

Pancreatic Tumor Organoid-Derived Factors from Cachectic Patients Disrupt Contractile Smooth Muscle Cells
 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Pancreatic Tumor Organoid Culture
2.2. Collection of Pancreatic Tumor Organoid Conditioned Medium
2.3. Smooth Muscle Cell Culture
2.4. Quantitative Real-Time PCR
2.5. Western Blotting
2.6. IncuCyte™ Cell Confluence Proliferation Assay
2.7. IncuCyte™ Scratch Wound Cell Migration Assay
2.8. Patient Cohort
2.9. Immunohistochemical Staining and Evaluation
2.10. Statistical Analysis
3. Results
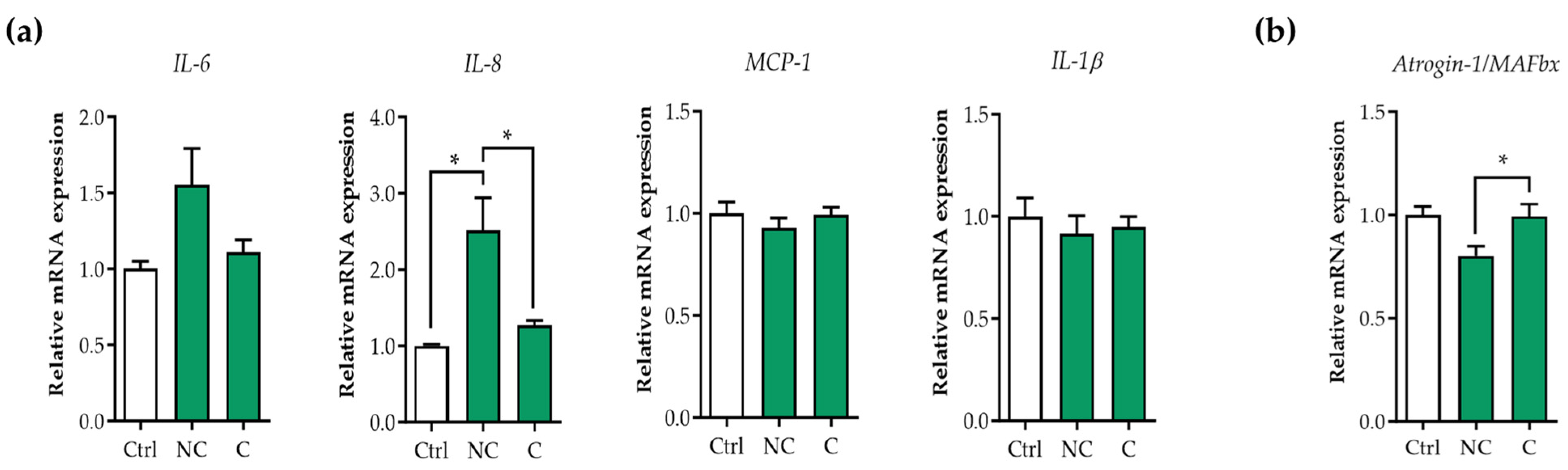
3.1. Tumor Organoid Factors from Non-Cachectic Pancreatic Cancer Patients Increase Pro-Inflammatory Cytokines in Human Visceral SMCs
3.2. E3 Ubiquitin Ligases Are Not Upregulated in Human Visceral SMCs Exposed to Pancreatic Tumor Organoid-Derived CM
3.3. Tumor Organoid Factors from Cachectic Pancreatic Cancer Patients Diminish Contractile Protein Levels in SMCs
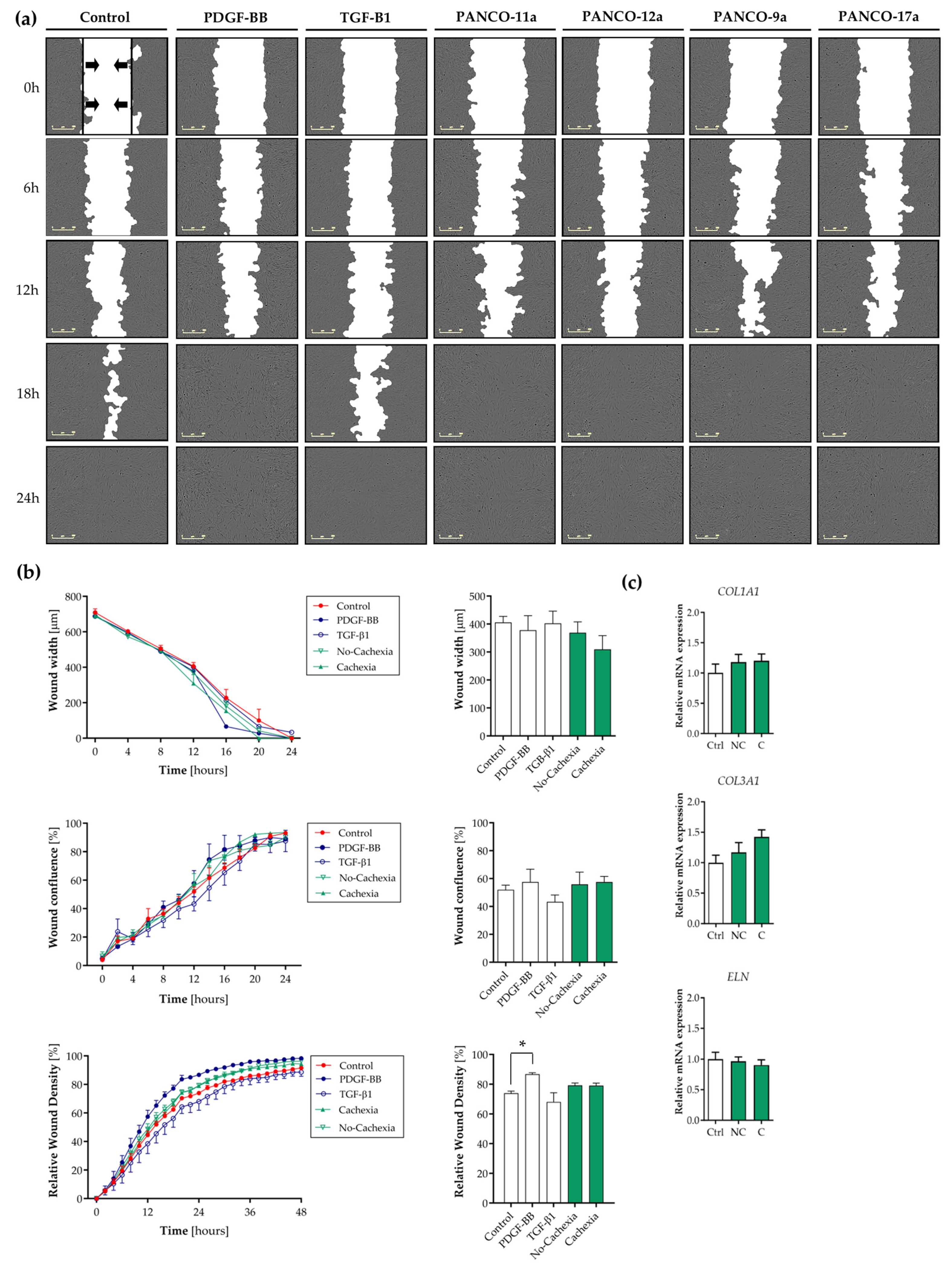
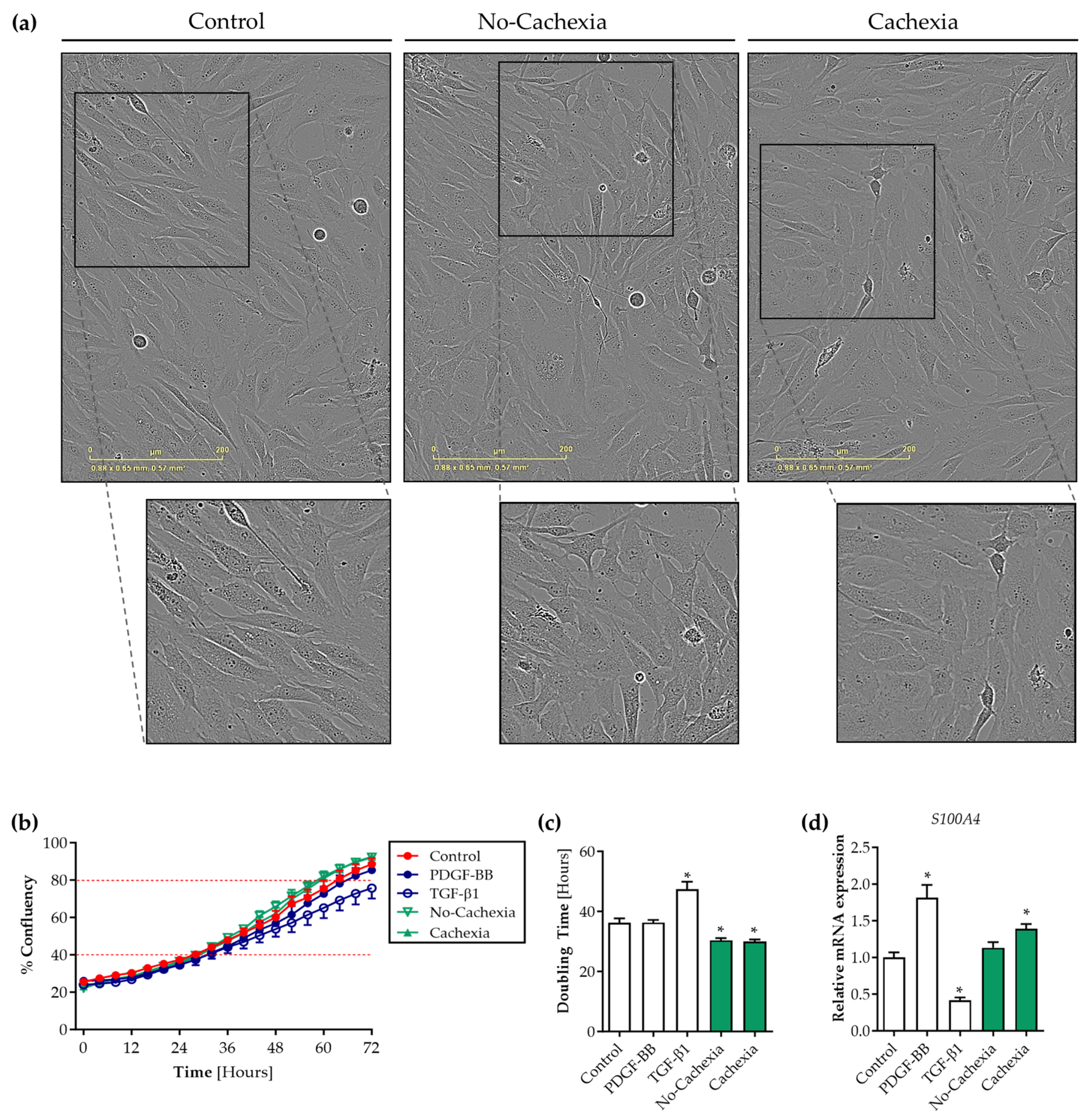
3.4. Tumor Organoid-Derived Factors Induce Proliferation but Not Migration of Contractile Human Visceral SMCs
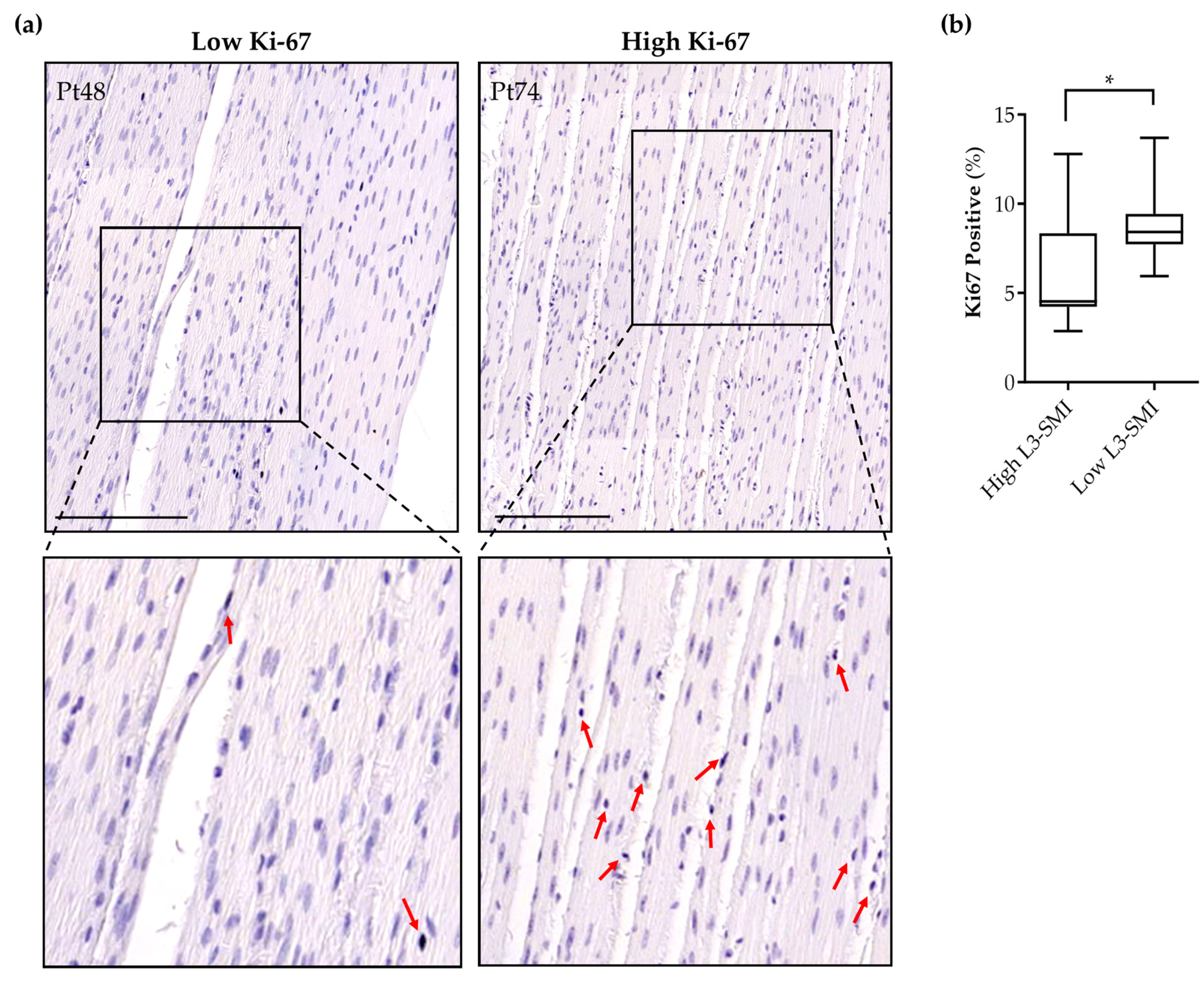
3.5. Sarcopenia in Pancreatic Cancer Patients Is Associated with Increased Intestinal Smooth Muscle Proliferation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primers 2018, 4, 17105. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef]
- Barkhudaryan, A.; Scherbakov, N.; Springer, J.; Doehner, W. Cardiac muscle wasting in individuals with cancer cachexia. ESC Heart Fail. 2017, 4, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Springer, J.; Tschirner, A.; Haghikia, A.; von Haehling, S.; Lal, H.; Grzesiak, A.; Kaschina, E.; Palus, S.; Potsch, M.; von Websky, K.; et al. Prevention of liver cancer cachexia-induced cardiac wasting and heart failure. Eur. Heart J. 2014, 35, 932–941. [Google Scholar] [CrossRef]
