

Targeting NPC1 in Renal Cell Carcinoma



Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. The Cells and Culture Conditions
2.2. Antibodies and Reagents
2.3. Western Blot Analysis
2.4. Cell Viability and Drug Interaction Analysis
2.5. siRNA Transfection
3. Results
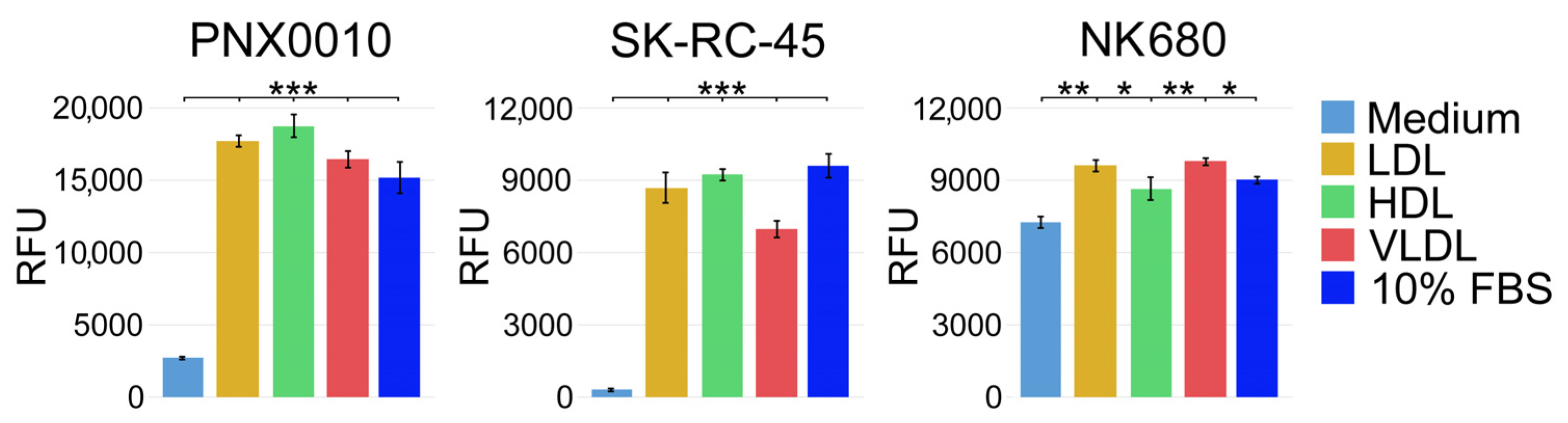
3.1. LDL, HDL, and VLDL Are Equally Effective in Supporting Viability of ccRCC Cells
3.2. LDL, HDL, and VLDL Compromise the Antitumor Activity of TKIs
3.3. Inhibition of Endosomal Cholesterol Trafficking Sensitizes ccRCC Cells to TKIs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shuch, B.; Amin, A.; Armstrong, A.J.; Eble, J.N.; Ficarra, V.; Lopez-Beltran, A.; Martignoni, G.; Rini, B.I.; Kutikov, A. Understanding pathologic variants of renal cell carcinoma: Distilling therapeutic opportunities from biologic complexity. Eur. Urol. 2015, 67, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Maines, F.; Caffo, O.; Veccia, A.; Trentin, C.; Tortora, G.; Galligioni, E.; Bria, E. Sequencing new agents after docetaxel in patients with metastatic castration-resistant prostate cancer. Crit. Rev. Oncol. Hematol. 2015, 96, 498–506. [Google Scholar] [CrossRef]
- Albiges, L.; Oudard, S.; Negrier, S.; Caty, A.; Gravis, G.; Joly, F.; Duclos, B.; Geoffrois, L.; Rolland, F.; Guillot, A.; et al. Complete remission with tyrosine kinase inhibitors in renal cell carcinoma. J. Clin. Oncol. 2012, 30, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Lee, J.L.; Park, I.; Park, S.; Ahn, Y.; Ahn, J.H.; Ahn, S.; Song, C.; Hong, J.H.; Kim, C.S.; et al. Comparative efficacy of vascular endothelial growth factor (VEGF) tyrosine kinase inhibitor (TKI) and mammalian target of rapamycin (mTOR) inhibitor as second-line therapy in patients with metastatic renal cell carcinoma after the failure of first-line VEGF TKI. Med. Oncol. 2012, 29, 3291–3297. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, M.; McDermott, D. Targeting PD-1/PD-L1 in the treatment of metastatic renal cell carcinoma. Ther. Adv. Urol. 2015, 7, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Gotink, K.J.; Broxterman, H.J.; Labots, M.; de Haas, R.R.; Dekker, H.; Honeywell, R.J.; Rudek, M.A.; Beerepoot, L.V.; Musters, R.J.; Jansen, G.; et al. Lysosomal sequestration of sunitinib: A novel mechanism of drug resistance. Clin. Cancer Res. 2011, 17, 7337–7346. [Google Scholar] [CrossRef]
- Kutikov, A.; Makhov, P.; Golovine, K.; Canter, D.J.; Sirohi, M.; Street, R.; Simhan, J.; Uzzo, R.G.; Kolenko, V.M. Interleukin-6: A Potential Biomarker of Resistance to Multitargeted Receptor Tyrosine Kinase Inhibitors in Castration-Resistant Prostate Cancer. Urology 2011, 8, 968.e7–968.e11. [Google Scholar]
- Makhov, P.B.; Golovine, K.; Kutikov, A.; Teper, E.; Canter, D.J.; Simhan, J.; Uzzo, R.G.; Kolenko, V.M. Modulation of Akt/mTOR Signaling Overcomes Sunitinib Resistance in Renal and Prostate Cancer Cells. Mol. Cancer Ther. 2012, 11, 1510–1517. [Google Scholar] [CrossRef]
