
Whole Exome-Wide Association Identifies Rare Variants in APC Associated with High-Risk Colorectal Cancer in the Middle East

 ,
,  ,
,
Simple Summary
Abstract
1. Introduction
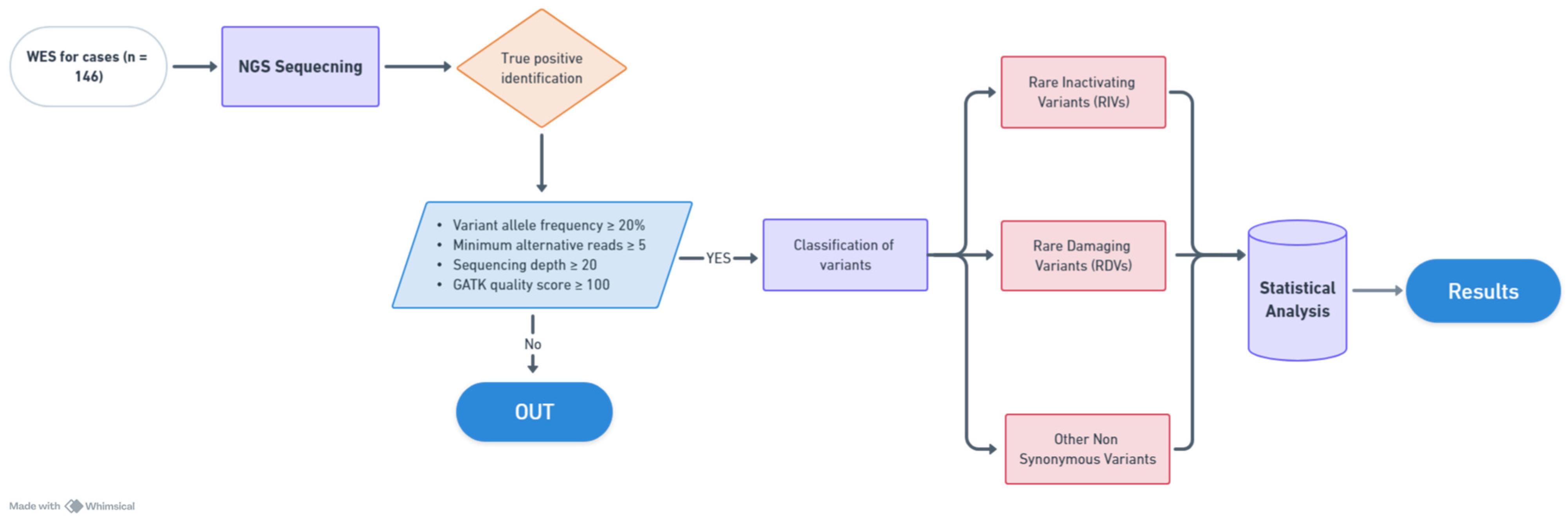
2. Materials and Methods
2.1. Patient Selection
2.2. DNA Extraction
2.3. Whole Exome Sequencing
2.4. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Council, S.H. Cancer Incidence Report 2020: Saudi Arabia; Saudi Cancer Registry: Riyadh, Saudi Arabia, 2020. [Google Scholar]
- Araghi, M.; Soerjomataram, I.; Bardot, A.; Ferlay, J.; Cabasag, C.J.; Morrison, D.S.; De, P.; Tervonen, H.; Walsh, P.M.; Bucher, O. Changes in colorectal cancer incidence in seven high-income countries: A population-based study. Lancet Gastroenterol. Hepatol. 2019, 4, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.E.; Hu, C.-Y.; You, Y.N.; Bednarski, B.K.; Rodriguez-Bigas, M.A.; Skibber, J.M.; Cantor, S.B.; Chang, G.J. Increasing disparities in the age-related incidences of colon and rectal cancers in the United States, 1975–2010. JAMA Surg. 2015, 150, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Yeo, H.; Betel, D.; Abelson, J.S.; Zheng, X.E.; Yantiss, R.; Shah, M.A. Early-onset colorectal cancer is distinct from traditional colorectal cancer. Clin. Color. Cancer 2017, 16, 293–299.e296. [Google Scholar] [CrossRef] [PubMed]
- Armelao, F.; de Pretis, G. Familial colorectal cancer: A review. World J. Gastroenterol. WJG 2014, 20, 9292. [Google Scholar]
- Chubb, D.; Broderick, P.; Frampton, M.; Kinnersley, B.; Sherborne, A.; Penegar, S.; Lloyd, A.; Ma, Y.P.; Dobbins, S.E.; Houlston, R.S. Genetic diagnosis of high-penetrance susceptibility for colorectal cancer (CRC) is achievable for a high proportion of familial CRC by exome sequencing. J. Clin. Oncol. 2015, 33, 426–432. [Google Scholar] [CrossRef]
- Lee, S.; Abecasis, G.R.; Boehnke, M.; Lin, X. Rare-variant association analysis: Study designs and statistical tests. Am. J. Hum. Genet. 2014, 95, 5–23. [Google Scholar] [CrossRef]
- Bu, R.; Siraj, A.K.; Azam, S.; Iqbal, K.; Qadri, Z.; Al-Rasheed, M.; Al-Sobhi, S.S.; Al-Dayel, F.; Al-Kuraya, K.S. Whole Exome-Wide Association Identifies Rare Variants in GALNT9 Associated with Middle Eastern Papillary Thyroid Carcinoma Risk. Cancers 2023, 15, 4235. [Google Scholar] [CrossRef]
- Grant, R.C.; Denroche, R.E.; Borgida, A.; Virtanen, C.; Cook, N.; Smith, A.L.; Connor, A.A.; Wilson, J.M.; Peterson, G.; Roberts, N.J. Exome-wide association study of pancreatic cancer risk. Gastroenterology 2018, 154, 719–722.e713. [Google Scholar] [CrossRef]
