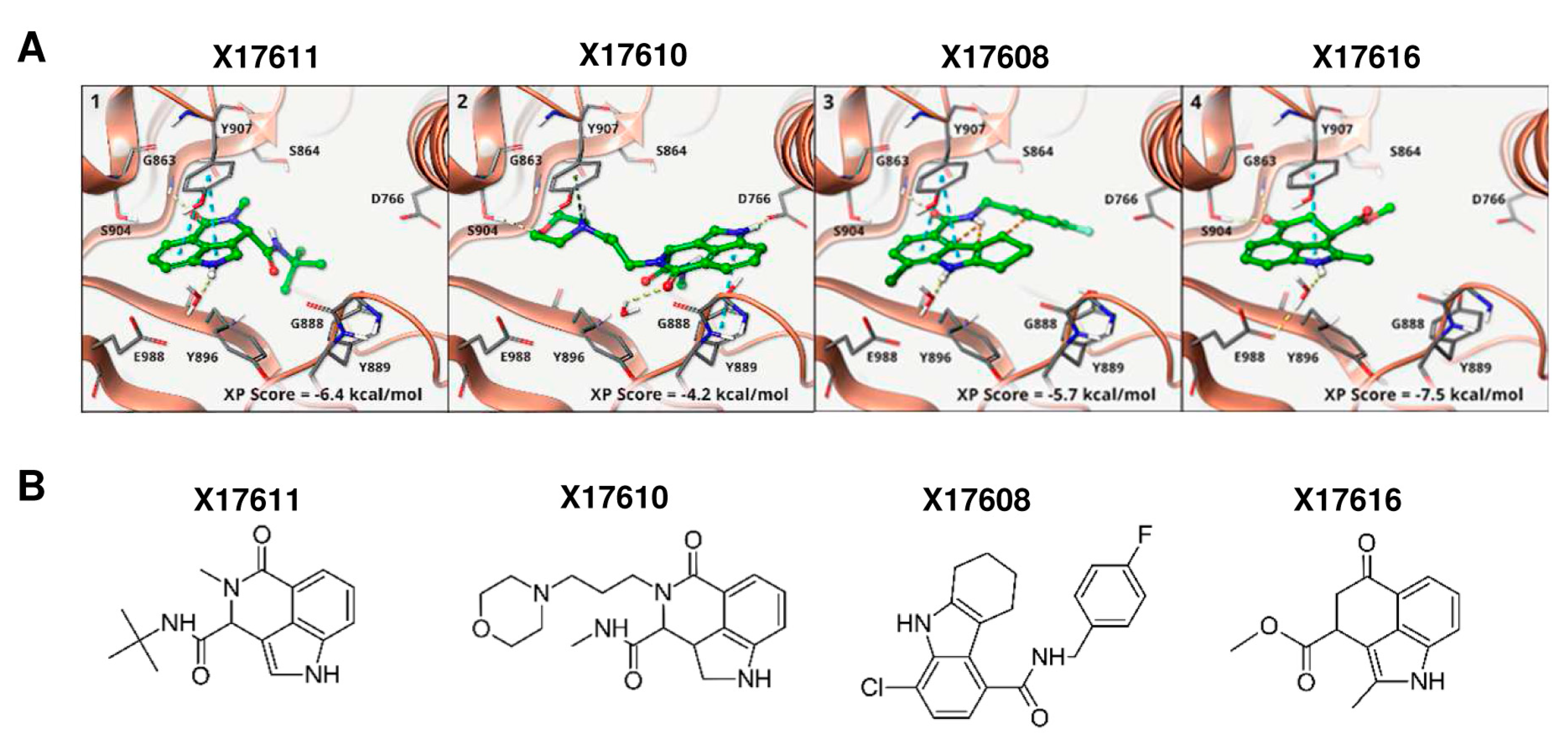
3.1. Identification of Putative PARPi Using Molecular Docking Studies
3.2. Activity Screening of PARP Inhibitors
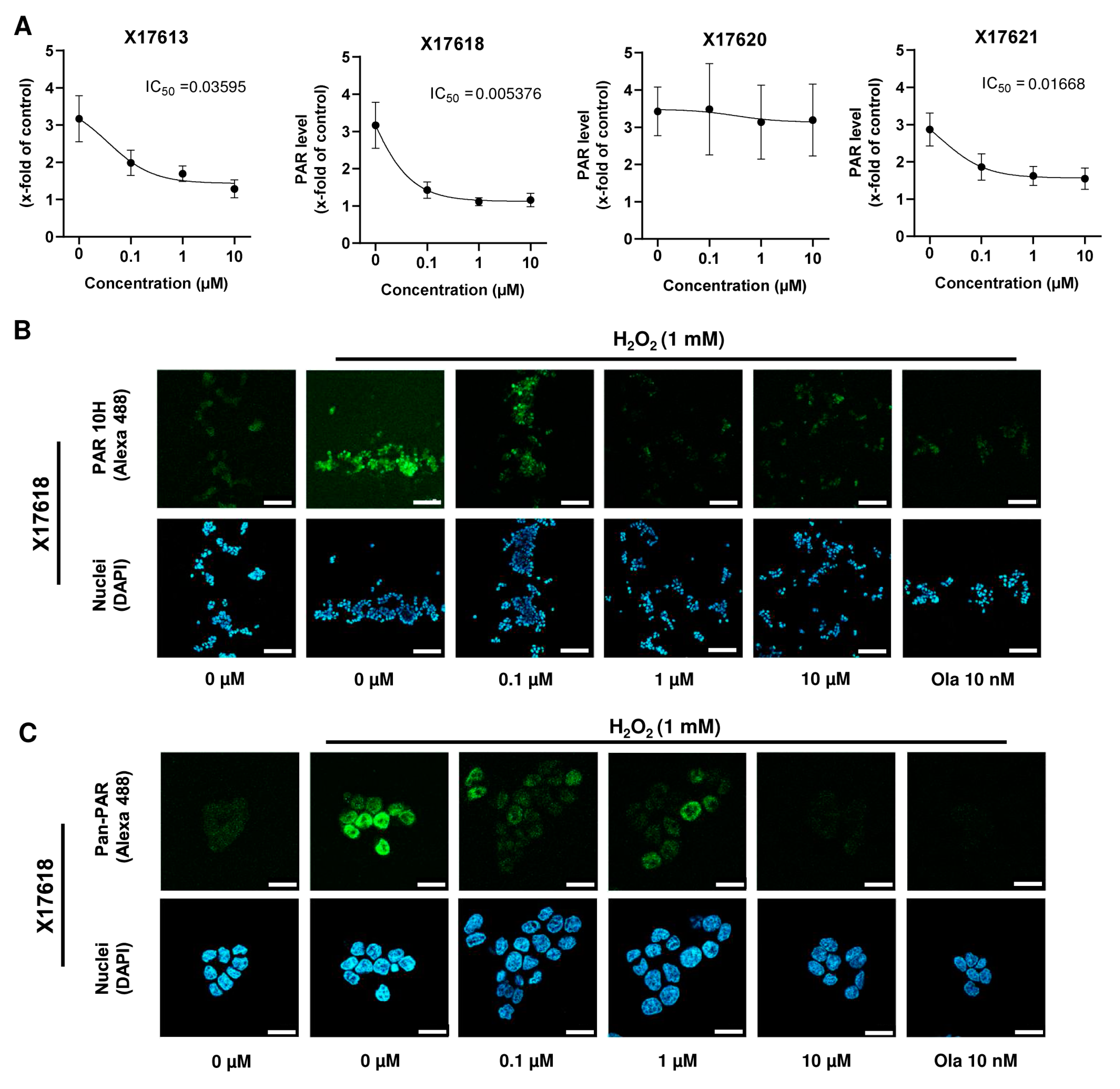
3.3. Effect of Selected Test Compounds on PAR Formation in HCT116 Cells
3.4. PARP-1 Trapping and Cytotoxicity of PARP Inhibitors
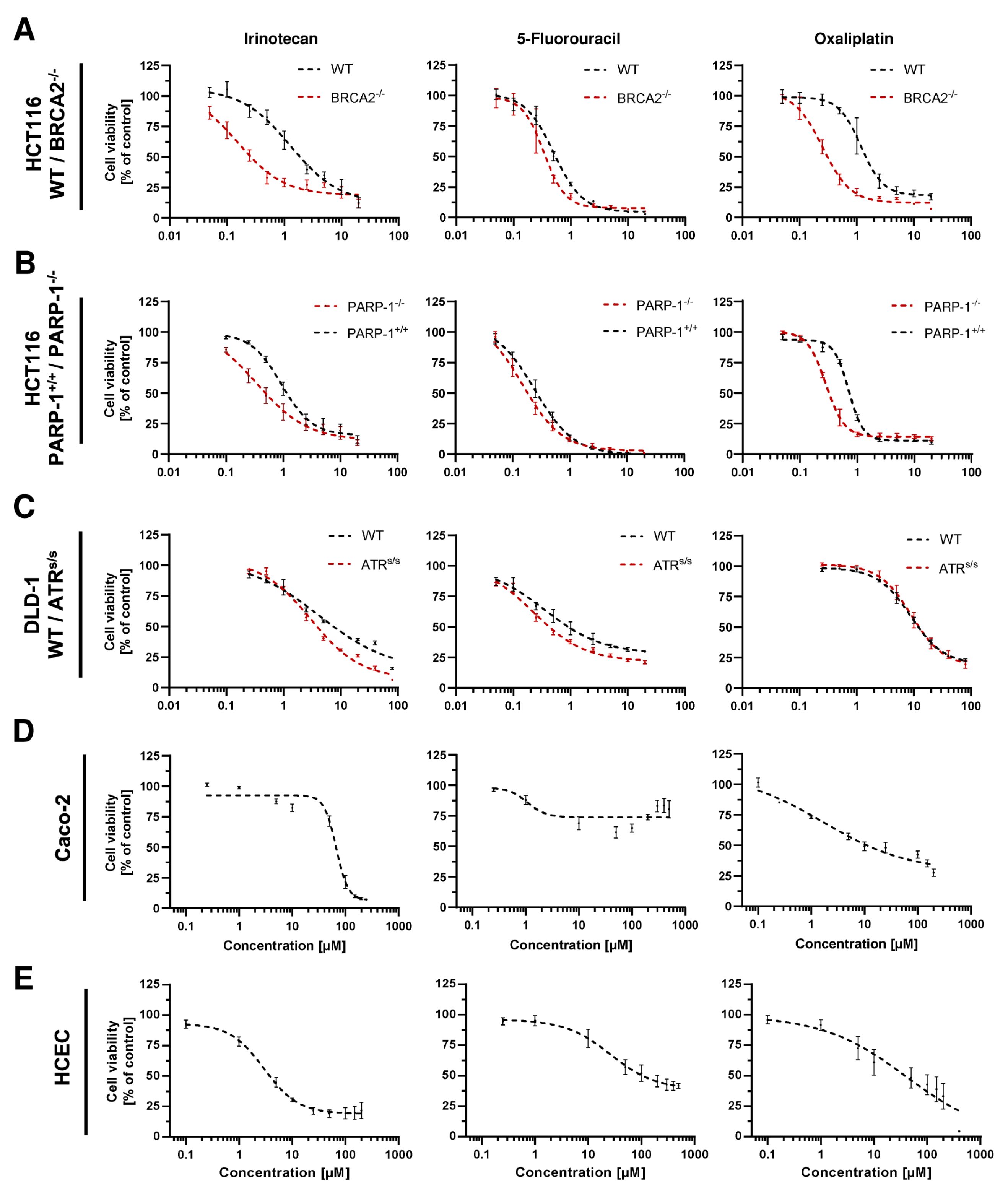
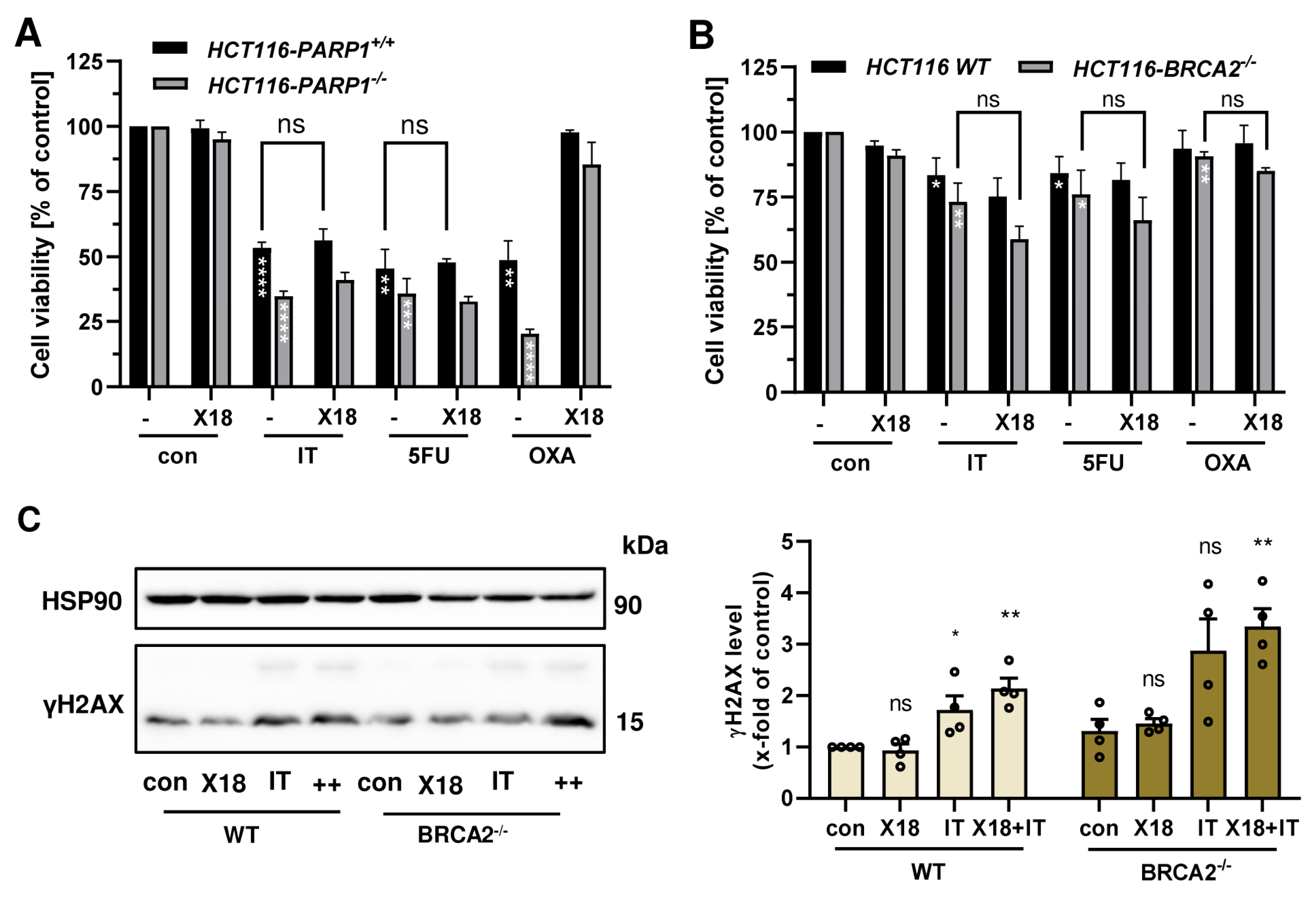
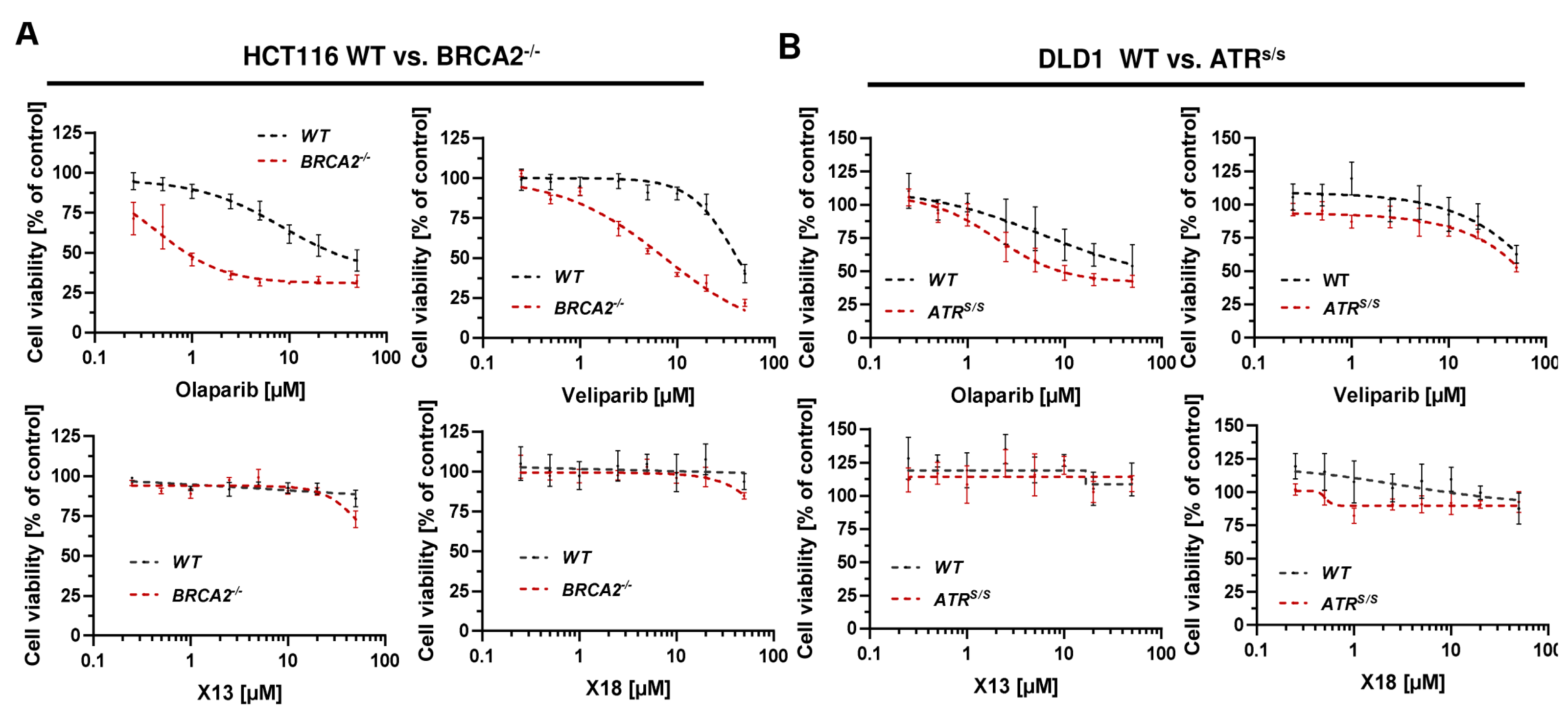
3.5. Cytotoxicity of PARPi Depending on the Cellular BRCA2 and ATR Status
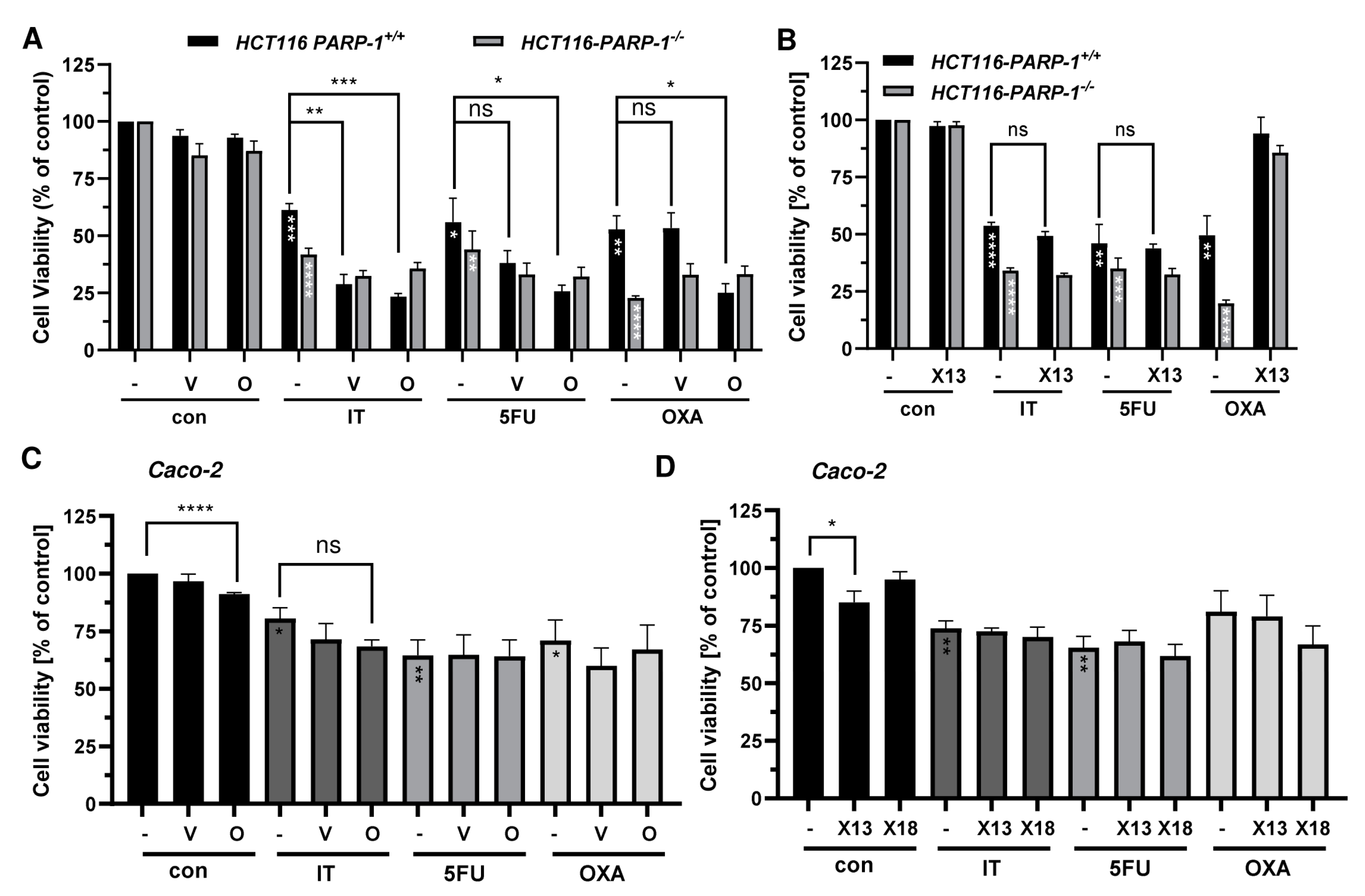
3.6. Combination of PARPi and Clinically Relevant Chemotherapeutic Drugs in CRC Cells
3.7. Impact of BRCA2 and ATR on the Potential Synergism of PARPi and Chemotherapeutics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


References
- Favoriti, P.; Carbone, G.; Greco, M.; Pirozzi, F.; Pirozzi, R.E.M.; Corcione, F. Worldwide burden of colorectal cancer: A review. Updates Surg. 2016, 68, 7–11. [Google Scholar] [CrossRef]
- Spaander, M.C.W.; Zauber, A.G.; Syngal, S.; Blaser, M.J.; Sung, J.J.; You, Y.N.; Kuipers, E.J. Young-onset colorectal cancer. Nat. Rev. Dis. Prim. 2023, 9, 21. [Google Scholar] [CrossRef]
- Biller, L.H.; Schrag, D. Diagnosis and Treatment of Metastatic Colorectal Cancer: A Review. JAMA 2021, 325, 669–685. [Google Scholar] [CrossRef]
- Hammond, W.A.; Swaika, A.; Mody, K. Pharmacologic resistance in colorectal cancer: A review. Ther. Adv. Med. Oncol. 2016, 8, 57–84. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef]
- Fahrer, J.; Kaina, B. O6-methylguanine-DNA methyltransferase in the defense against N-nitroso compounds and colorectal cancer. Carcinogenesis 2013, 34, 2435–2442. [Google Scholar] [CrossRef]
- Degrolard-Courcet, E.; Sokolowska, J.; Padeano, M.-M.; Guiu, S.; Bronner, M.; Chery, C.; Coron, F.; Lepage, C.; Chapusot, C.; Loustalot, C.; et al. Development of primary early-onset colorectal cancers due to biallelic mutations of the FANCD1/BRCA2 gene. Eur. J. Hum. Genet. 2014, 22, 979–987. [Google Scholar] [CrossRef]
- Phelan, C.M.; Iqbal, J.; Lynch, H.T.; Lubinski, J.; Gronwald, J.; Moller, P.; Ghadirian, P.; Foulkes, W.D.; Armel, S.; Eisen, A.; et al. Incidence of colorectal cancer in BRCA1 and BRCA2 mutation carriers: Results from a follow-up study. Br. J. Cancer 2014, 110, 530–534. [Google Scholar] [CrossRef]