- Xin, H.; Zhang, C.; Herrmann, A.; Du, Y.; Figlin, R.; Yu, H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009, 69, 2506–2513. [Google Scholar] [CrossRef]
- Adelaiye, R.; Ciamporcero, E.; Miles, K.M.; Sotomayor, P.; Bard, J.; Tsompana, M.; Conroy, D.; Shen, L.; Ramakrishnan, S.; Ku, S.Y.; et al. Sunitinib dose escalation overcomes transient resistance in clear cell renal cell carcinoma and is associated with epigenetic modifications. Mol. Cancer Ther. 2015, 14, 513–522. [Google Scholar] [CrossRef]
- Gebhard, R.L.; Clayman, R.V.; Prigge, W.F.; Figenshau, R.; Staley, N.A.; Reesey, C.; Bear, A. Abnormal cholesterol metabolism in renal clear cell carcinoma. J. Lipid Res. 1987, 28, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Llaverias, G.; Danilo, C.; Mercier, I.; Daumer, K.; Capozza, F.; Williams, T.M.; Sotgia, F.; Lisanti, M.P.; Frank, P.G. Role of cholesterol in the development and progression of breast cancer. Am. J. Pathol. 2011, 178, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Freeman, M.R.; Di Vizio, D.; Solomon, K.R. The Rafts of the Medusa: Cholesterol targeting in cancer therapy. Oncogene 2010, 29, 3745–3747. [Google Scholar] [CrossRef] [PubMed]
- Riscal, R.; Skuli, N.; Simon, M.C. Even Cancer Cells Watch Their Cholesterol! Mol. Cell 2019, 76, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Sundelin, J.P.; Stahlman, M.; Lundqvist, A.; Levin, M.; Parini, P.; Johansson, M.E.; Boren, J. Increased expression of the very low-density lipoprotein receptor mediates lipid accumulation in clear-cell renal cell carcinoma. PLoS ONE 2012, 7, e48694. [Google Scholar] [CrossRef] [PubMed]
- Gabitova, L.; Gorin, A.; Astsaturov, I. Molecular pathways: Sterols and receptor signaling in cancer. Clin. Cancer Res. 2014, 20, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Reinitz, F.; Youssef, M.; Hong, C.; Nathanson, D.; Akhavan, D.; Kuga, D.; Amzajerdi, A.N.; Soto, H.; Zhu, S.; et al. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 2011, 1, 442–456. [Google Scholar] [CrossRef]
- Pompey, S.; Zhao, Z.; Luby-Phelps, K.; Michaely, P. Quantitative fluorescence imaging reveals point of release for lipoproteins during LDLR-dependent uptake. J. Lipid Res. 2013, 54, 744–753. [Google Scholar] [CrossRef]
- Trigatti, B.L.; Krieger, M.; Rigotti, A. Influence of the HDL receptor SR-BI on lipoprotein metabolism and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1732–1738. [Google Scholar] [CrossRef]
- Rigotti, A.; Miettinen, H.E.; Krieger, M. The role of the high-density lipoprotein receptor SR-BI in the lipid metabolism of endocrine and other tissues. Endocr. Rev. 2003, 24, 357–387. [Google Scholar] [CrossRef]
- Xu, G.H.; Lou, N.; Shi, H.C.; Xu, Y.C.; Ruan, H.L.; Xiao, W.; Liu, L.; Li, X.; Xiao, H.B.; Qiu, B.; et al. Up-regulation of SR-BI promotes progression and serves as a prognostic biomarker in clear cell renal cell carcinoma. BMC Cancer 2018, 18, 88. [Google Scholar] [CrossRef]
- Liscum, L.; Dahl, N.K. Intracellular cholesterol transport. J. Lipid Res. 1992, 33, 1239–1254. [Google Scholar] [CrossRef]