- Liu, Y.; Xia, J.; McKay, J.; Tsavachidis, S.; Xiao, X.; Spitz, M.R.; Cheng, C.; Byun, J.; Hong, W.; Li, Y. Rare deleterious germline variants and risk of lung cancer. NPJ Precis. Oncol. 2021, 5, 12. [Google Scholar] [CrossRef]
- Li, J.; Zou, L.; Zhou, Y.; Li, L.; Zhu, Y.; Yang, Y.; Gong, Y.; Lou, J.; Ke, J.; Zhang, Y. A low-frequency variant in SMAD7 modulates TGF-β signaling and confers risk for colorectal cancer in Chinese population. Mol. Carcinog. 2017, 56, 1798–1807. [Google Scholar] [CrossRef]
- Chubb, D.; Broderick, P.; Dobbins, S.E.; Frampton, M.; Kinnersley, B.; Penegar, S.; Price, A.; Ma, Y.P.; Sherborne, A.L.; Palles, C. Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer. Nat. Commun. 2016, 7, 11883. [Google Scholar] [CrossRef] [PubMed]
- Esteban-Jurado, C.; Vila-Casadesús, M.; Garre, P.; Lozano, J.J.; Pristoupilova, A.; Beltran, S.; Muñoz, J.; Ocaña, T.; Balaguer, F.; López-Cerón, M. Whole-exome sequencing identifies rare pathogenic variants in new predisposition genes for familial colorectal cancer. Genet. Med. 2015, 17, 131–142. [Google Scholar] [CrossRef]
- Bouras, A.; Fabre, A.; Zattara, H.; Handallou, S.; Desseigne, F.; Kientz, C.; Prieur, F.; Peysselon, M.; Legrand, C.; Calavas, L. Hereditary Colorectal Cancer and Polyposis Syndromes Caused by Variants in Uncommon Genes. Genes Chromosomes Cancer 2024, 63, e23263. [Google Scholar] [CrossRef] [PubMed]
- Hassanin, E.; Spier, I.; Bobbili, D.R.; Aldisi, R.; Klinkhammer, H.; David, F.; Dueñas, N.; Hüneburg, R.; Perne, C.; Brunet, J. Clinically relevant combined effect of polygenic background, rare pathogenic germline variants, and family history on colorectal cancer incidence. BMC Med. Genom. 2023, 16, 42. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 233–254. [Google Scholar] [CrossRef]
- Siraj, A.K.; Masoodi, T.; Bu, R.; Parvathareddy, S.K.; Al-Badawi, I.A.; Al-Sanea, N.; Ashari, L.H.; Abduljabbar, A.; Alhomoud, S.; Al-Sobhi, S.S. Expanding the spectrum of germline variants in cancer. Hum. Genet. 2017, 136, 1431–1444. [Google Scholar] [CrossRef]
- Abubaker, J.; Jehan, Z.; Bavi, P.; Sultana, M.; Al-Harbi, S.; Ibrahim, M.; Al-Nuaim, A.; Ahmed, M.; Amin, T.; Al-Fehaily, M. Clinicopathological analysis of papillary thyroid cancer with PIK3CA alterations in a Middle Eastern population. J. Clin. Endocrinol. Metab. 2008, 93, 611–618. [Google Scholar] [CrossRef]
- Masoodi, T.; Siraj, A.K.; Siraj, S.; Azam, S.; Qadri, Z.; Parvathareddy, S.K.; Al-Sobhi, S.S.; AlDawish, M.; Alkuraya, F.S.; Al-Kuraya, K.S. Evolution and impact of subclonal mutations in papillary thyroid cancer. Am. J. Hum. Genet. 2019, 105, 959–973. [Google Scholar] [CrossRef]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef]