- Moretto, R.; Elliott, A.; Zhang, J.; Arai, H.; Germani, M.M.; Conca, V.; Xiu, J.; Stafford, P.; Oberley, M.; Abraham, J.; et al. Homologous Recombination Deficiency Alterations in Colorectal Cancer: Clinical, Molecular, and Prognostic Implications. J. Natl. Cancer Inst. 2022, 114, 271–279. [Google Scholar] [CrossRef]
- Gustavsson, B.; Carlsson, G.; Machover, D.; Petrelli, N.; Roth, A.; Schmoll, H.-J.; Tveit, K.-M.; Gibson, F. A Review of the Evolution of Systemic Chemotherapy in the Management of Colorectal Cancer. Clin. Color. Cancer 2015, 14, 1–10. [Google Scholar] [CrossRef]
- Mangerich, A.; Burkle, A. How to kill tumor cells with inhibitors of poly(ADP-ribosyl)ation. Int. J. Cancer 2011, 128, 251–265. [Google Scholar] [CrossRef]
- Martin-Hernandez, K.; Rodriguez-Vargas, J.M.; Schreiber, V.; Dantzer, F. Expanding functions of ADP-ribosylation in the maintenance of genome integrity. Semin. Cell Dev. Biol. 2017, 63, 92–101. [Google Scholar] [CrossRef]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Dörsam, B.; Seiwert, N.; Foersch, S.; Stroh, S.; Nagel, G.; Begaliew, D.; Diehl, E.; Kraus, A.; McKeague, M.; Minneker, V.; et al. PARP-1 protects against colorectal tumor induction, but promotes inflammation-driven colorectal tumor progression. Proc. Natl. Acad. Sci. USA 2018, 115, E4061–E4070. [Google Scholar] [CrossRef]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; de Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef]
- Cortesi, L.; Rugo, H.S.; Jackisch, C. An Overview of PARP Inhibitors for the Treatment of Breast Cancer. Target. Oncol. 2021, 16, 255–282. [Google Scholar] [CrossRef]
- Paulet, L.; Trecourt, A.; Leary, A.; Peron, J.; Descotes, F.; Devouassoux-Shisheboran, M.; Leroy, K.; You, B.; Lopez, J. Cracking the homologous recombination deficiency code: How to identify responders to PARP inhibitors. Eur. J. Cancer 2022, 166, 87–99. [Google Scholar] [CrossRef]
- Reilly, N.M.; Novara, L.; Di Nicolantonio, F.; Bardelli, A. Exploiting DNA repair defects in colorectal cancer. Mol. Oncol. 2019, 13, 681–700. [Google Scholar] [CrossRef]
- Pilié, P.G.; Gay, C.M.; Byers, L.A.; O’Connor, M.J.; Yap, T.A. PARP Inhibitors: Extending Benefit Beyond BRCA-Mutant Cancers. Clin. Cancer Res. 2019, 25, 3759–3771. [Google Scholar] [CrossRef]