- Bdolah, Y.; Segal, A.; Tanksale, P.; Karumanchi, S.A.; Lecker, S.H. Atrophy-related ubiquitin ligases atrogin-1 and MuRF-1 are associated with uterine smooth muscle involution in the postpartum period. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R971–R976. [Google Scholar] [CrossRef]
- Nakashima, K.; Ishida, A.; Katsumata, M. Atrogin-1/MAFbx, a muscle-specific ubiquitin ligase, is highly expressed in the smooth muscle of the chicken gizzard. Biosci. Biotechnol. Biochem. 2013, 77, 1092–1095. [Google Scholar] [CrossRef]
- Bouchelouche, K.; Alvarez, S.; Horn, T.; Nordling, J.; Bouchelouche, P. Human detrusor smooth muscle cells release interleukin-6, interleukin-8, and RANTES in response to proinflammatory cytokines interleukin-1β and tumor necrosis factor-α. Urology 2006, 67, 214–219. [Google Scholar] [CrossRef]
- Ng, E.K.; Panesar, N.; Longo, W.E.; Shapiro, M.J.; Kaminski, D.L.; Tolman, K.C.; Mazuski, J.E. Human intestinal epithelial and smooth muscle cells are potent producers of IL-6. Mediat. Inflamm. 2003, 12, 3–8. [Google Scholar] [CrossRef]
- Scirocco, A.; Matarrese, P.; Carabotti, M.; Ascione, B.; Malorni, W.; Severi, C. Cellular and Molecular Mechanisms of Phenotypic Switch in Gastrointestinal Smooth Muscle. J. Cell. Physiol. 2016, 231, 295–302. [Google Scholar] [CrossRef]
- Sanders, K.M.; Koh, S.D.; Ro, S.; Ward, S.M. Regulation of gastrointestinal motility-Insights from smooth muscle biology. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 633–645. [Google Scholar] [CrossRef]