- Laskowski, R.A. PDBsum new things. Nucleic Acids Res. 2009, 37, D355–D359. [Google Scholar] [CrossRef]
- Galiatsatos, P.; Foulkes, W.D. Familial adenomatous polyposis. Off. J. Am. Coll. Gastroenterol. ACG 2006, 101, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Burt, R.W.; Leppert, M.F.; Slattery, M.L.; Samowitz, W.S.; Spirio, L.N.; Kerber, R.A.; Kuwada, S.K.; Neklason, D.W.; DiSario, J.A.; Lyon, E. Genetic testing and phenotype in a large kindred with attenuated familial adenomatous polyposis. Gastroenterology 2004, 127, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.; Barnes, D.R.; Dunlop, J.; Barrowdale, D.; Antoniou, A.C.; Berg, J.N. Attenuated familial adenomatous polyposis manifests as autosomal dominant late-onset colorectal cancer. Eur. J. Hum. Genet. 2014, 22, 1330–1333. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, E.M.; Koeppe, E.; Everett, J.; Ulintz, P.; Kiel, M.; Osborne, J.; Williams, L.; Hanson, K.; Gruber, S.B.; Rozek, L.S. Germline genetic features of young individuals with colorectal cancer. Gastroenterology 2018, 154, 897–905.e891. [Google Scholar] [CrossRef] [PubMed]
- Samadder, N.J.; Giridhar, K.V.; Baffy, N.; Riegert-Johnson, D.; Couch, F.J. Hereditary cancer syndromes—A primer on diagnosis and management: Part 1: Breast-ovarian cancer syndromes. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2019; pp. 1084–1098. [Google Scholar]
- Frostberg, E.; Petersen, A.H.; Bojesen, A.; Rahr, H.B.; Lindebjerg, J.; Rønlund, K. The prevalence of pathogenic or likely pathogenic germline variants in a nationwide cohort of young colorectal cancer patients using a panel of 18 genes associated with colorectal cancer. Cancers 2021, 13, 5094. [Google Scholar] [CrossRef]
- Stanich, P.P.; Pearlman, R.; Hinton, A.; Gutierrez, S.; LaDuca, H.; Hampel, H.; Jasperson, K. Prevalence of germline mutations in polyposis and colorectal cancer–associated genes in patients with multiple colorectal polyps. Clin. Gastroenterol. Hepatol. 2019, 17, 2008–2015.e2003. [Google Scholar] [CrossRef]
- Heim, S.; Lage, H. Transcriptome analysis of different multidrug-resistant gastric carcinoma cells. In Vivo 2005, 19, 583–590. [Google Scholar]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Dai, J.; Li, Z.-X.; Zhang, Y.; Ma, J.-L.; Zhou, T.; You, W.-C.; Li, W.-Q.; Pan, K.-F. Whole genome messenger RNA profiling identifies a novel signature to predict gastric cancer survival. Clin. Transl. Gastroenterol. 2019, 10, e00004. [Google Scholar] [CrossRef]
- Shinriki, S.; Maeshiro, M.; Shimamura, K.; Kawashima, J.; Araki, E.; Ibusuki, M.; Yamamoto, Y.; Iwase, H.; Miyamoto, Y.; Baba, H. Evaluation of an amplicon-based custom gene panel for the diagnosis of hereditary tumors. Neoplasma 2020, 67, 898. [Google Scholar] [CrossRef]