- Giannini, G.; Ristori, E.; Cerignoli, F.; Rinaldi, C.; Zani, M.; Viel, A.; Ottini, L.; Crescenzi, M.; Martinotti, S.; Bignami, M.; et al. Human MRE11 is inactivated in mismatch repair-deficient cancers. EMBO Rep. 2002, 3, 248–254. [Google Scholar] [CrossRef]
- Vilar, E.; Bartnik, C.M.; Stenzel, S.L.; Raskin, L.; Ahn, J.; Moreno, V.; Mukherjee, B.; Iniesta, M.D.; Morgan, M.A.; Rennert, G.; et al. MRE11 Deficiency Increases Sensitivity to Poly(ADP-ribose) Polymerase Inhibition in Microsatellite Unstable Colorectal Cancers. Cancer Res. 2011, 71, 2632–2642. [Google Scholar] [CrossRef]
- Leichman, L.; Groshen, S.; O’Neil, B.H.; Messersmith, W.; Berlin, J.; Chan, E.; Leichman, C.G.; Cohen, S.J.; Cohen, D.; Lenz, H.-J.; et al. Phase II Study of Olaparib (AZD-2281) after Standard Systemic Therapies for Disseminated Colorectal Cancer. Oncologist 2016, 21, 172–177. [Google Scholar] [CrossRef]
- Wang, C.; Jette, N.; Moussienko, D.; Bebb, D.G.; Lees-Miller, S.P. ATM-Deficient Colorectal Cancer Cells Are Sensitive to the PARP Inhibitor Olaparib. Transl. Oncol. 2017, 10, 190–196. [Google Scholar] [CrossRef]
- Curtin, N.J.; Szabo, C. Poly(ADP-ribose) polymerase inhibition: Past, present and future. Nat. Rev. Drug Discov. 2020, 19, 711–736. [Google Scholar] [CrossRef]
- Arena, S.; Corti, G.; Durinikova, E.; Montone, M.; Reilly, N.M.; Russo, M.; Lorenzato, A.; Arcella, P.; Lazzari, L.; Rospo, G.; et al. A Subset of Colorectal Cancers with Cross-Sensitivity to Olaparib and Oxaliplatin. Clin. Cancer Res. 2020, 26, 1372–1384. [Google Scholar] [CrossRef]
- Keggenhoff, F.L.; Castven, D.; Becker, D.; Stojkovic, S.; Castven, J.; Zimpel, C.; Straub, B.K.; Gerber, T.; Langer, H.; Hahnel, P.; et al. PARP-1 selectively impairs KRAS-driven phenotypic and molecular features in intrahepatic cholangiocarcinoma. Gut 2024, 73, 1712–1724. [Google Scholar] [CrossRef]
- LaFargue, C.J.; Dal Molin, G.Z.; Sood, A.K.; Coleman, R.L. Exploring and comparing adverse events between PARP inhibitors. Lancet Oncol. 2019, 20, e15–e28. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Aoyagi-Scharber, M.; Gardberg, A.S.; Yip, B.K.; Wang, B.; Shen, Y.; Fitzpatrick, P.A. Structural basis for the inhibition of poly(ADP-ribose) polymerases 1 and 2 by BMN 673, a potent inhibitor derived from dihydropyridophthalazinone. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2014, 70, 1143–1149. [Google Scholar] [CrossRef]