- Zhou, T.; Yang, K.; Thapa, S.; Liu, H.; Wang, B.; Yu, S. Differences in Symptom Burden Among Cancer Patients With Different Stages of Cachexia. J. Pain Symptom Manag. 2017, 53, 919–926. [Google Scholar] [CrossRef]
- Vaes, R.D.W.; Welbers, T.T.J.; van Dijk, D.P.J.; Renspiess, D.; zur Hausen, A.; Olde Damink, S.W.M.; Rensen, S.S. Intestinal smooth muscle aberrations in pancreatic cancer patients with sarcopenia. JCSM Rapid Commun. 2021, 4, 187–196. [Google Scholar] [CrossRef]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef]
- Broutier, L.; Andersson-Rolf, A.; Hindley, C.J.; Boj, S.F.; Clevers, H.; Koo, B.K.; Huch, M. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nat. Protoc. 2016, 11, 1724–1743. [Google Scholar] [CrossRef]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef]
- Vaes, R.D.W.; van Dijk, D.P.J.; Welbers, T.T.J.; Blok, M.J.; Aberle, M.R.; Heij, L.; Boj, S.F.; Olde Damink, S.W.M.; Rensen, S.S. Generation and initial characterization of novel tumour organoid models to study human pancreatic cancer-induced cachexia. J. Cachexia Sarcopenia Muscle 2020, 11, 1509–1524. [Google Scholar] [CrossRef] [PubMed]
- Vaes, R.D.W.; van den Berk, L.; Boonen, B.; van Dijk, D.P.J.; Olde Damink, S.W.M.; Rensen, S.S. A novel human cell culture model to study visceral smooth muscle phenotypic modulation in health and disease. Am. J. Physiol. Cell Physiol. 2018, 315, C598–C607. [Google Scholar] [CrossRef] [PubMed]
- Perez-Reyes, N.; Halbert, C.L.; Smith, P.P.; Benditt, E.P.; McDougall, J.K. Immortalization of primary human smooth muscle cells. Proc. Natl. Acad. Sci. USA 1992, 89, 1224–1228. [Google Scholar] [CrossRef]
- Ruijter, J.M.; Ramakers, C.; Hoogaars, W.M.; Karlen, Y.; Bakker, O.; van den Hoff, M.J.; Moorman, A.F. Amplification efficiency: Linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009, 37, e45. [Google Scholar] [CrossRef] [PubMed]
