

Stem Cell Theory of Cancer: Clinical Implications for Cellular Metabolism and Anti-Cancer Metabolomics
 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Brief History

{kind=link}
| Name, Year | Origin/Nature of Cancer | Contribution | Reference |
|---|---|---|---|
| Virchow, 1863 | Stem-ness | Embryonal cells | [10] |
| Rous, 1911 | Genetic | Viral oncogenes | [5] |
| Farber, 1948 | Metabolic | Antifolate therapy of acute leukemia | [1] |
| Warburg, 1956 | Metabolic | Increased glycolysis and defective mitochondria | [2] |
| Stevens, 1964 | Stem-ness | Origin of cancer stem cells | [11] |
| Knudson, 1971 | Genetic | Tumor suppressor genes: 2-hit hypothesis | [6] |
| Vogelstein, 1988 | Genetic | Multistep carcinogenesis | [4] |
| Pierce, 1994 | Stem-ness | Maturation arrest of stem cells | [12,13] |
| Seyfried, 2010 | Metabolic | Nutrition and cancer | [3] |
3. Unified Theory of Cancer
3.1. Stem Cell Origin
3.2. Cellular Context
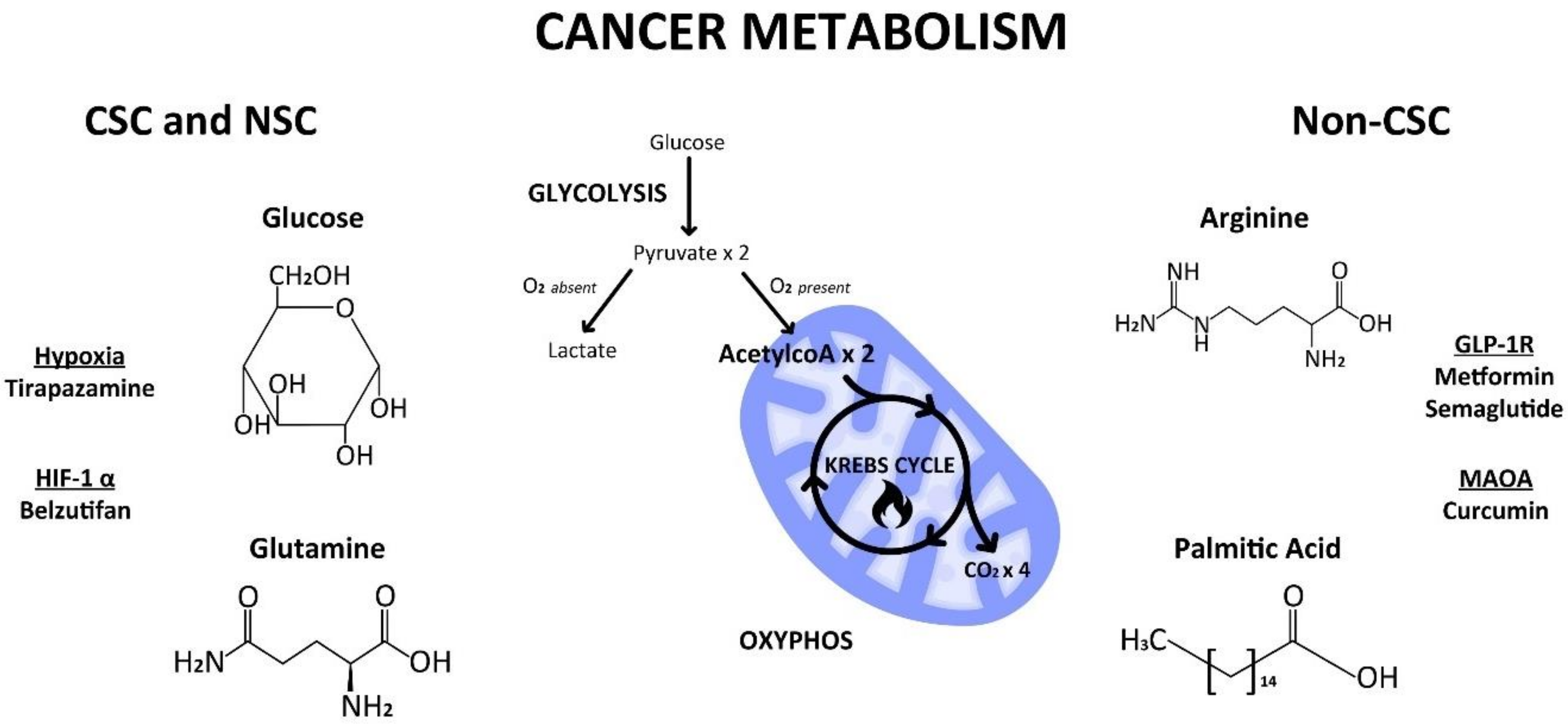
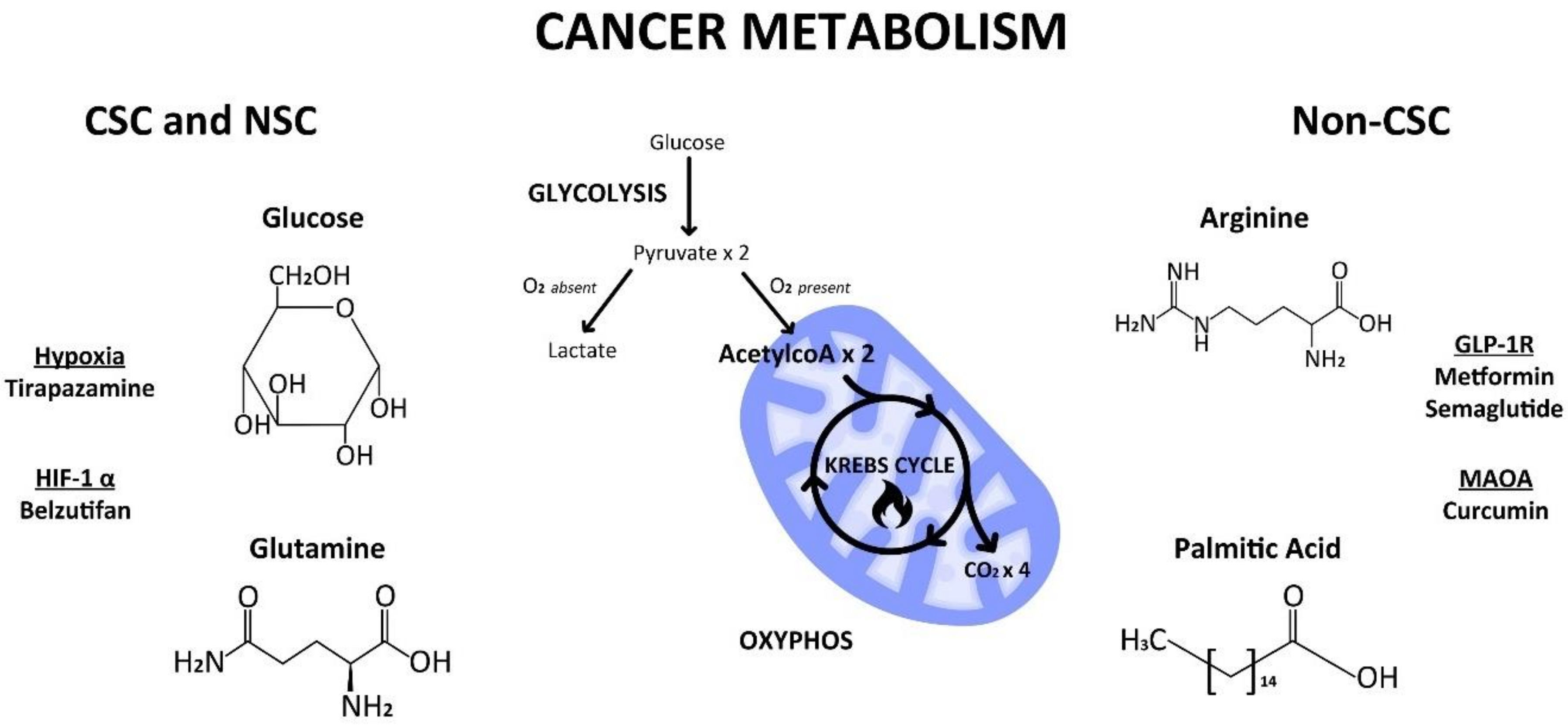
4. Cellular vs. Cancer Metabolism
4.1. Glycolysis
4.2. Mitochondrial Switch
4.3. HIF1α—Master Regulator
5. Metabolic Substrates
5.1. Glucose
5.2. Glutamine
5.3. Arginine
5.4. S-Adenosyl-L-methionine (SAM)