- Ogden, T.E.H.; Yang, J.-C.; Schimpl, M.; Easton, L.E.; Underwood, E.; Rawlins, P.B.; McCauley, M.M.; Langelier, M.-F.; Pascal, J.M.; Embrey, K.J.; et al. Dynamics of the HD regulatory subdomain of PARP-1; substrate access and allostery in PARP activation and inhibition. Nucleic Acids Res. 2021, 49, 2266–2288. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Xu, H.; Xian, J.; Vire, E.; McKinney, S.; Wei, V.; Wong, J.; Tong, R.; Kouzarides, T.; Caldas, C.; Aparicio, S. Up-regulation of the interferon-related genes in BRCA2 knockout epithelial cells. J. Pathol. 2014, 234, 386–397. [Google Scholar] [CrossRef]
- Hurley, P.J.; Wilsker, D.; Bunz, F. Human cancer cells require ATR for cell cycle progression following exposure to ionizing radiation. Oncogene 2007, 26, 2535–2542. [Google Scholar] [CrossRef]
- Roig, A.I.; Eskiocak, U.; Hight, S.K.; Kim, S.B.; Delgado, O.; Souza, R.F.; Spechler, S.J.; Wright, W.E.; Shay, J.W. Immortalized epithelial cells derived from human colon biopsies express stem cell markers and differentiate in vitro. Gastroenterology 2010, 138, 1012–1021.e5. [Google Scholar] [CrossRef]
- Seiwert, N.; Wecklein, S.; Demuth, P.; Hasselwander, S.; Kemper, T.A.; Schwerdtle, T.; Brunner, T.; Fahrer, J. Heme oxygenase 1 protects human colonocytes against ROS formation, oxidative DNA damage and cytotoxicity induced by heme iron, but not inorganic iron. Cell Death Dis. 2020, 11, 787. [Google Scholar] [CrossRef]
- Carlsson, M.J.; Vollmer, A.S.; Demuth, P.; Heylmann, D.; Reich, D.; Quarz, C.; Rasenberger, B.; Nikolova, T.; Hofmann, T.G.; Christmann, M.; et al. p53 triggers mitochondrial apoptosis following DNA damage-dependent replication stress by the hepatotoxin methyleugenol. Cell Death Dis. 2022, 13, 1009. [Google Scholar] [CrossRef] [PubMed]
- Demin, A.A.; Hirota, K.; Tsuda, M.; Adamowicz, M.; Hailstone, R.; Brazina, J.; Gittens, W.; Kalasova, I.; Shao, Z.; Zha, S.; et al. XRCC1 prevents toxic PARP1 trapping during DNA base excision repair. Mol. Cell 2021, 81, 3018–3030.e5. [Google Scholar] [CrossRef] [PubMed]
- Mimmler, M.; Peter, S.; Kraus, A.; Stroh, S.; Nikolova, T.; Seiwert, N.; Hasselwander, S.; Neitzel, C.; Haub, J.; Monien, B.H.; et al. DNA damage response curtails detrimental replication stress and chromosomal instability induced by the dietary carcinogen PhIP. Nucleic Acids Res. 2016, 44, 10259–10276. [Google Scholar] [CrossRef] [PubMed]
