
Unraveling the Genetic Heterogeneity of Acute Lymphoblastic Leukemia Based on NGS Applications

 , ,
, ,  ,
,  and
and
Simple Summary
Abstract
1. Introduction
| Panel A. Search strategy and selection criteria |
| Keywords such as “acute lymphoblastic Leukemia”, “high-throughput sequencing”, “massive sequencing”, “NGS”, “ALL risk factor”, “molecular classification”, “polymorphism”, “omics data”, and their combinations were used. Tags such as [tiab][res] or [intext: intittle] and quotation marks as search operators and (*) were used as wildcard characters or to narrow the search. The inclusion criteria were established using the following: 1. subject area and sub-areas (medicine, hematology, oncohematology, genetics), 2. type of study and publication (clinical and in vitro studies, clinical trials, observational studies, clinical practice guidelines), 3. period of analysis and language (the initial search was conducted between 2017 and 2023, and older references were also used where appropriate; only English or Spanish articles were included), and 4. availability: only open access documents were included. References include original articles, systematic reviews, and meta-analyses. The information retrieval was performed using Health in Science Descriptors (Medical Subject Headings of the National Library of Medicine of the USA-MeSH) and Hierarchical Code/thesauri, as well as databases such as Elsevier-Science Direct, Web of Science (Web of Knowledge-WoK), Scopus, and PubMed. |
2. Next-Generation Sequencing (NGS) Applications in ALL and New Insights
3. New Insights into ALL Genomic Profiling and Recent Classification Based on Molecular Features
4. Molecular Landscape of New and Emerging Genetic Subtypes of B-ALL
4.1. New Insights in Philadelphia ALL Patients (BCR::ABL1-Positive)
4.2. Phenocopies in B-ALL
4.2.1. BCR::ABL1-like (Ph-like)
4.2.2. ETV6::RUNX1-like
4.2.3. ZNF384r-like (Provisional Entity)
4.2.4. KMT2Ar-like (Provisional Entity)
4.3. Novel Gene Fusions in B-ALL
4.3.1. MYC-Rearranged
4.3.2. IGH Fusions, t(v;14q32)
4.3.3. TCF3-Rearranged (19p13.3)
4.3.4. DUX4-Rearranged
4.3.5. ZNF384-Rearranged (12p13.31)
4.3.6. MEF2D-Rearranged (1q22)
4.3.7. NUTM1-Rearranged (15q14)
4.3.8. UBTF::ATXN7L3/PAN3, CDX2 (Also Known as “CDX2/UBTF”)
4.4. Point Mutations
4.4.1. PAX5P80R
4.4.2. IKZF1N159Y
4.5. Other Emerging B-ALL Subtypes
4.5.1. PAX5-Altered
4.5.2. Non-Ph-like CRLF2r and “Double-Hit” BCL2/MYC (IGH/BCL2 and 8q24/MYC Rearrangement)
5. New Insights into Genomic Profiling of T-ALL
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Aleem, A.; Haque, A.R.; Roloff, G.W.; Griffiths, E.A. Application of Next-Generation Sequencing-Based Mutational Profiling in Acute Lymphoblastic Leukemia. Curr. Hematol. Malig. Rep. 2021, 16, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Lejman, M.; Chałupnik, A.; Chilimoniuk, Z.; Dobosz, M. Genetic Biomarkers and Their Clinical Implications in B-Cell Acute Lymphoblastic Leukemia in Children. Int. J. Mol. Sci. 2022, 23, 2755. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Greaves, M.; Mullighan, C.G. Acute lymphoblastic leukaemia. Lancet 2013, 381, 1943–1955. [Google Scholar] [CrossRef] [PubMed]
- Malard, F.; Mohty, M. Acute lymphoblastic leukaemia. Lancet 2020, 395, 1146–1162. [Google Scholar] [CrossRef]
- Raetz, E.A.; Teachey, D.T. T-cell acute lymphoblastic leukemia. Hematol. Am. Soc. Hematol. Educ. Program. 2016, 1, 580–588. [Google Scholar] [CrossRef]
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef]
- Schaaf, J.; Sedlmayr, M.; Schaefer, J.; Storf, H. Diagnosis of Rare Diseases: A scoping review of clinical decision support systems. Orphanet J. Rare Dis. 2020, 15, 263. [Google Scholar] [CrossRef]
- Katz, A.J.; Chia, V.M.; Schoonen, W.M.; Kelsh, M.A. Acute lymphoblastic leukemia: An assessment of international incidence, survival, and disease burden. Cancer Causes Control 2015, 26, 1627–1642. [Google Scholar] [CrossRef]
- Sasaki, K.; Jabbour, E.; Short, N.J.; Jain, N.; Ravandi, F.; Pui, C.; Kantarjian, H. Acute lymphoblastic leukemia: A population-based study of outcome in the United States based on the surveillance, epidemiology, and end results (SEER) database, 1980–2017. Am. J. Hematol. 2021, 96, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Downing, J.R.; Wilson, R.K.; Zhang, J.; Mardis, E.R.; Pui, C.-H.; Ding, L.; Ley, T.J.; Evans, W.E. The pediatric cancer genome project. Nat. Genet. 2012, 44, 619–622. [Google Scholar] [CrossRef]
- Hunger, S.P.; Mullighan, C.G. Acute lymphoblastic leukemia in children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [PubMed]
- Beneduce, G.; De Matteo, A.; Stellato, P.; Testi, A.M.; Bertorello, N.; Colombini, A.; Putti, M.C.; Rizzari, C.; Cesaro, S.; Cellini, M.; et al. Blinatumomab in Children and Adolescents with Relapsed/Refractory B Cell Precursor Acute Lymphoblastic Leukemia: A Real-Life Multicenter Retrospective Study in Seven AIEOP (Associazione Italiana di Ematologia e Oncologia Pediatrica) Centers. Cancers 2022, 14, 426. [Google Scholar] [CrossRef] [PubMed]
- Abdelmabood, S.; Fouda, A.E.; Boujettif, F.; Mansour, A. Treatment outcomes of children with acute lymphoblastic leukemia in a middle-income developing country: High mortalities, early relapses, and poor survival. J. Pediatr. (Rio J.) 2020, 96, 108–116. [Google Scholar] [CrossRef]
- Lennmyr, E.; Karlsson, K.; Ahlberg, L.; Garelius, H.; Hulegårdh, E.; Izarra, A.S.; Joelsson, J.; Kozlowski, P.; Moicean, A.; Tomaszewska-Toporska, B.; et al. Survival in adult acute lymphoblastic leukaemia (ALL): A report from the Swedish ALL Registry. Eur. J. Haematol. 2019, 103, 88–98. [Google Scholar] [CrossRef]
- Iacobucci, I.; Mullighan, C.G. Genetic basis of acute lymphoblastic leukemia. J. Clin. Oncol. 2017, 35, 975–983. [Google Scholar] [CrossRef]
- Tran, T.H.; Hunger, S.P. The genomic landscape of pediatric acute lymphoblastic leukemia and precision medicine opportunities. Semin. Cancer Biol. 2022, 84, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Akkari, Y.M.N.; Baughn, L.B.; Dubuc, A.M.; Smith, A.C.; Mallo, M. Guiding the global evolution of cytogenetic testing for hematologic malignancies. Blood 2020, 139, 2273–2284. [Google Scholar] [CrossRef]
- Moorman, A.V.; Harrison, C.J.; Buck, G.A.N.; Richards, S.M.; Secker-Walker, L.M.; Martineau, M.; Vance, G.H.; Cherry, A.M.; Higgins, R.R.; Fielding, A.K.; et al. Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): Analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial. Blood 2007, 109, 3189–3197. [Google Scholar] [CrossRef] [PubMed]
- Bendari, M.; Sraidi, S.; Khoubila, N. Genetic Abnormalities in ALL. In Cytogenetics-Classical and Molecular Strategies for Analysing Heredity Material. Cap 8; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Othman, M.A.K.; Melo, J.B.; Carreira, I.M.; Rincic, M.; Glaser, A.; Grygalewicz, B.; Gruhn, B.; Wilhelm, K.; Rittscher, K.; Meyer, B.; et al. High rates of submicroscopic aberrations in karyotypically normal acute lymphoblastic leukemia. Mol. Cytogenet. 2015, 8, 45. [Google Scholar] [CrossRef]
- Hunger, S.P.; Mullighan, C.G. Redefining ALL classification: Toward detecting high-risk ALL and implementing precision medicine. Blood 2015, 125, 3977–3987. [Google Scholar] [CrossRef]
- Brown, P.A.; Shah, B.; Advani, A.; Aoun, P.; Boyer, M.W.; Burke, P.W.; DeAngelo, D.J.; Dinner, S.; Fathi, A.T.; Gauthier, J.; et al. Acute lymphoblastic leukemia, version 2.2021. J. Natl. Compr. Cancer Netw. 2021, 19, 1079–1109. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Vosberg, S.; Schlee, C.; Heesch, S.; Schwartz, S.; Gökbuget, N.; Hoelzer, D.; Graf, A.; Krebs, S.; Bartram, I.; et al. Mutational spectrum of adult T-ALL. Oncotarget 2014, 6, 2754–2766. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-H.; Wang, H.-S.; Qian, X.-W.; Zhu, X.-H.; Miao, H.; Yu, Y.; Meng, J.-H.; Le, J.; Jiang, J.-Y.; Cao, P.; et al. Ras pathway mutation feature in the same individuals at diagnosis and relapse of childhood acute lymphoblastic leukemia. Transl. Pediatr. 2020, 9, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, S.; Kotur, N.; Stankovic, B.; Zukic, B.; Gasic, V.; Dokmanovic, L. Pharmacogenomic and pharmacotranscriptomic profiling of childhood acute lymphoblastic leukemia: Paving the way to personalized treatment. Genes 2019, 10, 191. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Ontiveros, E.P.; Griffiths, E.A.; Thompson, J.E.; Wang, E.S.; Wetzler, M. Pharmacogenetics predictive of response and toxicity in acute lymphoblastic leukemia therapy. Blood Rev. 2015, 29, 243–249. [Google Scholar] [CrossRef]