- Ablain, J.; Xu, M.; Rothschild, H.; Jordan, R.; Mito, J.; Daniels, B. SPRED1 Is a Tumor Suppressor in Mucosal Melanoma. Cancer Discov. 2018, 8, 1507. [Google Scholar]
- Ayanlaja, A.A.; Hong, X.; Cheng, B.; Zhou, H.; Kanwore, K.; Alphayo-Kambey, P.; Zhang, L.; Tang, C.; Adeyanju, M.M.; Gao, D. Susceptibility of cytoskeletal-associated proteins for tumor progression. Cell Mol. Life Sci. 2022, 79, 13. [Google Scholar] [CrossRef] [PubMed]
- Basak, A.J.; Maiti, S.; Hansda, A.; Mahata, D.; Duraivelan, K.; Kundapura, S.V.; Lee, W.; Mukherjee, G.; De, S.; Samanta, D. Structural insights into N-terminal IgV domain of BTNL2, a T cell inhibitory molecule, suggests a non-canonical binding interface for its putative receptors. J. Mol. Biol. 2020, 432, 5938–5950. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhao, H.; Li, M.; She, Q.; Liu, W.; Zhang, J.; Zhao, W.; Huang, S.; Wu, J. SHANK1 facilitates non-small cell lung cancer processes through modulating the ubiquitination of Klotho by interacting with MDM2. Cell Death Dis. 2022, 13, 403. [Google Scholar] [CrossRef] [PubMed]
- Chien, W.; Lee, K.; Ding, L.; Wuensche, P.; Kato, H.; Doan, N.; Poellinger, L.; Said, J.; Koeffler, H. PIAS4 is an activator of hypoxia signalling via VHL suppression during growth of pancreatic cancer cells. Br. J. Cancer 2013, 109, 1795–1804. [Google Scholar] [CrossRef]
- Chiu, C.-F.; Chang, H.-Y.; Huang, C.-Y.; Mau, C.-Z.; Kuo, T.-T.; Lee, H.-C.; Huang, S.-Y. Betulinic acid affects the energy-related proteomic profiling in pancreatic ductal adenocarcinoma cells. Molecules 2021, 26, 2482. [Google Scholar] [CrossRef]
- Fejzo, M.S.; Chen, H.-W.; Anderson, L.; McDermott, M.S.; Karlan, B.; Konecny, G.E.; Slamon, D.J. Analysis in epithelial ovarian cancer identifies KANSL1 as a biomarker and target gene for immune response and HDAC inhibition. Gynecol. Oncol. 2021, 160, 539–546. [Google Scholar] [CrossRef]
- Hanieh, H.; Ahmed, E.A.; Vishnubalaji, R.; Alajez, N.M. SOX4: Epigenetic regulation and role in tumorigenesis. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2010; pp. 91–104. [Google Scholar]
- Lapkina-Gendler, L.; Rotem, I.; Pasmanik-Chor, M.; Gurwitz, D.; Sarfstein, R. Identification of signaling pathways associated with cancer protection in Laron syndrome. Endocr. Relat. Cancer 2016, 23, 399–410. [Google Scholar] [CrossRef]
- Liot, S.; Aubert, A.; Hervieu, V.; El Kholti, N.; Schalkwijk, J.; Verrier, B.; Valcourt, U.; Lambert, E. Loss of Tenascin-X expression during tumor progression: A new pan-cancer marker. Matrix Biol. Plus 2020, 6, 100021. [Google Scholar] [CrossRef]