- Fahrer, J.; Huelsenbeck, J.; Jaurich, H.; Dorsam, B.; Frisan, T.; Eich, M.; Roos, W.P.; Kaina, B.; Fritz, G. Cytolethal distending toxin (CDT) is a radiomimetic agent and induces persistent levels of DNA double-strand breaks in human fibroblasts. DNA Repair 2014, 18, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Haince, J.F.; Poirier, G.G.; Kirkland, J.B. Nonisotopic methods for determination of poly(ADP-ribose) levels and detection of poly(ADP-ribose) polymerase. Curr. Protoc. Cell Biol. 2004, 21, Unit18.7. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Singh, K.; Almasan, A. Histone H2AX phosphorylation: A marker for DNA damage. Methods Mol. Biol. 2012, 920, 613–626. [Google Scholar] [CrossRef]
- Xiong, Y.; Guo, Y.; Liu, Y.; Wang, H.; Gong, W.; Liu, Y.; Wang, X.; Gao, Y.; Yu, F.; Su, D.; et al. Pamiparib is a potent and selective PARP inhibitor with unique potential for the treatment of brain tumor. Neoplasia 2020, 22, 431–440. [Google Scholar] [CrossRef]
- Zandarashvili, L.; Langelier, M.-F.; Velagapudi, U.K.; Hancock, M.A.; Steffen, J.D.; Billur, R.; Hannan, Z.M.; Wicks, A.J.; Krastev, D.B.; Pettitt, S.J.; et al. Structural basis for allosteric PARP-1 retention on DNA breaks. Science 2020, 368, eaax6367. [Google Scholar] [CrossRef]
- Hopkins, T.A.; Ainsworth, W.B.; Ellis, P.A.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Abraham, V.C.; Algire, M.A.; Shi, Y.; Olson, A.M.; et al. PARP1 Trapping by PARP Inhibitors Drives Cytotoxicity in Both Cancer Cells and Healthy Bone Marrow. Mol. Cancer Res. 2019, 17, 409–419. [Google Scholar] [CrossRef]
- Lawlor, D.; Martin, P.; Busschots, S.; Thery, J.; O’Leary, J.J.; Hennessy, B.T.; Stordal, B. PARP Inhibitors as P-glyoprotein Substrates. J. Pharm. Sci. 2014, 103, 1913–1920. [Google Scholar] [CrossRef]
- Arnold, C.; Demuth, P.; Seiwert, N.; Wittmann, S.; Boengler, K.; Rasenberger, B.; Christmann, M.; Huber, M.; Brunner, T.; Linnebacher, M.; et al. The Mitochondrial Disruptor Devimistat (CPI-613) Synergizes with Genotoxic Anticancer Drugs in Colorectal Cancer Therapy in a Bim-Dependent Manner. Mol. Cancer Ther. 2022, 21, 100–112. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Sandhu, D.; Antolin, A.A.; Cox, A.R.; Jones, A.M. Identification of different side effects between PARP inhibitors and their polypharmacological multi-target rationale. Br. J. Clin. Pharmacol. 2022, 88, 742–752. [Google Scholar] [CrossRef]