- Flegal, K.M.; Kit, B.K.; Graubard, B.I. Bias in Hazard Ratios Arising From Misclassification According to Self-Reported Weight and Height in Observational Studies of Body Mass Index and Mortality. Am. J. Epidemiol. 2018, 187, 125. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, D.P.; Bakens, M.J.; Coolsen, M.M.; Rensen, S.S.; van Dam, R.M.; Bours, M.J.; Weijenberg, M.P.; Dejong, C.H.; Olde Damink, S.W. Low skeletal muscle radiation attenuation and visceral adiposity are associated with overall survival and surgical site infections in patients with pancreatic cancer. J. Cachexia Sarcopenia Muscle 2017, 8, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Mourtzakis, M.; Prado, C.M.M.; Lieffers, J.R.; Reiman, T.; McCargar, L.J.; Baracos, V.E. A practical and precise approach to quantification of body composition in cancer patients using computed tomography images acquired during routine care. Appl. Physiol. Nutr. Metab. 2008, 33, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernandez, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [PubMed]
- Arnoldi, R.; Hiltbrunner, A.; Dugina, V.; Tille, J.C.; Chaponnier, C. Smooth muscle actin isoforms: A tug of war between contraction and compliance. Eur. J. Cell Biol. 2013, 92, 187–200. [Google Scholar] [CrossRef]
- Brisset, A.C.; Hao, H.; Camenzind, E.; Bacchetta, M.; Geinoz, A.; Sanchez, J.C.; Chaponnier, C.; Gabbiani, G.; Bochaton-Piallat, M.L. Intimal smooth muscle cells of porcine and human coronary artery express S100A4, a marker of the rhomboid phenotype in vitro. Circ. Res. 2007, 100, 1055–1062. [Google Scholar] [CrossRef]
- Chaabane, C.; Heizmann, C.W.; Bochaton-Piallat, M.L. Extracellular S100A4 induces smooth muscle cell phenotypic transition mediated by RAGE. Biochim. Biophys. Acta 2015, 1853, 2144–2157. [Google Scholar] [CrossRef]