- Mauri, G.; Patelli, G.; Roazzi, L.; Valtorta, E.; Amatu, A.; Marrapese, G.; Bonazzina, E.; Tosi, F.; Bencardino, K.; Ciarlo, G. Clinicopathological characterisation of MTAP alterations in gastrointestinal cancers. J. Clin. Pathol. 2024; ahead of print. [Google Scholar] [CrossRef]
- Ren, H.; Zhu, J.; Yu, H.; Bazhin, A.V.; Westphalen, C.B.; Renz, B.W.; Jacob, S.N.; Lampert, C.; Werner, J.; Angele, M.K. Angiogenesis-related gene expression signatures predicting prognosis in gastric cancer patients. Cancers 2020, 12, 3685. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Verma, S.; Angurana, S.L.; Tufail, Z.; Bhagat, V.; Nagyal, S.; Jamwal, R.S.; Sharma, B.; Shah, R.; Bhat, A. Exome sequencing identifies ADGRG4 G-protein-coupled receptors gene as a novel cancer biomarker in ovarian cancer patients from North India. J. Biochem. Mol. Toxicol. 2024, 38, e23672. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, W.; Guo, Y.; Zhang, Y.; Bai, X.; Xie, Y. Identification of critical prognosis signature associated with lymph node metastasis of stomach adenocarcinomas. World J. Surg. Oncol. 2023, 21, 61. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liu, Z.; Deng, D.; Fu, Z.; Chen, J.; Cui, Y.; Zhao, Z.; Zhang, X. Effect of MPP2 and its methylation levels on prognosis of colorectal cancer patients. World J. Surg. Oncol. 2024; in review. [Google Scholar] [CrossRef]
- Zhao, W.; Yang, L.; Chen, X.; Huang, W. Cardiac-Specific Gene TNNI3 as a Potential Oncogene for Kidney Cancer and Its Involvement in Wnt Signaling Pathway. Res. Sq. 2021; in review. [Google Scholar] [CrossRef]
- Zhu, L.; Li, Y.; Xie, X.; Zhou, X.; Gu, M.; Jie, Z.; Ko, C.-J.; Gao, T.; Hernandez, B.E.; Cheng, X. TBKBP1 and TBK1 form a growth factor signalling axis mediating immunosuppression and tumourigenesis. Nat. Cell Biol. 2019, 21, 1604–1614. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| N = 146 | |
|---|---|
| Age (years) | |
| Median (IQR), years | 41.0 (34.0–48.9) |
| <50 | 119 (81.5) |
| ≥50 | 27 (18.5) |
| Gender | |
| Female | 64 (43.8) |
| Male | 82 (56.2) |
| Family history of cancer | |
| Positive | 53 (36.3) |
| Negative | 93 (63.7) |
| Family history of colon cancer | |
| Positive | 28 (19.2) |
| Negative | 118 (80.8) |
| Body mass index (kg/m2) | |
| <30 | 92 (63.0) |
| ≥30 | 40 (27.4) |
| Unknown | 14 (9.6) |
| History of diabetes mellitus | |
| Present | 19 (13.0) |
| Absent | 97 (66.4) |
| Unknown | 30 (20.5) |
| Histologic subtype | |
| Adenocarcinoma | 127 (87.0) |
| Mucinous | 19 (13.0) |
| Tumor location | |
| Left colon | 113 (77.4) |