- Nordlund, J.; Syvänen, A.C. Epigenetics in pediatric acute lymphoblastic leukemia. Semin. Cancer Biol. 2018, 51, 129–138. [Google Scholar] [CrossRef]
- Drożak, P.; Bryliński, Ł.; Zawitkowska, J. A Comprehensive Overview of Recent Advances in Epigenetics in Pediatric Acute Lymphoblastic Leukemia. Cancers 2022, 14, 5384. [Google Scholar] [CrossRef]
- Gökbuget, N.; Boissel, N.; Chiaretti, S.; Dombret, H.; Doubek, M.; Fielding, A.K.; Foà, R.; Giebel, S.; Hoelzer, D.; Hunault, M.; et al. Management of ALL in adults: 2024 ELN recommendations from a European expert panel. Blood 2024, 143, 1903–1930. [Google Scholar] [CrossRef]
- Schwab, C.; Cranston, R.E.; Ryan, S.L.; Butler, E.; Winterman, E.; Hawking, Z.; Bashton, M.; Enshaei, A.; Russell, L.J.; Kingsbury, Z.; et al. Integrative genomic analysis of childhood acute lymphoblastic leukaemia lacking a genetic biomarker in the UKALL2003 clinical trial. Leukemia 2023, 37, 529–538. [Google Scholar] [CrossRef]
- Wu, C.; Li, W. Genomics and pharmacogenomics of pediatric acute lymphoblastic leukemia. Crit. Rev. Oncol. Hematol. 2018, 126, 100–111. [Google Scholar] [CrossRef]
- Abaji, R.; Krajinovic, M. Pharmacogenetics of asparaginase in acute lymphoblastic leukemia. Cancer Drug Resist. 2019, 2, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Short, N.J.; Jain, N.; Haddad, F.G.; Welch, M.A.; Ravandi, F.; Kantarjian, H. The evolution of acute lymphoblastic leukemia research and therapy at MD Anderson over four decades. J. Hematol. Oncol. 2023, 16, 22. [Google Scholar] [CrossRef]
- Pui, C.H.; Evans, W.E. A 50-year journey to cure childhood acute lymphoblastic leukemia. Semin. Hematol. 2013, 50, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Azzato, E.M.; Mullighan, C.G. Integration of next-generation sequencing to treat acute lymphoblastic leukemia with targetable lesions: The St. Jude Children’s Research Hospital approach. Front. Pediatr. 2017, 5, 258. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-F.; Dai, Y.-T.; Lilljebjörn, H.; Shen, S.-H.; Cui, B.-W.; Bai, L.; Liu, Y.-F.; Qian, M.-X.; Kubota, Y.; Kiyoi, H.; et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1223 cases. Proc. Natl. Acad. Sci. USA 2018, 115, E11711–E11720. [Google Scholar] [CrossRef]
- Passet, M.; Boissel, N.; Sigaux, F.; Saillard, C.; Bargetzi, M.; Ba, I.; Thomas, X.; Graux, C.; Chalandon, Y.; Leguay, T.; et al. PAX5 P80R mutation identifies a novel subtype of B-cellprecursor acute lymphoblastic leukemia with favorable outcome. Blood 2019, 133, 280–284. [Google Scholar] [CrossRef]
- Ishida, H.; Iguchi, A.; Aoe, M.; Takahashi, T.; Tamefusa, K.; Kanamitsu, K.; Fujiwara, K.; Washio, K.; Matsubara, T.; Tsukahara, H.; et al. Panel-based next-generation sequencing identifies prognostic and actionable genes in childhood acute lymphoblastic leukemia and is suitable for clinical sequencing. Ann. Hematol. 2019, 98, 657–668. [Google Scholar] [CrossRef]
- James, A.R.; Schroeder, M.P.; Neumann, M.; Bastian, L.; Eckert, C.; Gökbuget, N.; Tanchez, J.O.; Schlee, C.; Isaakidis, K.; Schwartz, S.; et al. Long non-coding RNAs defining major subtypes of B cell precursor acute lymphoblastic leukemia. J. Hematol. Oncol. 2019, 12, 8. [Google Scholar] [CrossRef]
- Fleur-Lominy, S.S.; Evensen, N.A.; Bhatla, T.; Sethia, G.; Narang, S.; Choi, J.H.; Ma, X.; Yang, J.J.; Kelly, S.; Raetz, E.; et al. Evolution of the epigenetic landscape in childhood B Acute lymphoblastic leukemia and its role in drug resistance. Cancer Res. 2020, 80, 5189–5202. [Google Scholar] [CrossRef]
- Erarslan-Uysal, B.; Kunz, J.B.; Rausch, T.; Richter-Pechańska, P.; van Belzen, I.A.; Frismantas, V.; Bornhauser, B.; Ordoñez-Rueada, D.; Paulsen, M.; Benes, V.; et al. Chromatin accessibility landscape of pediatric T-lymphoblastic leukemia and human T-cell precursors. EMBO Mol. Med. 2020, 12, e12104. [Google Scholar] [CrossRef]
- Navrkalova, V.; Plevova, K.; Hynst, J.; Pal, K.; Mareckova, A.; Reigl, T.; Jelinkova, H.; Vrzalova, Z.; Stranska, K.; Pavlova, S.; et al. LYmphoid NeXt-Generation Sequencing (LYNX) Panel: A Comprehensive Capture-Based Sequencing Tool for the Analysis of Prognostic and Predictive Markers in Lymphoid Malignancies. J. Mol. Diagn. 2021, 23, 959–974. [Google Scholar] [CrossRef] [PubMed]
- Hrabovsky, S.; Vrzalova, Z.; Stika, J.; Jelinkova, H.; Jarosova, M.; Navrkalova, V.; Martenek, J.; Folber, F.; Salek, C.; Horacek, J.M.; et al. Genomic landscape of B-other acute lymphoblastic leukemia in an adult retrospective cohort with a focus on BCR-ABL1-like subtype. Acta Oncol. 2021, 60, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Alsuwaidi, L.; Hachim, M.; Senok, A. Novel Markers in Pediatric Acute Lymphoid Leukemia: The Role of ADAM6 in B Cell Leukemia. Front. Cell Dev. Biol. 2021, 9, 706129. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.P.; Gazdova, J.; Darzenta, N.; Wren, D.; Proszek, P.; Fazio, G.; Songia, S.; Alcoceba, M.; Sarasquete, M.E.; Villarese, P.; et al. Validation of the EuroClonality-NGS DNA capture panel as an integrated genomic tool for lymphoproliferative disorders. Blood Adv. 2021, 5, 3188–3198. [Google Scholar] [CrossRef]
- van der Velden, V.H.; Brüggemann, M.; Cazzaniga, G.; Scheijen, B.; Tops, B.; Trka, J.; Pal, K.; Hänzelmann, S.; Fazio, G.; Songia, S.; et al. Potential and pitfalls of whole transcriptome-based immunogenetic marker identification in acute lymphoblastic leukemia; a EuroMRD and EuroClonality-NGS Working Group study. Leukemia 2021, 35, 924–928. [Google Scholar] [CrossRef]
- Hetzel, S.; Mattei, A.L.; Kretzmer, H.; Qu, C.; Chen, X.; Fan, Y.; Wu, G.; Roberts, K.G.; Luger, S.; Litzow, M.; et al. Acute lymphoblastic leukemia displays a distinct highly methylated genome. Nat. Cancer 2022, 3, 768–782. [Google Scholar] [CrossRef]
- Chaber, R.; Gurgul, A.; Tabarkiewicz, J.; Wróbel, G.; Szmatoła, T.; Jasielczuk, I.; Haus, O.; Lejman, M.; Rybka, B.; Ryczan-Krawczyk, R.; et al. MicroRNA gene methylation landscape in pediatric B-cell precursor acute lymphoblastic leukemia. Adv. Clin. Exp. Med. 2022, 31, 293–305. [Google Scholar] [CrossRef]
- Longjohn, M.N.; Squires, W.R.B.; Christian, S.L. Meta-analysis of microRNA profiling data does not reveal a consensus signature for B cell acute lymphoblastic leukemia. Gene 2022, 821, 146211. [Google Scholar] [CrossRef]
- Barnett, K.R.; Mobley, R.J.; Diedrich, J.D.; Bergeron, B.P.; Bhattarai, K.R.; Yang, W.; Crews, K.R.; Manring, C.S.; Jabbour, E.; Paietta, E.; et al. Epigenomic mapping in B-cell acute lymphoblastic leukemia identifies transcriptional regulators and noncoding variants promoting distinct chromatin architectures. bioRxiv 2023, 3, 1–28. [Google Scholar] [CrossRef]
- Levy, B.; Kanagal-Shamanna, R.; Sahajpal, N.S.; Neveling, K.; Rack, K.; Dewaele, B.; Weghuis, D.O.; Stevens-Kroef, M.; Puiggros, A.; Mallo, M.; et al. A framework for the clinical implementation of optical genome mapping in hematologic malignancies. Am. J. Hematol. 2024, 99, 642–661. [Google Scholar] [CrossRef]
- Sahajpal, N.S.; Mondal, A.K.; Tvrdik, T.; Hauenstein, J.; Shi, H.; Deeb, K.K.; Saxe, D.; Hastie, A.R.; Chaubey, A.; Savage, N.M.; et al. Clinical Validation and Diagnostic Utility of Optical Genome Mapping for Enhanced Cytogenomic Analysis of Hematological Neoplasms. J. Mol. Diagn. 2022, 24, 1279–1291. [Google Scholar] [CrossRef]
- Gordeeva, V.; Sharova, E.; Arapidi, G. Progress in Methods for Copy Number Variation Profiling. Int. J. Mol. Sci. 2022, 23, 2143. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Fang, Q.; Mi, Y. Prognostic significance of copy number variation in B-cell acute lymphoblastic leukemia. Front. Oncol. 2022, 12, 981036. [Google Scholar] [CrossRef] [PubMed]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.D.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- MLL Münchner Leukämielabor GmbH. MLL ALL Classification. 2022. Available online: https://www.mll.com/en/acute-lymphoblastic-leukemia-all/acute-lymphoblastic-leukemia-all#:~:text=ALL%3A%20Classification,lymphoblastic%20lymphoma%20(Swerdlow%20et%20al (accessed on 18 March 2024).