- Vaes, R.D.W.; van Dijk, D.P.J.; Olde Damink, S.W.M.; Rensen, S.S.; Langen, R. Human pancreatic tumor organoid-derived factors enhance myogenic differentiation. J. Cachexia Sarcopenia Muscle 2022, 13, 1302–1313. [Google Scholar] [CrossRef] [PubMed]
- Beamish, J.A.; He, P.; Kottke-Marchant, K.; Marchant, R.E. Molecular regulation of contractile smooth muscle cell phenotype: Implications for vascular tissue engineering. Tissue Eng. Part B Rev. 2010, 16, 467–491. [Google Scholar] [CrossRef] [PubMed]
- Niessen, P.; Rensen, S.; van Deursen, J.; De Man, J.; De Laet, A.; Vanderwinden, J.M.; Wedel, T.; Baker, D.; Doevendans, P.; Hofker, M.; et al. Smoothelin-a is essential for functional intestinal smooth muscle contractility in mice. Gastroenterology 2005, 129, 1592–1601. [Google Scholar] [CrossRef]
- Donthireddy, K.R.; Ailawadhi, S.; Nasser, E.; Schiff, M.D.; Nwogu, C.E.; Nava, H.R.; Javle, M.M. Malignant gastroparesis: Pathogenesis and management of an underrecognized disorder. J. Support. Oncol. 2007, 5, 355–363. [Google Scholar] [PubMed]
- Khan, M.A. Inflammation signals airway smooth muscle cell proliferation in asthma pathogenesis. Multidiscip. Respir. Med. 2013, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Nair, D.G.; Han, T.Y.; Lourenssen, S.; Blennerhassett, M.G. Proliferation modulates intestinal smooth muscle phenotype in vitro and in colitis in vivo. Am. J. Physiol. Gastrointest. Liver. Physiol. 2011, 300, G903–G913. [Google Scholar] [CrossRef] [PubMed]
- Nair, D.G.; Miller, K.G.; Lourenssen, S.R.; Blennerhassett, M.G. Inflammatory cytokines promote growth of intestinal smooth muscle cells by induced expression of PDGF-Rbeta. J. Cell. Mol. Med. 2014, 18, 444–454. [Google Scholar] [CrossRef]