| Right colon | 26 (17.8) |
| Transverse colon | 7 (4.8) |
| Histologic grade | |
| Well differentiated | 8 (5.5) |
| Moderately differentiated | 113 (77.4) |
| Poorly differentiated | 14 (9.6) |
| Unknown | 11 (7.5) |
| pT | |
| T1 | 4 (2.8) |
| T2 | 17 (11.7) |
| T3 | 90 (61.6) |
| T4 | 24 (16.4) |
| Unknown | 11 (7.5) |
| pN | |
| N0 | 56 (38.4) |
| N1/N2 | 79 (54.1) |
| Nx | 11 (7.5) |
| pM | |
| M0 | 111 (76.0) |
| M1 | 28 (19.2) |
| Mx | 7 (4.8) |
| TNM Stage | |
| I | 17 (11.7) |
| II | 38 (26.0) |
| III | 55 (37.7) |
| IV | 28 (19.2) |
| Unknown | 8 (5.4) |
| S No | Gene | Chr | Position | Ref | Alt | Variant Type | No. of Cases | % | No. of Controls | % | p-Value | Odds Ratio |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | APC | chr5 | 112,154,963 | C | T | Missense | 3 | 2.1 | 0 | 0.0 | 8.36 × 10−8 | 68.1 |
| 2 | APC | chr5 | 112,128,191 | C | T | Missense | 1 | 0.7 | 0 | 0.0 | 0.002 | 28.8 |
| 3 | APC | chr5 | 112,155,042 | G | C | Missense | 1 | 0.7 | 0 | 0.0 | 0.002 | 28.8 |
| 4 | APC | chr5 | 112,174,112 | G | T | Missense | 1 | 0.7 | 0 | 0.0 | 0.002 | 28.8 |
| 5 | APC | chr5 | 112,175,077 | T | - | Frameshift Deletion | 0 | 0.7 | 1 | 0.0 | 0.746 | −3.1 |
| 6 | RIMS1 | chr6 | 72,974,704 | T | G | Missense | 4 | 2.7 | 0 | 0.0 | 6.01 × 10−10 | 88.1 |
| 7 | RIMS1 | chr6 | 72,975,696 | - | TC | Frameshift Insertion | 1 | 0.7 | 0 | 0.0 | 0.002 | 28.8 |
| 8 | RIMS1 | chr6 | 72,945,397 | T | C | Missense | 0 | 0.0 | 1 | 0.1 | 0.746 | 3.2 |
| 9 | RIMS1 | chr6 | 72,984,083 | C | T | Missense | 0 | 0.0 | 1 | 0.1 | 0.746 | 3.2 |
| 10 | ST6GALNAC2 | chr17 | 74,566,661 | T | - | Frameshift Deletion | 2 | 1.4 | 1 | 0.1 | 0.001 | 19.4 |
| 11 | ST6GALNAC2 | chr17 | 74,568,782 | G | - | Frameshift Deletion | 1 | 0.7 | 1 | 0.1 | 0.050 | 9.6 |
| S No | Gene | No. of Cases | % Cases | No. of Controls | % Controls | p-Value | Odds Ratio |
|---|---|---|---|---|---|---|---|
| 1 | APC * | 6 | 4.1 | 1 | 0.1 | 5.08 × 10−12 | 59.7 |
| 2 | CD36 | 6 | 4.1 | 25 | 1.8 | 0.058 | 2.3 |
| 3 | RIMS1 * | 5 | 3.4 | 2 | 0.1 | 2.03 × 10−8 | 24.7 |
| 4 | ACOT4 | 3 | 2.1 | 4 | 0.3 | 0.003 | 7.3 |
| 5 | ST6GALNAC2 # | 3 | 2.1 | 2 | 0.1 | 1.12 × 10−4 | 14.6 |
| 6 | FSIP2 | 3 | 2.1 | 7 | 0.5 | 0.026 | 4.2 |
| 7 | PNPLA7 | 3 | 2.1 | 9 | 0.6 | 0.065 | 3.2 |
| 8 | TTN | 2 | 1.4 | 32 | 2.3 | 0.470 | 0.6 |
| 9 | TTLL10 | 2 | 1.4 | 5 | 0.4 | 0.084 | 3.9 |
| 10 | ITGA10 | 2 | 1.4 | 52 | 3.7 | 0.140 | 0.4 |
| S No | Gene | No. of Cases | % Cases | No. of Controls | % Controls | p-Value | Odds Ratio |
|---|---|---|---|---|---|---|---|
| 1 | TNXB | 10 | 6.8 | 1 | 0.1 | 0.00 × 10−0 | 102.5 |