- Duffield, A.S.; Mullighan, C.G.; Borowitz, M.J. International Consensus Classification of acute lymphoblastic leukemia/lymphoma. Virchows Archiv. 2023, 482, 11–26. [Google Scholar] [CrossRef]
- Nishiwaki, S.; Kim, J.H.; Ito, M.; Maeda, M.; Okuno, Y.; Koyama, D.; Ozawa, Y.; Gunji, M.; Osaki, M.; Kitamura, K.; et al. Multi-Lineage BCR-ABL Expression in Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia Is Associated With Improved Prognosis but No Specific Molecular Features. Front. Oncol. 2020, 10, 586567. [Google Scholar] [CrossRef]
- Rathana, K.; Clappier, E. Uncovering new layers of Ph+ ALL biology. Blood 2024, 143, 1322–1323. [Google Scholar] [CrossRef]
- Kim, J.C.; Chan-Seng-Yue, M.; Ge, S.; Zeng, A.G.X.; Ng, K.; Gan, O.I.; Garcia-Prat, L.; Flores-Figueroa, E.; Woo, T.; Zhang, A.X.W.; et al. Transcriptomic classes of BCR-ABL1 lymphoblastic leukemia. Nat. Genet. 2023, 55, 1186–1197. [Google Scholar] [CrossRef]
- Fedullo, A.L.; Messina, M.; Elia, L.; Piciocchi, A.; Gianfelici, V.; Lauretti, A.; Soddu, S.; Puzzolo, M.C.; Minotti, C.; Ferrara, F.; et al. Prognostic implications of additional genomic lesions in adult Philadelphia chromosomepositive acute lymphoblastic leukemia. Haematologica 2019, 104, 312–318. [Google Scholar] [CrossRef]
- Bastian, L.; Beder, T.; Barz, M.J.; Bendig, S.; Bartsch, L.; Walter, W.; Wolgast, N.; Brändl, B.; Rohrandt, C.; Hansen, B.-T.; et al. Developmental trajectories and cooperating genomic events define molecular subtypes of BCR::ABL1-positive ALL. Blood 2023, 143, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Roberts, K.G.; Jabbour, E.; Patel, K.; Eterovic, A.K.; Chen, K.; Zweidler-McKay, P.; Lu, X.; Fawcett, G.; Wang, S.A.; et al. Ph-like acute lymphoblastic leukemia: A high-risk subtype in adults. Blood 2017, 129, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.H.; Hunger, S.P. ABL-class fusion positive acute lymphoblastic leukemia: Can targeting ABL cure ALL? Haematologica 2020, 105, 1754–1757. [Google Scholar] [CrossRef]
- Den Boer, M.L.; Cario, G.; Moorman, A.V.; Boer, J.M.; de Groot-Kruseman, H.A.; Fiocco, M.; Escherich, G.; Imamura, T.; Yeoh, A.; Sutton, R.; et al. Outcomes of paediatric patients with B-cell acute lymphocytic leukaemia with ABL-class fusion in the pre-tyrosine-kinase inhibitor era: A multicentre, retrospective, cohort study. Lancet Haematol. 2021, 8, e55–e66. [Google Scholar] [CrossRef]
- Roberts, K.G.; Gu, Z.; Payne-Turner, D.; McCastlain, K.; Harvey, R.C.; Chen, I.-M.; Pei, D.; Iacobucci, I.; Valentine, M.; Pounds, S.B.; et al. High Frequency and Poor Outcome of Philadelphia Chromosome-Like Acute Lymphoblastic Leukemia in Adults. J. Clin. Oncol. 2017, 35, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Norvilas, R.; Dirse, V.; Semaskeviciene, R.; Mickeviciute, O.; Gineikiene, E.; Stoskus, M.; Vaitkeviciene, G.; Rascon, J.; Griskevicius, L. Low incidence of ABL-class and JAK-STAT signaling pathway alterations in uniformly treated pediatric and adult B-cell acute lymphoblastic leukemia patients using MRD risk-directed approach—A population-based study. BMC Cancer 2021, 21, 326. [Google Scholar] [CrossRef]
- Reshmi, S.C.; Harvey, R.C.; Roberts, K.G.; Stonerock, E.; Smith, A.; Jenkins, H.; Chen, I.-M.; Valentine, M.; Liu, Y.; Li, Y.; et al. Targetable kinase gene fusions in high-risk B-ALL: A study from the Children’s Oncology Group. Blood 2017, 129, 3352–3361. [Google Scholar] [CrossRef]
- Downes, C.E.; McClure, B.J.; McDougal, D.P.; Heatley, S.L.; Bruning, J.B.; Thomas, D.; Yeung, D.T.; White, D.L. JAK2 Alterations in Acute Lymphoblastic Leukemia: Molecular Insights for Superior Precision Medicine Strategies. Front. Cell Dev. Biol. 2022, 10, 942053. [Google Scholar] [CrossRef]
- Cario, G.; Leoni, V.; Conter, V.; Baruchel, A.; Schrappe, M.; Biondi, A. BCR-ABL1-like acute lymphoblastic leukemia in childhood and targeted therapy. Haematologica 2020, 105, 2200–2204. [Google Scholar] [CrossRef]
- Płotka, A.; Lewandowski, K. BCR/ABL1-Like Acute Lymphoblastic Leukemia: From Diagnostic Approaches to Molecularly Targeted Therapy. Acta Haematol. 2022, 145, 122–130. [Google Scholar] [CrossRef]
- Roberts, K.G.; Janke, L.J.; Zhao, Y.; Seth, A.; Ma, J.; Finkelstein, D.; Smith, S.; Ebata, K.; Tuch, B.B.; Hunger, S.P.; et al. Etv6-ntrk3 induces aggressive acute lymphoblastic leukemia highly sensitive to selective trk inhibition. Blood 2018, 132, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Malouf, C.; Ottersbach, K. Molecular processes involved in B cell acute lymphoblastic leukaemia. Cell. Mol. Life Sci. 2018, 75, 417–446. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Camponeschi, A.; Nordlund, J.; Marincevic-Zuniga, Y.; Abrahamsson, J.; Lönnerholm, G.; Fogelstrand, L.; Mårtensson, I. RAG1 co-expression signature identifies ETV6-RUNX1-like B-cell precursor acute lymphoblastic leukemia in children. Cancer Med. 2021, 10, 3997–4003. [Google Scholar] [CrossRef]
- Lilljebjörn, H.; Henningsson, R.; Hyrenius-Wittsten, A.; Olsson, L.; Orsmark-Pietras, C.; Von Palffy, S.; Askmyr, M.; Rissler, M.; Schrappe, M.; Cario, G.; et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 11790. [Google Scholar] [CrossRef] [PubMed]
- Zaliova, M.; Kotrova, M.; Bresolin, S.; Stuchly, J.; Stary, J.; Hrusak, O.; Kronnie, G.T.; Trka, J.; Zuna, J.; Vaskova, M. ETV6/RUNX1-like acute lymphoblastic leukemia: A novel B-cell precursor leukemia subtype associated with the CD27/CD44 immunophenotype. Genes. Chromosomes Cancer 2017, 56, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G. Genetics and prognosis of ALL in chlidren vs adults. Hematol. Am. Soc. Hematol. Educ. Program. 2018, 1, 137–145. [Google Scholar] [CrossRef]
- Ryan, S.L.; Peden, J.F.; Kingsbury, Z.; Schwab, C.J.; James, T.; Polonen, P.; Mijuskovic, M.; Becq, J.; Yim, R.; Cranston, R.E.; et al. Whole genome sequencing provides comprehensive genetic testing in childhood B-cell acute lymphoblastic leukaemia. Leukemia 2023, 37, 518–528. [Google Scholar] [CrossRef]
- Lee, S.H.R.; Li, Z.; Tai, S.T.; Oh, B.L.Z.; Yeoh, A.E.J. Genetic alterations in childhood acute lymphoblastic leukemia: Interactions with clinical features and treatment response. Cancers 2021, 13, 4068. [Google Scholar] [CrossRef]
- Jeha, S.; Choi, J.; Roberts, K.G.; Pei, D.; Coustan-Smith, E.; Inaba, H.; Rubnitz, J.E.; Ribeiro, R.C.; Gruber, T.A.; Raimondi, S.C.; et al. Clinical Significance of Novel Subtypes of Acute Lymphoblastic Leukemia in the Context of Minimal Residual Disease–Directed Therapy. Blood Cancer Discov. 2021, 2, 326–337. [Google Scholar] [CrossRef]
- Schmidt, B.M.; Brown, L.M.; Ryland, G.L.; Lonsdale, A.; Kosasih, H.J.; Ludlow, L.E.; Majewski, I.J.; Blombery, P.; Ekert, P.G.; Davidson, N.M.; et al. ALLSorts: An RNA-Seq subtype classifier for B-cell acute lymphoblastic leukemia. Blood Adv. 2022, 6, 4093–4097. [Google Scholar] [CrossRef]
- Novakova, M.; Zaliova, M.; Fiser, K.; Vakrmanova, B.; Slamova, L.; Musilova, A.; Brüggemann, M.; Ritgen, M.; Fronkova, E.; Kalina, T.; et al. DUX4r, ZNF384r and PAX5-P80R mutated B-cell precursor acute lymphoblastic leukemia frequently undergo monocytic switch. Haematologica 2021, 106, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, I.; Kimura, S.; Mullighan, C.G. Biologic and Therapeutic Implications of Genomic Alterations in Acute Lymphoblastic Leukemia. J. Clin. Med. 2021, 10, 3792. [Google Scholar] [CrossRef]