- Owens, M.W.; Grisham, M.B. Cytokines increase proliferation of human intestinal smooth muscle cells: Possible role in inflammation-induced stricture formation. Inflammation 1993, 17, 481–487. [Google Scholar] [CrossRef]
- Stamatiou, R.; Paraskeva, E.; Gourgoulianis, K.; Molyvdas, P.A.; Hatziefthimiou, A. Cytokines and growth factors promote airway smooth muscle cell proliferation. ISRN Inflamm. 2012, 2012, 731472. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Gao, H.; Zhao, H.; Bhatia, M.; Zeng, Y. Roles of airway smooth muscle dysfunction in chronic obstructive pulmonary disease. J. Transl. Med. 2018, 16, 262. [Google Scholar] [CrossRef]
- Stanzel, R.D.; Lourenssen, S.; Nair, D.G.; Blennerhassett, M.G. Mitogenic factors promoting intestinal smooth muscle cell proliferation. Am. J. Physiol. Cell Physiol. 2010, 299, C805–C817. [Google Scholar] [CrossRef]
- Cole, C.L.; Kleckner, I.R.; Jatoi, A.; Schwarz, E.M.; Dunne, R.F. The Role of Systemic Inflammation in Cancer-Associated Muscle Wasting and Rationale for Exercise as a Therapeutic Intervention. JCSM Clin. Rep. 2018, 3, e00065. [Google Scholar] [CrossRef]
- Lappas, M. The IL-1β signalling pathway and its role in regulating pro-inflammatory and pro-labour mediators in human primary myometrial cells. Reprod. Biol. 2017, 17, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Xiao, H.T.; Zhang, Y.; Tong, R.S.; Zhang, L.J.; Bian, Y.; He, X. IL-1β: A key modulator in asthmatic airway smooth muscle hyper-reactivity. Expert Rev. Respir. Med. 2015, 9, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Parpaleix, A.; Amsellem, V.; Houssaini, A.; Abid, S.; Breau, M.; Marcos, E.; Sawaki, D.; Delcroix, M.; Quarck, R.; Maillard, A.; et