- Wojtanik, K.M.; Liscum, L. The transport of low density lipoprotein-derived cholesterol to the plasma membrane is defective in NPC1 cells. J. Biol. Chem. 2003, 278, 14850–14856. [Google Scholar] [CrossRef] [PubMed]
- Pfisterer, S.G.; Peranen, J.; Ikonen, E. LDL-cholesterol transport to the endoplasmic reticulum: Current concepts. Curr. Opin. Lipidol. 2016, 27, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Abi-Mosleh, L.; Wang, M.L.; Deisenhofer, J.; Goldstein, J.L.; Brown, M.S.; Infante, R.E. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 2009, 137, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Peake, K.B.; Vance, J.E. Defective cholesterol trafficking in Niemann-Pick C-deficient cells. FEBS Lett. 2010, 584, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Soffientini, U.; Graham, A. Intracellular cholesterol transport proteins: Roles in health and disease. Clin. Sci. 2016, 130, 1843–1859. [Google Scholar] [CrossRef] [PubMed]
- Lange, Y.; Ye, J.; Rigney, M.; Steck, T. Cholesterol movement in Niemann-Pick type C cells and in cells treated with amphiphiles. J. Biol. Chem. 2000, 275, 17468–17475. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, K.I.; Kuo, L.W.; Williams, M.M.; Lind, H.; Crump, L.S.; Hammond, N.G.; Spoelstra, N.S.; Caino, M.C.; Richer, J.K. NPC1 Confers Metabolic Flexibility in Triple Negative Breast Cancer. Cancers 2022, 14, 3543. [Google Scholar] [CrossRef] [PubMed]
- Burns, V.E.; Kerppola, T.K. ATR-101 inhibits cholesterol efflux and cortisol secretion by ATP-binding cassette transporters, causing cytotoxic cholesterol accumulation in adrenocortical carcinoma cells. Br. J. Pharmacol. 2017, 174, 3315–3332. [Google Scholar] [CrossRef]
- Do, H.T.; Bruelle, C.; Tselykh, T.; Jalonen, P.; Korhonen, L.; Lindholm, D. Reciprocal regulation of very low density lipoprotein receptors (VLDLRs) in neurons by brain-derived neurotrophic factor (BDNF) and Reelin: Involvement of the E3 ligase Mylip/Idol. J. Biol. Chem. 2013, 288, 29613–29620. [Google Scholar] [CrossRef]
- Hong, C.; Duit, S.; Jalonen, P.; Out, R.; Scheer, L.; Sorrentino, V.; Boyadjian, R.; Rodenburg, K.W.; Foley, E.; Korhonen, L.; et al. The E3 ubiquitin ligase IDOL induces the degradation of the low density lipoprotein receptor family members VLDLR and ApoER2. J. Biol. Chem. 2010, 285, 19720–19726. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Jiang, T.; Zhao, G.J. Quercetin induces the selective uptake of HDL-cholesterol via promoting SR-BI expression and the activation of the PPARgamma/LXRalpha pathway. Food Funct. 2018, 9, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Venkateswaran, A.; Laffitte, B.A.; Joseph, S.B.; Mak, P.A.; Wilpitz, D.C.; Edwards, P.A.; Tontonoz, P. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proc. Natl. Acad. Sci. USA 2000, 97, 12097–12102. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.W.; Wang, Q.; Ma, X.; Li, X.X.; Liu, X.H.; Xiao, J.; Liao, D.F.; Xiang, J.; Tang, C.K. TGF-beta1 up-regulates expression of ABCA1, ABCG1 and SR-BI through liver X receptor alpha signaling pathway in THP-1 macrophage-derived foam cells. J. Atheroscler. Thromb. 