- Langelier, M.F.; Lin, X.; Zha, S.; Pascal, J.M. Clinical PARP inhibitors allosterically induce PARP2 retention on DNA. Sci. Adv. 2023, 9, eadf7175. [Google Scholar] [CrossRef]
- Antolin, A.A.; Ameratunga, M.; Banerji, U.; Clarke, P.A.; Workman, P.; Al-Lazikani, B. The kinase polypharmacology landscape of clinical PARP inhibitors. Sci. Rep. 2020, 10, 2585. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Kulke, M.H.; Fuchs, C.S.; Allen, B.A.; Uno, H.; Hornick, J.L.; Ukaegbu, C.I.; Brais, L.K.; McNamara, P.G.; Mayer, R.J.; et al. Cancer Susceptibility Gene Mutations in Individuals With Colorectal Cancer. J. Clin. Oncol. 2017, 35, 1086–1095. [Google Scholar] [CrossRef]
- Tahara, M.; Inoue, T.; Sato, F.; Miyakura, Y.; Horie, H.; Yasuda, Y.; Fujii, H.; Kotake, K.; Sugano, K. The Use of Olaparib (AZD2281) Potentiates SN-38 Cytotoxicity in Colon Cancer Cells by Indirect Inhibition of Rad51-Mediated Repair of DNA Double-Strand Breaks. Mol. Cancer Ther. 2014, 13, 1170–1180. [Google Scholar] [CrossRef]
- Murai, J.; Zhang, Y.; Morris, J.; Ji, J.; Takeda, S.; Doroshow, J.H.; Pommier, Y.G. Rationale for PARP inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J. Pharmacol. Exp. Ther. 2014, 349, 408–416. [Google Scholar] [CrossRef]
- Augustine, T.; Maitra, R.; Zhang, J.; Nayak, J.; Goel, S. Sensitization of colorectal cancer to irinotecan therapy by PARP inhibitor rucaparib. Investig. New Drugs 2019, 37, 948–960. [Google Scholar] [CrossRef]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef]
- Innocenti, F.; Undevia, S.D.; Iyer, L.; Chen, P.X.; Das, S.; Kocherginsky, M.; Karrison, T.; Janisch, L.; Ramirez, J.; Rudin, C.M.; et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J. Clin. Oncol. 2004, 22, 1382–1388. [Google Scholar] [CrossRef]
- Thomas, A.; Pommier, Y. Targeting Topoisomerase I in the Era of Precision Medicine. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 6581–6589. [Google Scholar] [CrossRef]
- Marzi, L.; Szabova, L.; Gordon, M.; Weaver Ohler, Z.; Sharan, S.K.; Beshiri, M.L.; Etemadi, M.; Murai, J.; Kelly, K.; Pommier, Y. The Indenoisoquinoline TOP1 Inhibitors Selectively Target Homologous Recombination-Deficient and Schlafen 11-Positive Cancer Cells and Synergize with Olaparib. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 6206–6216. [Google Scholar] [CrossRef]