al. Role of interleukin-1 receptor 1/MyD88 signalling in the development and progression of pulmonary hypertension. Eur. Respir. J. 2016, 48, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Chamley-Campbell, J.; Campbell, G.R.; Ross, R. The smooth muscle cell in culture. Physiol. Rev. 1979, 59, 1–61. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total (n = 22) | High L3-SMI (n = 11) | Low L3-SMI (n = 11) | p-Value | |
|---|---|---|---|---|
| Male (n,%) | 10 (45.5%) | 5 (45.5%) | 5 (45.5%) | p = 1.00 |
| Age (years) | 67.8 (±8.5) | 67.8 (±7.2) | 67.8 (±10.0) | p = 1.00 |
| BMI (kg/m2) | 23.9 (±2.9) | 25.3 (±3.0) | 22.5 (±1.9) | p = 0.015 |
| Weight loss * (%) | 9.4 (±7.4) | 8.2 (±7.0) | 10.7 (±7.9) | p = 0.44 |
| MUST score ≥ 2# (n,%) | 8 (36.4%) | 3 (30.0%) | 5 (45.5%) | p = 0.66 |
| L3 skeletal muscle index (cm2/m2) | ||||
| Male | 49.3 (±4.3) | 52.4 (±3.6) | 46.1 (±1.9) | p = 0.0084 |
| Female | 41.9 (±8.0) | 48.1 (±5.5) | 35.7 (±4.1) | p = 0.0013 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaes, R.D.W.; van Bijnen, A.A.; Damink, S.W.M.O.; Rensen, S.S. Pancreatic Tumor Organoid-Derived Factors from Cachectic Patients Disrupt Contractile Smooth Muscle Cells. Cancers 2024, 16, 542. https://doi.org/10.3390/cancers16030542
Vaes RDW, van Bijnen AA, Damink SWMO, Rensen SS. Pancreatic Tumor Organoid-Derived Factors from Cachectic Patients Disrupt Contractile Smooth Muscle Cells. Cancers. 2024; 16(3):542. https://doi.org/10.3390/cancers16030542
Chicago/Turabian StyleVaes, Rianne D. W., Annemarie A. van Bijnen, Steven W. M. Olde Damink, and Sander S. Rensen. 2024. "Pancreatic Tumor Organoid-Derived Factors from Cachectic Patients Disrupt Contractile Smooth Muscle Cells" Cancers 16, no. 3: 542. https://doi.org/10.3390/cancers16030542