- Zaliova, M.; Winkowska, L.; Stuchly, J.; Fiser, K.; Triska, P.; Zwyrtkova, M.; Hrusak, O.; Starkova, J.; Sramkova, L.; Stary, J.; et al. A novel class of ZNF384 aberrations in acute leukemia. Blood Adv. 2021, 5, 4393–4397. [Google Scholar] [CrossRef]
- Russell, L.J.; Enshaei, A.; Jones, L.; Erhorn, A.; Masic, D.; Bentley, H.; Laczko, K.S.; Fielding, A.K.; Goldstone, A.H.; Goulden, N.; et al. IGH Translocations Are Prevalent in Teenagers and Young Adults With Acute Lymphoblastic Leukemia and Are Associated With a Poor Outcome. J. Clin. Oncol. 2014, 32, 1453–1462. [Google Scholar] [CrossRef]
- Roberts, K.G.; Mullighan, C.G. Genomics in acute lymphoblastic leukaemia: Insights and treatment implications. Nat. Rev. Clin. Oncol. 2015, 12, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Parolini, M.; Mecucci, C.; Matteucci, C.; Giussani, U.; Intermesoli, T.; Tosi, M.; Rambaldi, A.; Bassan, R. Highly aggressive T-cell acute lymphoblastic leukemia with t(8;14)(q24;q11): Extensive genetic characterization and achievement of early molecular remission and long-term survival in an adult patient. Blood Cancer J. 2014, 4, e176. [Google Scholar] [CrossRef]
- Bomken, S.; Enshaei, A.; Schwalbe, E.C.; Mikulasova, A.; Dai, Y.; Zaka, M.; Fung, K.T.; Bashton, M.; Lim, H.; Jones, L.; et al. Molecular characterization and clinical outcome of B-cell precursor acute lymphoblastic leukemia with IG-MYC rearrangement. Haematologica 2023, 108, 717–731. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Pan, S.; Xie, T.; Liu, H. MYC in T-cell acute lymphoblastic leukemia: Functional implications and targeted strategi. Blood Sci. 2021, 3, 65–70. [Google Scholar] [CrossRef]
- Bhojwani, D.; Yang, J.J.; Pui, C.H. Biology of childhood acute lymphoblastic leukemia. Pediatr. Clin. N. Am. 2015, 62, 47–60. [Google Scholar] [CrossRef]
- Schwab, C.J.; Murdy, D.; Butler, E.; Enshaei, A.; Winterman, E.; Cranston, R.E.; Ryan, S.; Barretta, E.; Hawking, Z.; Murray, J.; et al. Genetic characterisation of childhood B-other-acute lymphoblastic leukaemia in UK patients by fluorescence in situ hybridisation and Multiplex Ligation-dependent Probe Amplification. Br. J. Haematol. 2022, 196, 753–763. [Google Scholar] [CrossRef]
- Nigro, L.L. Biology of Childhood Acute Lymphoblastic Leukemia. J. Pediatr. Hematol. Oncol. 2013, 35, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.W.; Roberts, K.G.; Gu, Z.; Shi, L.; Pounds, S.; Pei, D.; Cheng, C.; Dai, Y.; Devidas, M.; Qu, C.; et al. The genomic landscape of pediatric acute lymphoblastic leukemia. Nat. Genet. 2022, 54, 1376–1389. [Google Scholar] [CrossRef] [PubMed]
- Herold, T.; Schneider, S.; Metzeler, K.H.; Neumann, M.; Hartmann, L.; Roberts, K.G.; Konstandin, N.P.; Greif, P.A.; Bräundl, K.; Ksienzyk, B.; et al. Adults with philadelphia chromosome–like acute lymphoblastic leukemia frequently have igh-CRLF2 and JAK2 mutations, persistence of minimal residual disease and poor prognosis. Haematologica 2017, 102, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.-L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable Kinase-Activating Lesions in Ph-like Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Ghazavi, F.; Lammens, T.; Van Roy, N.; Poppe, B.; Speleman, F.; Benoit, Y.; Van Vlierberghe, P.; De Moerloose, B. Molecular basis and clinical significance of genetic aberrations in B-cell precursor acute lymphoblastic leukemia. Exp. Hematol. 2015, 43, 640–653. [Google Scholar] [CrossRef]
- Kubota-Tanaka, M.; Osumi, T.; Miura, S.; Tsujimoto, H.; Imamura, T.; Nishimura, A.; Oki, K.; Nakamura, K.; Miyamoto, S.; Inoue, K.; et al. B-lymphoblastic lymphoma with TCF3-PBX1 fusion gene. Haematologica 2019, 104, e35–e37. [Google Scholar] [CrossRef]
- Rowsey, R.A.; Smoley, S.A.; Williamson, C.M.; Vasmatzis, G.; Smadbeck, J.B.; Ning, Y.; Greipp, P.T.; Hoppman, N.L.; Baughn, L.B.; Ketterling, R.P.; et al. Characterization of TCF3 rearrangements in pediatric B-lymphoblastic leukemia/lymphoma by mate-pair sequencing (MPseq) identifies complex genomic rearrangements and a novel TCF3/TEF gene fusion. Blood Cancer J. 2019, 9, 81. [Google Scholar] [CrossRef]
- Tirado, C.A.; Shabsovich, D.; Yeh, L.; Pullarkat, S.T.; Yang, L.; Kallen, M.; Rao, N. A (1;19) translocation involving TCF3-PBX1 fusion within the context of a hyperdiploid karyotype in adult B-ALL: A case report and review of the literature. Biomark. Res. 2015, 3, 4. [Google Scholar] [CrossRef]
- Lehnertz, B.; Chagraoui, J.; MacRae, T.; Tomellini, E.; Corneau, S.; Mayotte, N.; Boivin, I.; Durand, A.; Gracias, D.; Sauvageau, G. HLF expression defines the human hematopoietic stem cell state. Blood 2021, 138, 2642–2654. [Google Scholar] [CrossRef]
- Paulsson, K.; Jonson, T.; Øra, I.; Olofsson, T.; Panagopoulos, I.; Johansson, B. Characterisation of genomic translocation breakpoints and identification of an alternative TCF3/PBX1 fusion transcript in t(1;19)(q23;p13)-positive acute lymphoblastic leukaemias. Br. J. Haematol. 2007, 138, 196–201. [Google Scholar] [CrossRef]
- Lin, N.; Yan, X.; Cai, D.; Wang, L. Leukemia With TCF3-ZNF384 Rearrangement as a Distinct Subtype of Disease With Distinct Treatments: Perspectives From A Case Report and Literature Review. Front. Oncol. 2021, 11, 709036. [Google Scholar] [CrossRef] [PubMed]
- Fischer, U.; Forster, M.; Rinaldi, A.; Risch, T.; Sungalee, S.; Warnatz, H.-J.; Bornhauser, B.; Gombert, M.; Kratsch, C.; Stütz, A.M.; et al. Genomics and drug profiling of fatal TCF3-HLFâ ’positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat. Genet. 2015, 47, 1020–1029. [Google Scholar] [CrossRef]
- Takeda, R.; Yokoyama, K.; Ogawa, M.; Kawamata, T.; Fukuyama, T.; Kondoh, K.; Takei, T.; Nakamura, S.; Ito, M.; Yusa, N.; et al. The first case of elderly TCF3-HLF-positive B-cell acute lymphoblastic leukemia. Leuk Lymphoma 2019, 60, 2821–2824. [Google Scholar] [CrossRef]
- Lejman, M.; Włodarczyk, M.; Zawitkowska, J.; Kowalczyk, J.R. Comprehensive chromosomal aberrations in a case of a patient with TCF3-HLF-positive BCP-ALL. BMC Med. Genom. 2020, 13, 58. [Google Scholar] [CrossRef]
- Mouttet, B.; Vinti, L.; Ancliff, P.; Bodmer, N.; Brethon, B.; Cario, G.; Chen-Santel, C.; Elitzur, S.; Hazar, V.; Kunz, J.; et al. Durable remissions in TCF3-HLF positive acute lymphoblastic leukemia with blinatumomab and stem cell transplantation. Haematologica 2019, 104, e244–e247. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wan, X.; Yang, F.; Shi, W.; Liu, R.; Ding, L.; Tang, Y.; Luo, C.; Yang, X.; Ma, Y.; et al. Successful Treatment of TCF3-HLF–positive Childhood B-ALL with Chimeric Antigen Receptor T-Cell Therapy. Clin. Lymphoma Myeloma Leuk. 2021, 21, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lee, S.H.R.; Ni Chin, W.H.; Lu, Y.; Jiang, N.; Lim, E.H.H.; Coustan-Smith, E.; Chiew, K.H.; Oh, B.L.Z.; Koh, G.S.; et al. Distinct clinical characteristics of DUX4- And PAX5-altered childhood B-lymphoblastic leukemia. Blood Adv. 2021, 5, 5226–5238. [Google Scholar] [CrossRef]
- Paietta, E.; Roberts, K.G.; Wang, V.; Gu, Z.; Buck, G.A.N.; Pei, D.; Cheng, C.; Levine, R.L.; Abdel-Wahab, O.; Cheng, Z.; et al. Molecular classification improves risk assessment in adult BCR-ABL1–negative B-ALL. Blood 2021, 138, 948. [Google Scholar] [CrossRef]
- Yasuda, T.; Tsuzuki, S.; Kawazu, M.; Hayakawa, F.; Kojima, S.; Ueno, T.; Imoto, N.; Kohsaka, S.; Kunita, A.; Doi, K.; et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat. Genet. 2016, 48, 569–574. [Google Scholar] [CrossRef]