- Deng, S.; Vlatkovic, T.; Li, M.; Zhan, T.; Veldwijk, M.R.; Herskind, C. Targeting the DNA Damage Response and DNA Repair Pathways to Enhance Radiosensitivity in Colorectal Cancer. Cancers 2022, 14, 4874. [Google Scholar] [CrossRef]
- Glynne-Jones, R.; Wyrwicz, L.; Tiret, E.; Brown, G.; Rodel, C.; Cervantes, A.; Arnold, D.; Committee, E.G. Rectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, iv22–iv40. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Ji, Z.; Zhai, E.; Xu, K.; Zhang, Y.; Li, Q.; Jing, H.; Wang, X.; Song, X. PARP inhibitor olaparib enhances the efficacy of radiotherapy on XRCC2-deficient colorectal cancer cells. Cell Death Dis. 2022, 13, 505. [Google Scholar] [CrossRef]
- Carter, R.; Cheraghchi-Bashi, A.; Westhorpe, A.; Yu, S.; Shanneik, Y.; Seraia, E.; Ouaret, D.; Inoue, Y.; Koch, C.; Wilding, J.; et al. Identification of anticancer drugs to radiosensitise BRAF-wild-type and mutant colorectal cancer. Cancer Biol. Med. 2019, 16, 234–246. [Google Scholar] [CrossRef]
- Czito, B.G.; Deming, D.A.; Jameson, G.S.; Mulcahy, M.F.; Vaghefi, H.; Dudley, M.W.; Holen, K.D.; DeLuca, A.; Mittapalli, R.K.; Munasinghe, W.; et al. Safety and tolerability of veliparib combined with capecitabine plus radiotherapy in patients with locally advanced rectal cancer: A phase 1b study. Lancet Gastroenterol. Hepatol. 2017, 2, 418–426. [Google Scholar] [CrossRef]
- Gorbunova, V.; Beck, J.T.; Hofheinz, R.-D.; Garcia-Alfonso, P.; Nechaeva, M.; Cubillo Gracian, A.; Mangel, L.; Elez Fernandez, E.; Deming, D.A.; Ramanathan, R.K.; et al. A phase 2 randomised study of veliparib plus FOLFIRI±bevacizumab versus placebo plus FOLFIRI±bevacizumab in metastatic colorectal cancer. Br. J. Cancer 2019, 120, 183–189. [Google Scholar] [CrossRef]
- Combès, E.; Andrade, A.F.; Tosi, D.; Michaud, H.-A.; Coquel, F.; Garambois, V.; Desigaud, D.; Jarlier, M.; Coquelle, A.; Pasero, P.; et al. Inhibition of Ataxia-Telangiectasia Mutated and RAD3-Related (ATR) Overcomes Oxaliplatin Resistance and Promotes Antitumor Immunity in Colorectal Cancer. Cancer Res. 2019, 79, 2933–2946. [Google Scholar] [CrossRef]
- Shkundina, I.S.; Gall, A.A.; Dick, A.; Cocklin, S.; Mazin, A.V. New RAD51 Inhibitors to Target Homologous Recombination in Human Cells. Genes 2021, 12, 920. [Google Scholar] [CrossRef] [PubMed]