2010, 17, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Golovine, K.; Makhov, P.; Naito, S.; Raiyani, H.; Tomaszewski, J.; Mehrazin, R.; Tulin, A.; Kutikov, A.; Uzzo, R.G.; Kolenko, V.M. Piperlongumine and its analogs down-regulate expression of c-Met in renal cell carcinoma. Cancer Biol. Ther. 2015, 16, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Naito, S.; Makhov, P.; Astsaturov, I.; Golovine, K.; Tulin, A.; Kutikov, A.; Uzzo, R.G.; Kolenko, V.M. LDL cholesterol counteracts the antitumour effect of tyrosine kinase inhibitors against renal cell carcinoma. Br. J. Cancer 2017, 116, 1203–1207. [Google Scholar] [CrossRef] [PubMed]
- Golovine, K.; Makhov, P.; Uzzo, R.G.; Shaw, T.; Kunkle, D.; Kolenko, V.M. Overexpression of the zinc uptake transporter hZIP1 inhibits nuclear factor-kappaB and reduces the malignant potential of prostate cancer cells in vitro and in vivo. Clin. Cancer Res. 2008, 14, 5376–5384. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Makhov, P.; Sohn, J.A.; Serebriiskii, I.G.; Fazliyeva, R.; Khazak, V.; Boumber, Y.; Uzzo, R.G.; Kolenko, V.M. CRISPR/Cas9 genome-wide loss-of-function screening identifies druggable cellular factors involved in sunitinib resistance in renal cell carcinoma. Br. J. Cancer 2020, 123, 1749–1756. [Google Scholar] [CrossRef]
- Parinaud, J.; Perret, B.; Ribbes, H.; Chap, H.; Pontonnier, G.; Douste-Blazy, L. High density lipoprotein and low density lipoprotein utilization by human granulosa cells for progesterone synthesis in serum-free culture: Respective contributions of free and esterified cholesterol. J. Clin. Endocrinol. Metab. 1987, 64, 409–417. [Google Scholar] [CrossRef]
- Canter, D.; Kutikov, A.; Golovine, K.; Makhov, P.; Simhan, J.; Uzzo, R.G.; Kolenko, V.M. Are all multi-targeted tyrosine kinase inhibitors created equal? An in vitro study of sunitinib and pazopanib in renal cell carcinoma cell lines. Can. J. Urol. 2011, 18, 5819–5825. [Google Scholar]
- Kirsanov, K.I.; Kotova, E.; Makhov, P.; Golovine, K.; Lesovaya, E.A.; Kolenko, V.M.; Yakubovskaya, M.G.; Tulin, A.V. Minor grove binding ligands disrupt PARP-1 activation pathways. Oncotarget 2014, 5, 428–437. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thomas, C.; Ji, Y.; Lodhi, N.; Kotova, E.; Pinnola, A.D.; Golovine, K.; Makhov, P.; Pechenkina, K.; Kolenko, V.; Tulin, A.V. Non-NAD-Like poly(ADP-Ribose) Polymerase-1 Inhibitors effectively Eliminate Cancer in vivo. EBioMedicine 2016, 13, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Trinh, M.N.; Lu, F.; Li, X.; Das, A.; Liang, Q.; De Brabander, J.K.; Brown, M.S.; Goldstein, J.L. Triazoles inhibit cholesterol export from lysosomes by binding to NPC1. Proc. Natl. Acad. Sci. USA 2017, 114, 89–94. [Google Scholar] [CrossRef]