| 2 | GPR112 | 7 | 4.8 | 0 | 0.0 | 0.00 × 10−0 | 150.1 |
| 3 | COL11A2 | 5 | 3.4 | 0 | 0.0 | 4.42 × 10−12 | 108.5 |
| 4 | ANKRD33B | 4 | 2.7 | 1 | 0.1 | 6.91 × 10−8 | 39.3 |
| 5 | TBKBP1 | 5 | 3.4 | 2 | 0.1 | 2.03 × 10−8 | 24.7 |
| 6 | OR5K4 | 5 | 3.4 | 2 | 0.1 | 2.03 × 10−8 | 24.7 |
| 7 | MTAP | 5 | 3.4 | 2 | 0.1 | 2.03 × 10−8 | 24.7 |
| 8 | SHANK1 | 10 | 6.8 | 13 | 0.9 | 2.02 × 10−8 | 7.8 |
| 9 | SPRED1 | 4 | 2.7 | 1 | 0.1 | 6.91 × 10−8 | 39.3 |
| 10 | MPP2 | 4 | 2.7 | 1 | 0.1 | 6.91 × 10−8 | 39.3 |
| 11 | KANSL1 | 4 | 2.7 | 1 | 0.1 | 6.91 × 10−8 | 39.3 |
| 12 | PIAS4 | 4 | 2.7 | 1 | 0.1 | 6.91 × 10−8 | 39.3 |
| 13 | TNNI3 | 4 | 2.7 | 1 | 0.1 | 6.91 × 10−8 | 39.3 |
| 14 | BTNL2 | 4 | 2.7 | 1 | 0.1 | 6.91 × 10−8 | 39.3 |
| 15 | CAPZA1 | 4 | 2.7 | 2 | 0.1 | 1.64 × 10−6 | 19.6 |
| 16 | OSTC | 4 | 2.7 | 2 | 0.1 | 1.64 × 10−6 | 19.6 |
| 17 | SOX4 | 4 | 2.7 | 2 | 0.1 | 1.64 × 10−6 | 19.6 |
| S No | Gene | No. of Cases | % Cases | No. of Controls | % Controls | p-Value | Odds Ratio |
|---|---|---|---|---|---|---|---|
| 1 | TNXB | 16 | 11.0 | 1 | 0.1 | 0.00 × 10−0 | 171.6 |
| 2 | TAP2 | 8 | 5.5 | 0 | 0.0 | 0.00 × 10−0 | 171.3 |
| 3 | GPSM3 | 7 | 4.8 | 0 | 0.0 | 0.00 × 10−0 | 150.1 |
| 4 | ADGRG4 | 7 | 4.8 | 0 | 0.0 | 0.00 × 10−0 | 150.1 |
| 5 | TMEM229A | 8 | 5.5 | 4 | 0.3 | 1.11 × 10−11 | 20.2 |
| 6 | ANKRD33B | 6 | 4.1 | 1 | 0.1 | 5.08 × 10−12 | 59.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siraj, A.K.; Bu, R.; Azam, S.; Qadri, Z.; Iqbal, K.; Parvathareddy, S.K.; Al-Dayel, F.; Al-Kuraya, K.S. Whole Exome-Wide Association Identifies Rare Variants in APC Associated with High-Risk Colorectal Cancer in the Middle East. Cancers 2024, 16, 3720. https://doi.org/10.3390/cancers16213720
Siraj AK, Bu R, Azam S, Qadri Z, Iqbal K, Parvathareddy SK, Al-Dayel F, Al-Kuraya KS. Whole Exome-Wide Association Identifies Rare Variants in APC Associated with High-Risk Colorectal Cancer in the Middle East. Cancers. 2024; 16(21):3720. https://doi.org/10.3390/cancers16213720
Chicago/Turabian StyleSiraj, Abdul Khalid, Rong Bu, Saud Azam, Zeeshan Qadri, Kaleem Iqbal, Sandeep Kumar Parvathareddy, Fouad Al-Dayel, and Khawla S. Al-Kuraya. 2024. "Whole Exome-Wide Association Identifies Rare Variants in APC Associated with High-Risk Colorectal Cancer in the Middle East" Cancers 16, no. 21: 3720. https://doi.org/10.3390/cancers16213720
APA StyleSiraj, A. K., Bu, R., Azam, S., Qadri, Z., Iqbal, K., Parvathareddy, S. K., Al-Dayel, F., & Al-Kuraya, K. S. (2024). Whole Exome-Wide Association Identifies Rare Variants in APC Associated with High-Risk Colorectal Cancer in the Middle East. Cancers, 16(21), 3720. https://doi.org/10.3390/cancers16213720