- Liu, Y.-F.; Wang, B.-Y.; Zhang, W.-N.; Huang, J.-Y.; Li, B.-S.; Zhang, M.; Jiang, L.; Li, J.-F.; Wang, M.-J.; Dai, Y.-J.; et al. Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. EBioMedicine 2016, 8, 173–183. [Google Scholar] [CrossRef]
- Zhang, J.; McCastlain, K.; Yoshihara, H.; Xu, B.; Chang, Y.; Churchman, M.L.; Wu, G.; Li, Y.; Wei, L.; Iacobucci, I.; et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat. Genet. 2016, 48, 1481–1489. [Google Scholar] [CrossRef]
- Lilljebjörn, H.; Fioretos, T. New oncogenic subtypes in pediatric B-cell precursor acute lymphoblastic leukemia. Blood 2017, 130, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, S.; Butler, E.; Ohki, K.; Kiyokawa, N.; Bergmann, A.K.; Boer, J.M.; Cavé, H.; Cazzaniga, G.; Yeoh, A.E.J.; Imamura, T.; et al. Acute Lymphoblastic Leukemia with Zinc-Finger Protein 384 (ZNF384)-Related Rearrangements: A Retrospective Analysis from the Ponte Di Legno Childhood ALL Working Group. Blood 2019, 134, 652. [Google Scholar] [CrossRef]
- Qin, Y.-Z.; Jiang, Q.; Xu, L.-P.; Wang, Y.; Jiang, H.; Dao, F.-T.; Chen, W.-M.; Zhao, X.-S.; Liu, Y.-R.; Zhang, X.-H.; et al. The Prognostic Significance of ZNF384 Fusions in Adult Ph-Negative B-Cell Precursor Acute Lymphoblastic Leukemia: A Comprehensive Cohort Study From a Single Chinese Center. Front. Oncol. 2021, 11, 632532. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Bai, W.; Cheng, Q.; Fang, J. ZNF384-Related Fusion Genes in Acute Lymphoblastic Leukemia. Cancer Control 2023, 30, 1–7. [Google Scholar] [CrossRef]
- Roberts, K.G.; Mullighan, C.G. The biology of B-progenitor acute lymphoblastic leukemia. Cold Spring Harb. Perspect. Med. 2020, 10, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Shinsuke, H.; Kentaro, O.; Kazuhiko, N.; Hitoshi, I.; Yukihide, M.; Kohji, O.; Akinori, Y.; Kazuki, T.; Yuya, S.; Ai, Y.; et al. ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica 2017, 102, 118–129. [Google Scholar] [CrossRef]
- Heavey, B.; Charalambous, C.; Cobaleda, C.; Busslinger, M. Myeloid lineage switch of Pax5 mutant but not wild-type B cell progenitors by C/EBPα and GATA factors. EMBO J. 2003, 22, 3887–3897. [Google Scholar] [CrossRef]
- Kansal, R. Diagnosis and Molecular Pathology of Lymphoblastic Leukemias and Lymphomas in the Era of Genomics and Precision Medicine: Historical Evolution and Current Concepts—Part 2: B-/T-Cell Acute Lymphoblastic Leukemias. Lymphatics 2023, 1, 118–154. [Google Scholar] [CrossRef]
- Qian, M.; Zhang, H.; Kham, S.K.-Y.; Liu, S.; Jiang, C.; Zhao, X.; Lu, Y.; Goodings, C.; Lin, T.-N.; Zhang, R.; et al. Whole-transcriptome sequencing identifies a distinct subtype of acute lymphoblastic leukemia with predominant genomic abnormalities of EP300 and CREBBP. Genome Res. 2017, 27, 185–195. [Google Scholar] [CrossRef]
- Ohki, K.; Kiyokawa, N.; Saito, Y.; Hirabayashi, S.; Nakabayashi, K.; Ichikawa, H.; Momozawa, Y.; Okamura, K.; Yoshimi, A.; Ogata-Kawata, H.; et al. Clinical and molecular characteristics of MEF2D fusion-positive B-cell precursor acute lymphoblastic leukemia in childhood, including a novel translocation resulting in MEF2D-HNRNPH1 gene fusion. Haematologica 2019, 104, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Churchman, M.; Roberts, K.; Li, Y.; Liu, Y.; Harvey, R.C.; McCastlain, K.; Reshmi, S.C.; Payne-Turner, D.; Iacobucci, I.; et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 13331. [Google Scholar] [CrossRef] [PubMed]
- Ohki, K.; Butler, E.R.; Kiyokawa, N.; Hirabayashi, S.; Bergmann, A.K.; Möricke, A.; Boer, J.M.; Cavé, H.; Cazzaniga, G.; Yeoh, A.E.J.; et al. Clinical characteristics and outcomes of B-cell precursor ALL with MEF2D rearrangements: A retrospective study by the Ponte di Legno Childhood ALL Working Group. Leukemia 2023, 37, 212–216. [Google Scholar] [CrossRef]
- Leongamornlert, D.; Gutiérrez-Abril, J.; Lee, S.; Barretta, E.; Creasey, T.; Gundem, G.; Levine, M.F.; Arango-Ossa, J.E.; Liosis, K.; Medina-Martinez, J.S.; et al. Diagnostic utility of whole genome sequencing in adults with B-other acute lymphoblastic leukemia. Blood Adv. 2023, 7, 3862–3873. [Google Scholar] [CrossRef]
- Van Outersterp, I.; Hormann, F.M.; Hoogkamer, A.Q.; Boeree, A.; Broek, S.A.V.D.; Boer, M.L.D.; Boer, J.M. Characterization of a novel MEF2D-BCL9 fusion-positive acute lymphoblastic leukemia cell line. Haematologica 2023, 108, 2859–2864. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, H.; Li, Z.; Bai, L.; Wang, Q.; Li, J.; Jiang, M.; Xue, Q.; Cheng, N.; Zhang, W.; et al. Functional, structural, and molecular characterizations of the leukemogenic driver MEF2D-HNRNPUL1 fusion. Blood 2022, 140, 1390–1407. [Google Scholar] [CrossRef] [PubMed]
- Boer, J.M.; Valsecchi, M.G.; Hormann, F.M.; Antić, Ž.; Zaliova, M.; Schwab, C.; Cazzaniga, G.; Arfeuille, C.; Cavé, H.; Attarbaschi, A.; et al. Favorable outcome of NUTM1-rearranged infant and pediatric B cell precursor acute lymphoblastic leukemia in a collaborative international study. Leukemia 2021, 35, 2978–2982. [Google Scholar] [CrossRef]
- Hormann, F.M.; Hoogkamer, A.Q.; Beverloo, H.B.; Boeree, A.; Dingjan, I.; Wattel, M.M.; Stam, R.W.; Escherich, G.; Pieters, R.; den Boer, M.L.; et al. NUTM1 is a recurrent fusion gene partner in B-cell precursor acute lymphoblastic leukemia associated with increased expression of genes on chromosome band 10p12.31-12.2. Haematologica 2019, 104, 455–5459. [Google Scholar] [CrossRef]
- Charlab, R.; Racz, R. The expanding universe of NUTM1 fusions in pediatric cancer. Clin. Transl. Sci. 2023, 16, 1331–1339. [Google Scholar] [CrossRef]
- Boer, J.; Valsecchi, M.; Hormann, F.; Antic, Z.; Zaliova, M.; Schwab, C.; Cazzaniga, G.; Arfeuille, C.; Cavé, H.; Attarbaschi, A.; et al. NUTM1-Rearranged Infant and Pediatric B Cell Precursor Acute Lymphoblastic Leukemia: A Good Prognostic Subtype Identified in a Collaborative International Study. Blood 2020, 136, 25–26. [Google Scholar] [CrossRef]
- McClure, B.J.; Pal, M.; Heatley, S.L.; Rehn, J.; Schutz, C.; Breen, J.; Venn, N.C.; Sutton, R.; Khaw, S.L.; Yeung, D.T.; et al. Two novel cases of NUTM1-rearranged B-cell acute lymphoblastic leukaemia presenting with high-risk features. Br. J. Haematol. 2022, 196, 1407–1411. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Montefiori, L.; Iacobucci, I.; Zhao, Y.; Gao, Q.; Paietta, E.M.; Haferlach, C.; Laird, A.D.; Mead, P.E.; Gu, Z.; et al. Enhancer retargeting of CDX2 and UBTF::ATXN7L3 define a subtype of high-risk B-progenitor acute lymphoblastic leukemia. Blood 2022, 136, 3519–3531. [Google Scholar] [CrossRef] [PubMed]
- Passet, M.; Kim, R.; Gachet, S.; Sigaux, F.; Chaumeil, J.; Galland, A.; Sexton, T.; Quentin, S.; Hernandez, L.; Larcher, L.; et al. Concurrent CDX2 cis-deregulation and UBTF::ATXN7L3 fusion define a novel high-risk subtype of B-cell ALL. Blood 2022, 136, 3505–3518. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Safavi, S.; Woodward, E.L.; Duployez, N.; Olsson-Arvidsson, L.; Ungerbäck, J.; Sigvardsson, M.; Zaliova, M.; Zuna, J.; Fioretos, T.; et al. 13q12.2 deletions in acute lymphoblastic leukemia lead to upregulation of FLT3 through enhancer hijacking. Blood 2020, 136, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Bastian, L.; Hartmann, A.M.; Beder, T.; Hänzelmann, S.; Kässens, J.; Bultmann, M.; Hoeppner, M.P.; Franzenburg, S.; Wittig, M.; Franke, A.; et al. UBTF::ATXN7L3 gene fusion defines novel B cell precursor ALL subtype with CDX2 expression and need for intensified treatment. Leukemia 2022, 36, 1676–1680. [Google Scholar] [CrossRef]