- Li, Y.; Schwabe, R.F.; DeVries-Seimon, T.; Yao, P.M.; Gerbod-Giannone, M.C.; Tall, A.R.; Davis, R.J.; Flavell, R.; Brenner, D.A.; Tabas, I. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-alpha and interleukin-6: Model of NF-kappaB- and map kinase-dependent inflammation in advanced atherosclerosis. J. Biol. Chem. 2005, 280, 21763–21772. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Dang, Y.J.; Ren, Y.R.Z.; Liu, J.O. Cholesterol trafficking is required for mTOR activation in endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 4764–4769. [Google Scholar] [CrossRef] [PubMed]
- Morales, M.L.; Arenas, A.; Ortiz-Ruiz, A.; Leivas, A.; Rapado, I.; Rodriguez-Garcia, A.; Castro, N.; Zagorac, I.; Quintela-Fandino, M.; Gomez-Lopez, G.; et al. MEK inhibition enhances the response to tyrosine kinase inhibitors in acute myeloid leukemia. Sci. Rep. 2019, 9, 18630. [Google Scholar] [CrossRef] [PubMed]
- Blakely, C.M.; Pazarentzos, E.; Olivas, V.; Asthana, S.; Yan, J.J.; Tan, I.; Hrustanovic, G.; Chan, E.; Lin, L.; Neel, D.S.; et al. NF-kappaB-activating complex engaged in response to EGFR oncogene inhibition drives tumor cell survival and residual disease in lung cancer. Cell Rep. 2015, 11, 98–110. [Google Scholar] [CrossRef]
- Bedke, J.; Stuhler, V.; Stenzl, A.; Brehmer, B. Immunotherapy for kidney cancer: Status quo and the future. Curr. Opin. Urol. 2018, 28, 8–14. [Google Scholar] [CrossRef]
- Mosillo, C.; Ciccarese, C.; Bimbatti, D.; Fantinel, E.; Volta, A.D.; Bisogno, I.; Zampiva, I.; Santoni, M.; Massar, F.; Brunelli, M.; et al. Renal cell carcinoma in one year: Going inside the news of 2017—A report of the main advances in RCC cancer research. Cancer Treat. Rev. 2018, 67, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Makhov, P.; Joshi, S.; Ghatalia, P.; Kutikov, A.; Uzzo, R.G.; Kolenko, V.M. Resistance to Systemic Therapies in Clear Cell Renal Cell Carcinoma: Mechanisms and Management Strategies. Mol. Cancer Ther. 2018, 17, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Plimack, E.R.; Puzanov, I.; Fishman, M.N.; McDermott, D.F.; Cho, D.C.; Vaishampayan, U.; George, S.; Olencki, T.E.; Tarazi, J.C.; et al. Axitinib in combination with pembrolizumab in patients with advanced renal cell cancer: A non-randomised, open-label, dose-finding, and dose-expansion phase 1b trial. Lancet Oncol. 2018, 19, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Calvo, E.; Porta, C.; Grunwald, V.; Escudier, B. The Current and Evolving Landscape of First-Line Treatments for Advanced Renal Cell Carcinoma. Oncologist 2019, 24, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Fuca, G.; de Braud, F.; Di Nicola, M. Immunotherapy-based combinations: An update. Curr. Opin. Oncol. 2018, 30, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Vano, Y.A.; Ladoire, S.; Elaidi, R.; Dermeche, S.; Eymard, J.C.; Falkowski, S.; Gross-Goupil, M.; Malouf, G.; Narciso, B.; Sajous, C.; et al. First-Line Treatment of Metastatic Clear Cell Renal Cell Carcinoma: What Are the Most Appropriate Combination Therapies? Cancers 2021, 13, 5548. [Google Scholar] [CrossRef] [PubMed]