- Durinikova, E.; Reilly, N.M.; Buzo, K.; Mariella, E.; Chila, R.; Lorenzato, A.; Dias, J.M.L.; Grasso, G.; Pisati, F.; Lamba, S.; et al. Targeting the DNA Damage Response Pathways and Replication Stress in Colorectal Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 3874–3889. [Google Scholar] [CrossRef] [PubMed]
- Petropoulos, M.; Karamichali, A.; Rossetti, G.G.; Freudenmann, A.; Iacovino, L.G.; Dionellis, V.S.; Sotiriou, S.K.; Halazonetis, T.D. Transcription-replication conflicts underlie sensitivity to PARP inhibitors. Nature 2024, 628, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Potlitz, F.; Link, A. Chemotion Repository 2024. Available online: https://dx.doi.org/10.14272/collection/CWG_2024-02-14 (accessed on 14 February 2024).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| X17613 | X17618 | Olaparib | Veliparib | IT | 5-FU | OXA | |
|---|---|---|---|---|---|---|---|
| HCT116 WT | - | - | 8.40 | - | 1.28 | 0.50 | 1.19 |
| HCT116 BRCA2−/− | - | - | 0.46 | 7.45 | 0.16 | 0.34 | 0.25 |
| HCT116 PARP1+/+ | - | - | 11.21 | - | 0.93 | 0.24 | 0.71 |
| HCT116 PARP1−/− | - | - | - | - | 0.35 | 0.15 | 0.28 |
| DLD-1 WT | - | - | 5.88 | - | 4.19 | 0.35 | 8.77 |
| DLD-1 ATRs/s | - | - | 1.99 | - | 2.99 | 0.22 | 9.08 |
| Caco-2 | - | - | - | - | 66.52 | - | 11.18 |
| HCEC | - | - | - | - | 2.95 | 26.23 | 40.71 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demuth, P.; Thibol, L.; Lemsch, A.; Potlitz, F.; Schulig, L.; Grathwol, C.; Manolikakes, G.; Schade, D.; Roukos, V.; Link, A.; et al. Targeting PARP-1 and DNA Damage Response Defects in Colorectal Cancer Chemotherapy with Established and Novel PARP Inhibitors. Cancers 2024, 16, 3441. https://doi.org/10.3390/cancers16203441
Demuth P, Thibol L, Lemsch A, Potlitz F, Schulig L, Grathwol C, Manolikakes G, Schade D, Roukos V, Link A, et al. Targeting PARP-1 and DNA Damage Response Defects in Colorectal Cancer Chemotherapy with Established and Novel PARP Inhibitors. Cancers. 2024; 16(20):3441. https://doi.org/10.3390/cancers16203441
Chicago/Turabian StyleDemuth, Philipp, Lea Thibol, Anna Lemsch, Felix Potlitz, Lukas Schulig, Christoph Grathwol, Georg Manolikakes, Dennis Schade, Vassilis Roukos, Andreas Link, and et al. 2024. "Targeting PARP-1 and DNA Damage Response Defects in Colorectal Cancer Chemotherapy with Established and Novel PARP Inhibitors" Cancers 16, no. 20: 3441. https://doi.org/10.3390/cancers16203441