- Galland, A.; Gourain, V.; Habbas, K.; Güler, Y.; Martin, E.; Ebel, C.; Tavian, M.; Vallat, L.; Chenard, M.; Mauvieux, L.; et al. CDX2 expression in the hematopoietic lineage promotes leukemogenesis via TGFβ inhibition. Mol. Oncol. 2021, 15, 2318–2329. [Google Scholar] [CrossRef]
- Darvishi, M.; Mashati, P.; Khosravi, A. The clinical significance of CDX2 in leukemia: A new perspective for leukemia research. Leuk. Res. 2018, 72, 45–51. [Google Scholar] [CrossRef]
- Jung, M.; Schieck, M.; Hofmann, W.; Tauscher, M.; Lentes, J.; Bergmann, A.; Stelter, M.; Möricke, A.; Alten, J.; Schlegelberger, B.; et al. Frequency and prognostic impact of PAX5 p.P80R in pediatric acute lymphoblastic leukemia patients treated on an AIEOP-BFM acute lymphoblastic leukemia protocol. Genes. Chromosomes Cancer 2020, 59, 667–671. [Google Scholar] [CrossRef]
- Jia, Z.; Gu, Z. PAX5 alterations in B-cell acute lymphoblastic leukemia. Front. Oncol. 2022, 12, 1023606. [Google Scholar] [CrossRef]
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 2019, 51, 296–307. [Google Scholar] [CrossRef]
- Bastian, L.; Schroeder, M.P.; Eckert, C.; Schlee, C.; Tanchez, J.O.; Kämpf, S.; Wagner, D.L.; Schulze, V.; Isaakidis, K.; Lázaro-Navarro, J.; et al. PAX5 biallelic genomic alterations define a novel subgroup of B-cell precursor acute lymphoblastic leukemia. Leukemia 2019, 33, 1895–1909. [Google Scholar] [CrossRef]
- Østergaard, A.; Boer, J.M.; van Leeuwen, F.N.; Pieters, R.; Den Boer, M.L. IKZF1 in acute lymphoblastic leukemia: The rise before the fall? Leuk. Lymphoma 2024, 29, 2396046. [Google Scholar] [CrossRef] [PubMed]
- Stanulla, M.; Cavé, E.E.; Cavé, C.; Moorman, A.V. IKZF1 deletions in pediatric acute lymphoblastic leukemia: Still a poor prognostic marker? Blood 2020, 135, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.K.; Gogakos, T.; Lip, V.; Tsai, J.; Li, Y.D.; Fisch, A.; Weiss, J.; Grimmett, L.; Tran, T.H.; Caron, M.; et al. Outlier expression of isoforms by targeted RNA sequencing as clinical markers of genomic variants in B lymphoblastic leukemia and other tumor types. medRxiv 2022. [Google Scholar] [CrossRef]
- Fazio, G.; Biondi, A.; Cazzaniga, G. The Role of PAX5 in ALL. Nov. Asp. Acute Lymphoblstic Leuk. 2011, 1, 211–234. [Google Scholar]
- Fazio, G.; Bresolin, S.; Silvestri, D.; Quadri, M.; Saitta, C.; Vendramini, E.; Buldini, B.; Palmi, C.; Bardini, M.; Grioni, A.; et al. PAX5 fusion genes are frequent in poor-risk childhood acute lymphoblastic leukaemia and can be targeted with BIBF1120. EBioMedicine 2022, 83, 104224. [Google Scholar] [CrossRef]
- Schwab, C.; Nebral, K.; Chilton, L.; Leschi, C.; Waanders, E.; Boer, J.M.; Žaliová, M.; Sutton, R.; Öfverholm, I.I.; Ohki, K.; et al. Intragenic amplification of PAX5: A novel subgroup in B-cell precursor acute lymphoblastic leukemia? Blood Adv. 2017, 1, 1473–1477. [Google Scholar] [CrossRef]
- Jean, J.; Kovach, A.E.; Doan, A.; Oberley, M.J.; Ji, J.; Schmidt, R.J.; Biegel, J.A.; Bhojwani, D.; Raca, G. Characterization of PAX5 intragenic tandem multiplication in pediatric B-lymphoblastic leukemia by optical genome mapping. Blood Adv. 2022, 6, 3343–3346. [Google Scholar] [CrossRef]
- He, L.; Li, Z.; Fan, X.; Chen, J.; Wu, H.; Fu, Y. ‘Double hit’ B-lymphoblastic lymphoma with concurrent IGH/BCL2 and 8q24/MYC translocations: A case report. Transl. Cancer Res. 2021, 10, 1594–1598. [Google Scholar] [CrossRef]
- Genescà, E.; la Starza, R. Early T-Cell Precursor ALL and Beyond: Immature and Ambiguous Lineage T-ALL Subsets. Cancers 2022, 14, 1873. [Google Scholar] [CrossRef]
- Coustan-Smith, E.; Mullighan, C.G.; Onciu, M.; Behm, F.G.; Raimondi, S.C.; Pei, D.; Cheng, C.; Su, X.; Rubnitz, J.E.; Basso, G.; et al. Early T-cell precursor leukaemia: A subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009, 10, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Lamb, A.V.; O’brien, S.; Ravandi, F.; Konopleva, M.; Jabbour, E.; Zuo, Z.; Jorgensen, J.; Lin, P.; Pierce, S.; et al. Early T-cell precursor acute lymphoblastic leukemia/lymphoma (ETP-ALL/LBL) in adolescents and adults: A high-risk subtype. Blood 2016, 127, 1863–1869. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Nayernama, A.; Jones, S.C.; de Claro, R.A.; Waldron, P.E. A novel germline variant in PIK3R1 results in SHORT syndrome associated withTAL/LMO T-cell acute lymphoblastic leukemia. Am. J. Hematol. 2020, 95, E335–E336. [Google Scholar] [CrossRef]
- Bardelli, V.; Arniani, S.; Pierini, V.; Di Giacomo, D.; Pierini, T.; Gorello, P.; Mecucci, C.; La Starza, R. T-cell acute lymphoblastic leukemia: Biomarkers and their clinical usefulness. Genes 2021, 12, 1118. [Google Scholar] [CrossRef]
- Tasian, S.K.; Hunger, S.P. Genomic characterization of paediatric acute lymphoblastic leukaemia: An opportunity for precision medicine therapeutics. Br. J. Haematol. 2017, 176, 867–882. [Google Scholar] [CrossRef]
- Mroczek, A.; Zawitkowska, J.; Kowalczyk, J.; Lejman, M. Comprehensive overview of gene rearrangements in childhood T-cell acute lymphoblastic leukaemia. Int. J. Mol. Sci. 2021, 22, 808. [Google Scholar] [CrossRef]
- Papenhausen, P.; Kelly, C.A.; Zhang, Z.; Tepperberg, J.; Burnside, R.D.; Schwartz, S. Multidisciplinary analysis of pediatric T-ALL: 9q34 gene fusions. Cancer Genet. 2019, 1, 231–232. [Google Scholar] [CrossRef]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef]
- Mansur, M.B.; Furness, C.L.; Nakjang, S.; Enshaei, A.; Alpar, D.; Colman, S.M.; Minto, L.; Irving, J.; Poole, B.V.; Noronha, E.P.; et al. The genomic landscape of teenage and young adult T-cell acute lymphoblastic leukemia. Cancer Med. 2021, 10, 4864–4873. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, Y.; Huang, X.; Zhang, Y.; Qian, J.; Li, J.; Li, X.; Li, C.; Lou, Y.; Mai, W.; et al. CDKN2A deletions are associated with poor outcomes in 101 adults with T-cell acute lymphoblastic leukemia. Am. J. Hematol. 2021, 96, 312–319. [Google Scholar] [CrossRef] [PubMed]
- De Bie, J.; Quessada, J.; Tueur, G.; Lefebvre, C.; Luquet, I.; Toujani, S.; Cuccuini, W.; Lafage-Pochitaloff, M.; Michaux, L. Cytogenetics in the management of T-cell acute lymphoblastic leukemia (T-ALL): Guidelines from the Groupe Francophone de Cytogénétique Hématologique (GFCH). Curr. Res. Transl. Med. 2023, 71, 103431. [Google Scholar] [CrossRef]
- Coustan-Smith, E.; Conter, V. Closing the circle for ETP ALL. Blood 2023, 142, 2039–2040. [Google Scholar] [CrossRef] [PubMed]
- Sin, C.F.; Man, P.H.M. Early T-Cell Precursor Acute Lymphoblastic Leukemia: Diagnosis, Updates in Molecular Pathogenesis, Management, and Novel Therapies. Front. Oncol. 2021, 11, 750789. [Google Scholar] [CrossRef]
- Li, Z.; Song, Y.; Zhang, Y.; Li, C.; Wang, Y.; Xue, W.; Lu, L.; Jin, M.; Zhou, Z.; Wang, X.; et al. Genomic and outcome analysis of adult T-cell lymphoblastic lymphoma. Haematologica 2020, 105, E107–E110. [Google Scholar] [CrossRef]
- Khabirova, E.; Jardine, L.; Coorens, T.H.H.; Webb, S.; Treger, T.D.; Engelbert, J.; Porter, T.; Prigmore, E.; Collord, G.; Piapi, A.; et al. Single-cell transcriptomics reveals a distinct developmental state of KMT2A-rearranged infant B-cell acute lymphoblastic leukemia. Nat. Med. 2022, 28, 743–751. [Google Scholar] [CrossRef]
- Iacobucci, I.; Witkowski, M.T.; Mullighan, C.G. Single-cell analysis of acute lymphoblastic and linage-ambiguous leukemia: Approaches and molecular insights. Blood 2023, 141, 356–368. [Google Scholar] [CrossRef]