- Massari, F.; Rizzo, A.; Mollica, V.; Rosellini, M.; Marchetti, A.; Ardizzoni, A.; Santoni, M. Immune-based combinations for the treatment of metastatic renal cell carcinoma: A meta-analysis of randomised clinical trials. Eur. J. Cancer 2021, 154, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Colhoun, H.M.; Leiter, L.A.; Muller-Wieland, D.; Cariou, B.; Ray, K.K.; Tinahones, F.J.; Domenger, C.; Letierce, A.; Israel, M.; Samuel, R.; et al. Effect of alirocumab on individuals with type 2 diabetes, high triglycerides, and low high-density lipoprotein cholesterol. Cardiovasc. Diabetol. 2020, 19, 14. [Google Scholar] [CrossRef]
- Mombelli, G.; Castelnuovo, S.; Pavanello, C. Potential of PCSK9 as a new target for the management of LDL cholesterol. Res. Rep. Clin. Cardiol. 2015, 6, 73–86. [Google Scholar] [CrossRef][Green Version]
- Sabatine, M.S.; Giugliano, R.P.; Wiviott, S.D.; Raal, F.J.; Blom, D.J.; Robinson, J.; Ballantyne, C.M.; Somaratne, R.; Legg, J.; Wasserman, S.M.; et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 2015, 372, 1500–1509. [Google Scholar] [CrossRef]
- Moride, Y.; Hegele, R.A.; Langer, A.; McPherson, R.; Miller, D.B.; Rinfret, S. Clinical and public health assessment of benefits and risks of statins in primary prevention of coronary events: Resolved and unresolved issues. Can. J. Cardiol. 2008, 24, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Hajar, R. PCSK 9 Inhibitors: A Short History and a New Era of Lipid-lowering Therapy. Heart Views 2019, 20, 74–75. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, S.I.; Muniyappa, R.; Francisco, R.; Sowers, J.R. Clinical review 145: Pleiotropic effects of statins: Lipid reduction and beyond. J. Clin. Endocrinol. Metab. 2002, 87, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, E.J.; Asztalos, B.F. The effects of statins on high-density lipoproteins. Curr. Atheroscler. Rep. 2006, 8, 41–49. [Google Scholar] [CrossRef]
- Barter, P.J.; Brandrup-Wognsen, G.; Palmer, M.K.; Nicholls, S.J. Effect of statins on HDL-C: A complex process unrelated to changes in LDL-C: Analysis of the VOYAGER Database. J. Lipid Res. 2010, 51, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- McTaggart, F.; Jones, P. Effects of statins on high-density lipoproteins: A potential contribution to cardiovascular benefit. Cardiovasc. Drugs Ther. 2008, 22, 321–338. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Tsubakio-Yamamoto, K.; Ohama, T.; Nakagawa-Toyama, Y.; Nishida, M. Molecular mechanisms of HDL-cholesterol elevation by statins and its effects on HDL functions. J. Atheroscler. Thromb. 2010, 17, 436–451. [Google Scholar] [CrossRef] [PubMed]
- Teramoto, T.; Kobayashi, M.; Tasaki, H.; Yagyu, H.; Higashikata, T.; Takagi, Y.; Uno, K.; Baccara-Dinet, M.T.; Nohara, A. Efficacy and Safety of Alirocumab in Japanese Patients With Heterozygous Familial Hypercholesterolemia or at High Cardiovascular Risk with Hypercholesterolemia Not Adequately Controlled With Statins- ODYSSEY JAPAN Randomized Controlled Trial. Circ. J. 2016, 80, 1980–1987. [Google Scholar] [CrossRef]
- Brusselmans, K.; Timmermans, L.; Van de Sande, T.; Van Veldhoven, P.P.; Guan, G.; Shechter, I.; Claessens, F.; Verhoeven, G.; Swinnen, J.V. Squalene synthase, a determinant of Raft-associated cholesterol and modulator of cancer cell proliferation. J. Biol. Chem. 2007, 282, 18777–18785. [Google Scholar] [CrossRef]
- Ribas, V.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondria, cholesterol and cancer cell metabolism. Clin. Transl. Med. 2016, 5, 22. [Google Scholar] [CrossRef]
- Huetsch, J.C.; Suresh, K.; Shimoda, L.A. When higher cholesterol is better: Membrane cholesterol loss and endothelial Ca2+ signaling. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H780–H783. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | ED25 | IC50 | IC75 | IC90 |
|---|---|---|---|---|
| 786-O | 2.1 | 2.8 | 3.7 | 4.9 |
| SK-RC-45 | 0.09 | 0.4 | 1.8 | 8.2 |
| PNX0010 | 5.2 | 10.4 | 15.9 | 31.3 |
| NK686 | 30.7 | 54.8 | 97.9 | 174.7 |
| NKE | 26.5 | 52.9 | 105.9 | 211 |
| RWPE-1 | 21.8 | 30.3 | 42.1 | 58.5 |
| PZ-HPV-7 | 13.7 | 28.0 | 57 | 116.2 |
| HUVEC | 19.7 | 34.1 | 59.4 | 103.6 |
| Drug | Cell Line | ED25 | ED50 | ED75 | ED90 |
|---|---|---|---|---|---|
| U18666A | 786-O (N) | 1.5 | 2.3 | 3.5 | 5.4 |
| 786-O (H) | 0.7 | 1.2 | 2.1 | 3.6 | |
| PNX0010 (N) | 2.2 | 3.3 | 4.9 | 7.2 | |
| PNX0010 (H) | 0.4 | 0.9 | 1.9 | 3.8 | |
| Posaconazole | 786-O (N) | 4.7 | 10.1 | 21.6 | 46.2 |
| 786-O (H) | 1.3 | 3.0 | 6.7 | 15.3 | |
| PNX0010 (N) | 4.6 | 8.0 | 14.2 | 24.9 | |
| PNX0010 (H) | 0.9 | 1.9 | 4.2 | 9.2 |
| Drug | Cell Line | ED25 | ED50 | ED75 | ED90 |
|---|---|---|---|---|---|
| Sunitinib | 786-O | 0.56 | 0.52 | 0.47 | 0.41 |
| SK-RC-45 | 0.45 | 0.41 | 0.37 | 0.32 | |
| PNX0010 | 0.50 | 0.46 | 0.42 | 0.39 | |
| Cabozantinib | 786-O | 0.44 | 0.40 | 0.35 | 0.30 |
| SK-RC-45 | 0.39 | 0.33 | 0.27 | 0.22 | |
| PNX0010 | 0.52 | 0.45 | 0.41 | 0.38 | |
| Pazopanib | 786-O | 0.37 | 0.31 | 0.25 | 0.20 |
| SK-RC-45 | 0.42 | 0.36 | 0.29 | 0.23 | |
| PNX0010 | 0.48 | 0.40 | 0.33 | 0.26 |
| Drug | Cell Line | ED25 | ED50 | ED75 | ED90 |
|---|---|---|---|---|---|
| Axitinib | 786-O | 5.4 | 7.4 | 10.1 | 13.9 |
| 786-O-NPC1KD | 1.3 | 2.5 | 5.0 | 9.9 | |
| PNX0010 | 4.1 | 6.9 | 11.5 | 19.3 | |
| PNX0010-NPC1KD | 1.5 | 2.6 | 4.7 | 8.5 | |
| Cabozantinib | 786-O | 5.6 | 8.5 | 12.8 | 19.3 |
| 786-O-NPC1KD | 2.5 | 4.0 | 6.4 | 10.4 | |
| PNX0010 | 4.6 | 6.8 | 10.0 | 14.9 | |
| PNX0010-NPC1KD | 2.7 | 3.9 | 5.7 | 8.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fazliyeva, R.; Makhov, P.; Uzzo, R.G.; Kolenko, V.M. Targeting NPC1 in Renal Cell Carcinoma. Cancers 2024, 16, 517. https://doi.org/10.3390/cancers16030517
Fazliyeva R, Makhov P, Uzzo RG, Kolenko VM. Targeting NPC1 in Renal Cell Carcinoma. Cancers. 2024; 16(3):517. https://doi.org/10.3390/cancers16030517
Chicago/Turabian StyleFazliyeva, Rushaniya, Peter Makhov, Robert G. Uzzo, and Vladimir M. Kolenko. 2024. "Targeting NPC1 in Renal Cell Carcinoma" Cancers 16, no. 3: 517. https://doi.org/10.3390/cancers16030517
APA StyleFazliyeva, R., Makhov, P., Uzzo, R. G., & Kolenko, V. M. (2024). Targeting NPC1 in Renal Cell Carcinoma. Cancers, 16(3), 517. https://doi.org/10.3390/cancers16030517