- Zhang, J.; Zeng, L.; Wang, Y.; Pan, J.; Li, X.; Feng, B.; Yang, Q. Gene Mutations Related to Glucocorticoid Resistance in Pediatric Acute Lymphoblastic Leukemia. Front. Pediatr. 2022, 10, 831229. [Google Scholar] [CrossRef] [PubMed]
- Waanders, E.; Gu, Z.; Dobson, S.M.; Antić, Ž.; Crawford, J.C.; Ma, X.; Edmonson, M.N.; Payne-Turner, D.; van de Vorst, M.; Jongmans, M.C.; et al. Mutational Landscape and Patterns of Clonal Evolution in Relapsed Pediatric Acute Lymphoblastic Leukemia. Blood Cancer Discov. 2020, 1, 96–111. [Google Scholar] [CrossRef]
- van der Ham, C.G.; Suurenbroek, L.C.; Kleisman, M.M.; Antić, Ž.; Lelieveld, S.H.; Yeong, M.; Westera, L.; Sonneveld, E.; Hoogerbrugge, P.M.; van der Velden, V.H.J.; et al. Mutational mechanisms in multiply relapsed pediatric acute lymphoblastic leukemia. Leukemia 2024, 38, 2366–2375. [Google Scholar] [CrossRef]
- Ching, T.; Duncan, M.E.; Newman-Eerkes, T.; McWhorter, M.M.E.; Tracy, J.M.; Steen, M.S.; Brown, R.P.; Venkatasubbarao, S.; Akers, N.K.; Vignali, M.; et al. Analytical evaluation of the clonoSEQ Assay for establishing measurable (minimal) residual disease in acute lymphoblastic leukemia, chronic lymphocytic leukemia, and multiple myeloma. BMC Cancer 2020, 20, 612. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.V.; Such, E.; Sargas, C.; Simarro, J.; Miralles, A.; Pérez, G.; de Juan, I.; Palanca, S.; Avetisyan, G.; Santiago, M.; et al. Design and Validation of a Custom Next-Generation Sequencing Panel in Pediatric Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2023, 24, 4440. [Google Scholar] [CrossRef] [PubMed]
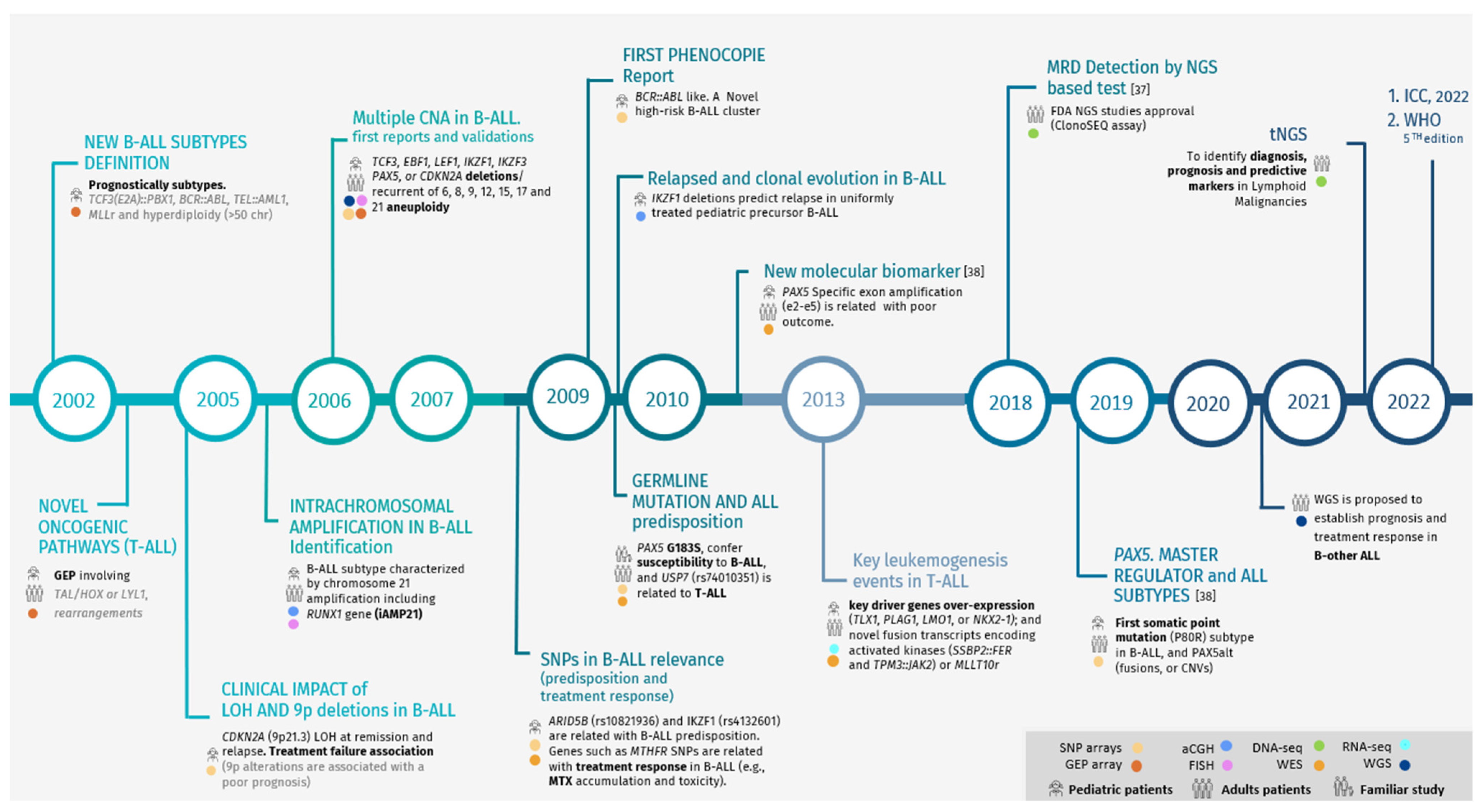
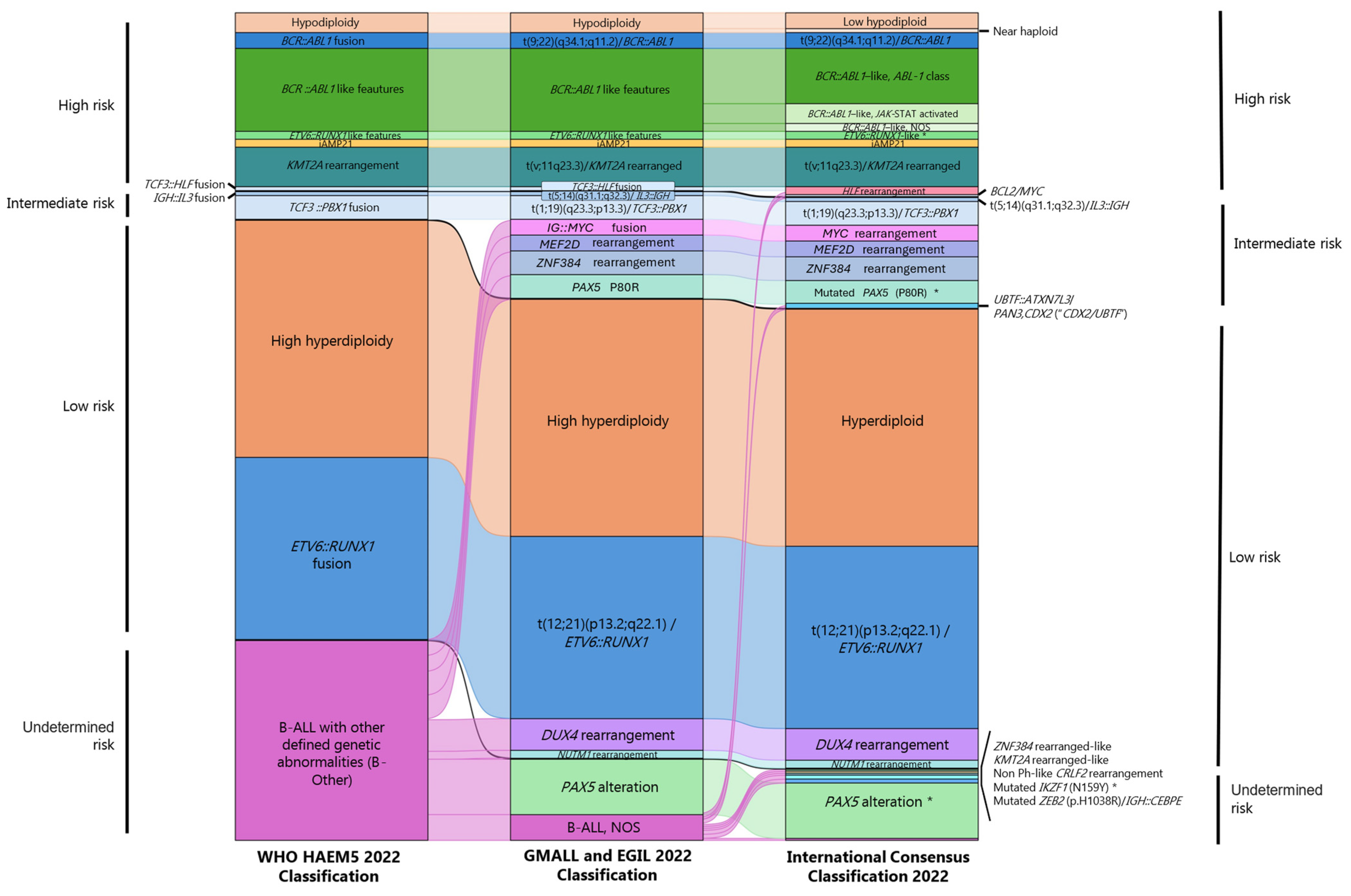
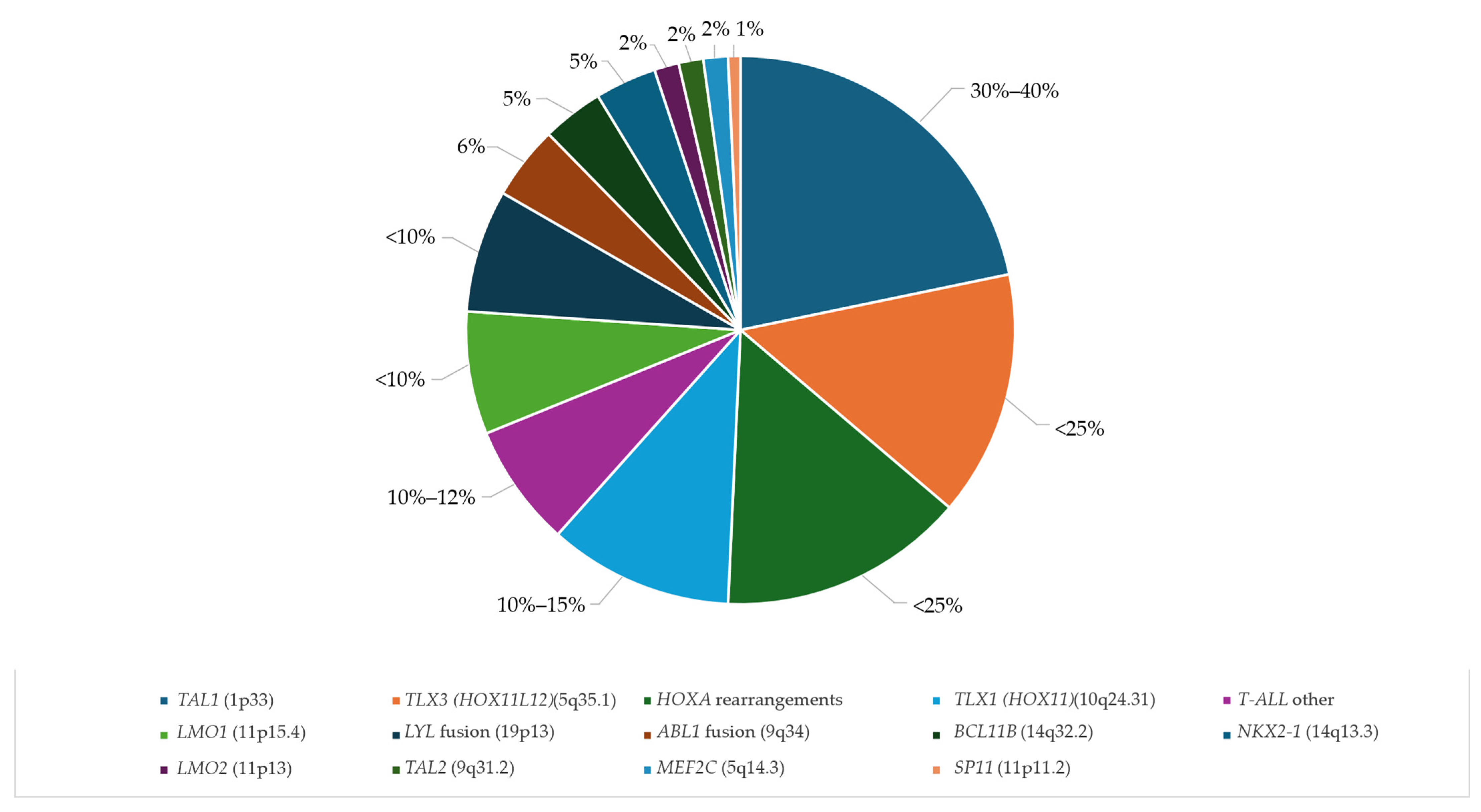

{kind=link}
{kind=link}
{kind=link}
| Year | NGS | Sample Size | Application | Main Findings |
|---|---|---|---|---|
| 2017 | WES | 1000 cases (B-ALL and T-ALL) | Therapeutic targets Identification | Clinical and molecular data at diagnosis and remission of ALL patients, including MRD are currently being collected. They will include the identification of somatic or germline variants associated with response to treatment, assessment of diversity, clonal evolution, etc. This study proposes to use NGS in diagnosis alongside morphological, immunophenotyping, and other molecular approaches, including scNGS [35]. |
| 2018 | RNAseq | 1223 B-ALL samples | Characterization of molecular subtypes for diagnostic accuracy and risk estimation | This study proposes fourteen molecular subtypes based on transcriptional profiling, including six previously undescribed subtypes: (i) PAX5 or CRLF2 fusions, (ii) PAX5 mutation (p.P80R), (iii) IKZF1 mutation (p.N159Y), (iv) ZEB2 mutation (p.H1038R) or IGH/CEBPE fusion, (v) HLF rearrangements, and (vi) NUTM1 rearrangements [36]. |
| 2019 | RNAseq | 170 B-ALL samples | Biomarker identification | Report on the first oncogenic subtype defined by a point mutation in ALL (PAX5 P80R) that may harbor a second PAX5 allele inactivation, biallelic CDKN2A deletion, and RAS signaling alterations [37]. |
| 2019 | tNGS | 17 childhood B-ALL samples | Translational research for diagnosis and monitoring of ALL patients | Matched diagnostic, remission, and relapse samples allowed for the detection of “potentially actionable gene variants” such as CDKN2A/B deletions and KRAS, ABL1, FLT3, and NOTCH mutations, as well as the detection of germline alterations such as TP53 R248Q [38]. |
| 2019 | RNA-seq | 40 diagnoses and 42 relapsed BCP-ALL samples | Insights into epigenomic changes | At diagnosis, DUX4-specific lncRNAs were associated with the TGF-beta and p53 signaling pathways. For the Ph-like subtype, lncRNAs were correlated with genes involved in the JAK-STAT and PIK3/AKT/mTOR signaling pathways. For all subtypes, lncRNAs were found to be differentially expressed at diagnosis and relapse. About 80% of the lncRNAs were downregulated at relapse, e.g., TCL6 in the DUX4r subtype, and, LINC00312 and miR-17-92a-1 in the DUX4r and Ph-like subtypes [39]. |
| 2020 | WGS, ChIP-seq RNA-seq RRBS-Seq | 34 B-ALL Samples | Epigenome in clonal evolution and treatment failure | This study provides the first reports of the role of epigenetic mechanisms in driving clonal evolution in B-ALL. Changes in gene expression were directly correlated with epigenetic alterations with changes in H3K27ac, and PRC2 (Polycomb complex) hypermethylation was detected at relapse in many patients [40]. |
| 2020 | ATAC-Seq RNA-Seq | Primary cells of T-ALL patients and patient-derived xenograft models | Insights into epigenomic changes | Two distinct groups of T-ALL were identified based on accessible transcription factor (TF)-binding-motifs profiles. The first group was characterized by highly accessible SPI1-binding sites, and the second lacked hyper-accessible SPI1-binding sites and DAB1 expression [41]. |
| 2021 | RNAseq tNGS | 58 AYAs and adult B-ALL samples | ALL diagnosis and stratification | This study introduces the first custom panel (LYmphoid Next-Generation Sequencing-LYNX Panel) as a versatile tool for the simultaneous analysis of genomic markers (mutations, indels, chromosomal aberrations, and IG/TR rearrangements) in most common lymphoid neoplasms (70 lymphoma-related genes) with a sensitivity of 5% variant allele frequency, as well as reliable identification of large genome-wide (≥6 Mb) and recurrent chromosomal aberrations (≥300 kb). This panel allowed for the description of novel variants classified as pathogenic or probable pathogenic in TP53, NRAS, JAK2, PAX5, CREBBP, NF1, FLT3, ATM, KRAS, and RUNX1 genes, as well as IKZF1plus profile identification [42,43]. |
| 2021 | in silico analysis | 1978 samples of childhood B-ALL | Biomarker identification and prognostic association | ADAM6 may be a novel genetic biomarker. Homozygous ADAM6 deletion was detected around 60% of cases and was associated with higher total leukocyte count, higher first relapse rate, MRD-positive status, and higher mutation variants. It was more common in the TCF3::PBX1 and ETV6::RUNX1 subtypes [44]. |
| 2021 | DNA-seq ddPCR | 376 LPDs and 14 LPD cell lines | Technique Performance/Translational research | Assays were designed to capture the diversity and functional variation of joining genes across 1243 regions included in a tNGS panel. Sensitivity for detecting IG/TCR clonality ranged from 96.5 to 97.3%. The detection of CNAs was dependent on the locus, with high sensitivities being reported for trisomy 12 and del(11q) and lower sensitivities for del(17p) and del(13q) [45]. |
| 2021 | DNA-seq RNA-seq | 165 ALL patients at diagnosis | Technique performance/translational research | EuroClonality-NGS captures validation as a tNGS assay for SNVs, CNVs, Indels, Translocations, and IGH and TRC rearrangements. Less than half of the markers were also found in the RNAseq data (44%). The assay performance for somatic mutations was 100% reproducible for both sensitivity and specificity at >4% VAF [46]. |
| 2022 | WGBS WGS WES | 82 B-ALL patients | Insights into the epigenome | Local hypermethylation was observed in the global DNA methylation landscape of ALL and, more specifically, in T-ALL patients [47]. |
| 2022 | Methyla-tionEPIC BeadChip Arrays | 38 children with B-ALL pediatrics cases and controls | Methylation profile and microRNA changes landscape | Among specific genetic subtypes (ETV6::RUNX1, TCF3::PBX1, IGH rearrangement, hyperdiploidy), miRNAs with differentially methylated sites were found. The results showed that MIR326, MIR200c, MIR125B, MIR203, and MIR181A were significantly differentially expressed in B-ALL cases compared with healthy controls. This study was complemented by meta-analysis and in silico analysis to identify differentially expressed miRNAs in B-ALL. Although no consensus dataset was found among the studies, miR-181b, miR-181a, miR-128, miR-128a, miR-181c, miR-155, miR-142-3p, and miR-451 were postulated as biomarkers associated with B-ALL [48,49]. |
| 2023 | ATAC-Seq ChIP-Seq Hi-C | 154 B-ALL | Insights into epigenomic changes | This was the first large-scale analysis of chromatin accessibility in the B-ALL genome across extensive B-ALL subtypes. The percentage of differentially accessible chromatin sites (DAS) was associated with each molecular subtype. It was higher in the KMT2Ar, TCF3-PBX1 fusion, and ZNF384r subtypes. Analysis of bound TF motif footprint prevalence identified several ETS family TFs (EHF, ELF3, SPI1, and SPIB), zinc finger TFs (ZNF263, ZNF460, ZNF740, and ZNF148), and CTCF as the most altered motifs leading to differences in chromatin accessibility [50]. Subtypes: BCR::ABL1, DUX4r, ETV6::RUNX1, high hyperdiploidy, low hypodiploidy, KMT2Ar, Ph-like, PAX5alt, TCF3::PBX1, ZNF384r, B-other samples, and B-ALL cell lines (697, BALL1, Nalm6, REH, RS411, SEM, and SUPB15) |
| Method | Variants Detected | Genetic Subtypes | Limitations |
|---|---|---|---|
| WGS | Point mutations, aneuploidy, SV, CNVs, and BCR/TCR rearrangements. Useful for detecting germinal variants and polymorphism related to predisposition and treatment response, as well as GWAS | B-ALL: HeH, Hypodiploidy, BCR::ABL1, ETV6::RUNX1; TCF3::PBX1, KMT2Ar, DUX4r; MEF2Dr; ZNF384r, NUTM1r; HLFr, BCL2::MYC; PAX5 (P80R); IKZF1 (N159Y); other related CNVs. | 1. A considerable number of unknown significance or likely pathogenic status 2. Currently, this is considered a high-cost method based on sequencing-platform acquisition, data storage, and analysis. |
| T-ALL: BCL11B; TAL/LMO rearrangements, HOXA; SPI1; NKX2-1; TAL1 mutations | |||
| WES | Point mutations, SV, CNVs, and aneuploidies. Useful for detecting novel fusions | B-ALL: PAX5 (P80R), IKZF1 (N159Y), and mutations in the Ph-like group (ABL1, JAK) | 1. Detection of phenocopies 2. Limitation of the analysis to the detection of variants in coding regions 3. Less coverage (less sensitive) |
| Target Sequencing (DNA/RNA) | Point mutations, aneuploidy, SV (InDels and gene fusions), and CNVs. Useful for increasing sensitivity (greater depth of coverage compared with WGS studies) for the detection of variants with low VAF; as well as variants of intron- and splicing-related regions variants | Defined target variants, such as BCR::ABL1, ETV6::RUNX1; TCF3::PBX1, BCL2::MYC; intrachromosomal amplifications (iAMP21, PAX5AMP), and other variants, such as CNVs (IKZF1, CDKN2A/2B, BTG1, PAX5); or mutations including PAX5 (P80R); or IKZF1 (N159Y) | Non-targeted alterations |
| WTS | Point mutations, CNVs, gene fusion, GEP, alternative splicing analysis, and BCR/TCR rearrangements | Subtypes defined by distinct gene expression profiles (phenocopies). Target variants such as BCR::ABL1, ETV6::RUNX1, TCF3::PBX1, and BCL2::MYC, novel fusions, PAX5 alteration and others | Ploidy alterations and NOS T-ALL or B-other ALL |
| T-ALL: SET::NUP214, PICALM::MLLT10, NUP98::RAP1GDS1, TAL/LMO rearrangements | |||
| OGM * | SV, aneuploidies, CNV, and balanced shifts in position | B-ALL: HeH, hypodiploidy, BCR-ABL1, ETV6::RUNX1; TCF3::PBX1, KMT2Ar, DUX4r, MEF2Dr; ZNF384r, NUTM1r, HLFr, BCL2::MYC; and other novel fusions | 1. Point mutation detection 2. Only fresh or preserved samples that guarantee adequate uHMW DNA integrity can be used. |
| T-ALL: TAL/LMO rearrangements KMT2A::PRDM10 and other novel fusions |
| WHO-HAEM5, 2022 Classification [55] | MLL, GMALL, and EGIL, 2022 Classification [57] | International Consensus Classification (ICC), 2022 [58] |
|---|---|---|
| B-acute lymphoblastic leukemia (B-ALL) | ||
| Aneuploidies | ||
| High hyperdiploidy | High hyperdiploidy | Hyperdiploidy |
| Hypodiploidy | Hypodiploidy | Low hypodiploidy |
| Near haploid | ||
| Gene fusion | ||
| BCR::ABL1 fusion | t(9;22)(q34.1;q11.2)/BCR::ABL1 | t(9;22)(q34.1;q11.2)/BCR::ABL1 |
| with lymphoid only involvement | ||
| with multilineage involvement | ||
| ETV6::RUNX1 fusion | t(12;21)(p13.2;q22.1)/ETV6::RUNX1 | t(12;21)(p13.2;q22.1)/ETV6::RUNX1 |
| IGH::IL3 fusion | t(5;14)(q31.1;q32.3)/IL3::IGH | t(5;14)(q31.1;q32.3)/IL3::IGH |
| TCF3::PBX1 fusion | t(1;19)(q23.3;p13.3)/TCF3::PBX1 | t(1;19)(q23.3;p13.3)/TCF3::PBX1 |
| TCF3::HLF fusion | TCF3::HLF fusion | |
| BCL2/MYC | ||
| Phenocopies | ||
| BCR::ABL1-like features | BCR::ABL1–like | BCR::ABL1–like, ABL-1 class rearranged |
| BCR::ABL1–like, JAK-STAT activated | ||
| BCR::ABL1–like, NOS | ||
| ETV6::RUNX1-like features | ETV6::RUNX1-like | ETV6::RUNX1-like (provisional entity) |
| ZNF384 rearranged-like (provisional entity) | ||
| KMT2A rearranged-like (provisional entity) | ||
| Intrachromosomal amplifications | ||
| iAMP21 | iAMP21 | iAMP21 |
| Other specified rearrangements | ||
| KMT2A rearrangement | t(v;11q23.3)/KMT2A rearranged | t(v;11q23.3)/KMT2A rearranged |
| IG::MYC fusions | MYC rearrangement | |
| B-Other | DUX4 rearrangement | DUX4 rearrangement |
| MEF2D rearrangement | MEF2D rearrangement | |
| ZNF384(362) rearrangement | ZNF384(362) rearrangement | |
| NUTM1 rearrangement | NUTM1 rearrangement | |
| HLF rearrangement | ||
| UBTF::ATXN7L3/PAN3,CDX2 (“CDX2/UBTF”) | ||
| Non Ph-like CRLF2 rearrangement | ||
| Point mutations | ||
| PAX5 P80R | Mutated PAX5 (P80R) (provisional entity) | |
| Mutated IKZF1 (N159Y) | ||
| Mutated ZEB2 (p.H1038R)/IGH::CEBPE (provisional entity) | ||
| Other B-ALL subtypes | ||
| PAX5 alteration | PAX5 alteration (provisional entity) | |
| B-ALL, NOS | B-ALL, NOS | |
| T-acute lymphoblastic leukemia (T-ALL) | ||
| Early T-cell precursor ALL, NOS | Early T-cell precursor ALL, NOS | Early T-cell precursor ALL, BCL11B-activated |
| T-ALL, NOS | T-ALL, NOS | Early T-cell precursor ALL, NOS |
| T-ALL, NOS | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramírez Maldonado, V.; Navas Acosta, J.; Maldonado Marcos, I.; Villaverde Ramiro, Á.; Hernández-Sánchez, A.; Hernández Rivas, J.M.; Benito Sánchez, R. Unraveling the Genetic Heterogeneity of Acute Lymphoblastic Leukemia Based on NGS Applications. Cancers 2024, 16, 3965. https://doi.org/10.3390/cancers16233965
Ramírez Maldonado V, Navas Acosta J, Maldonado Marcos I, Villaverde Ramiro Á, Hernández-Sánchez A, Hernández Rivas JM, Benito Sánchez R. Unraveling the Genetic Heterogeneity of Acute Lymphoblastic Leukemia Based on NGS Applications. Cancers. 2024; 16(23):3965. https://doi.org/10.3390/cancers16233965
Chicago/Turabian StyleRamírez Maldonado, Valentina, Josgrey Navas Acosta, Iván Maldonado Marcos, Ángela Villaverde Ramiro, Alberto Hernández-Sánchez, Jesús M. Hernández Rivas, and Rocío Benito Sánchez. 2024. "Unraveling the Genetic Heterogeneity of Acute Lymphoblastic Leukemia Based on NGS Applications" Cancers 16, no. 23: 3965. https://doi.org/10.3390/cancers16233965
APA StyleRamírez Maldonado, V., Navas Acosta, J., Maldonado Marcos, I., Villaverde Ramiro, Á., Hernández-Sánchez, A., Hernández Rivas, J. M., & Benito Sánchez, R. (2024). Unraveling the Genetic Heterogeneity of Acute Lymphoblastic Leukemia Based on NGS Applications. Cancers, 16(23), 3965. https://doi.org/10.3390/cancers16233965