6. Clinical Implications
6.1. Aspartame Saga
6.2. Lipid Phobia
6.3. Ketogenic Diet
6.4. Weight Loss Shots or Pills
7. More Therapeutic Implications
7.1. An Exemplary Anti-CSC Drug
Metformin
7.2. A Neglected Anti-CSC Drug
MAOA Inhibitors
8. Drug vs. Therapy Development
8.1. Tirapazamine
8.2. Belzutifan
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Farber, S.; Diamond, L.K. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Shelton, L.M. Cancer as a metabolic disease. Nutr. Metab. 2010, 7, 7. [Google Scholar] [CrossRef]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef]
- Rous, P. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J. Exp. Med. 1911, 13, 397–411. [Google Scholar] [CrossRef]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sanchez, R.; Rodriguez-Enriquez, S.; Marin-Hernandez, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [PubMed]
- Werely, C.J.; Heyns, C.F.; van Velden, D.J.; van Helden, P.D. DNA fingerprint detection of somatic mutations in benign prostatic hyperplasia and prostatic adenocarcinoma. Genes Chromosomes Cancer 1996, 17, 31–36. [Google Scholar] [CrossRef]
- Firestein, G.S.; Echeverri, F.; Yeo, M.; Zvaifler, N.J.; Green, D.R. Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc. Natl. Acad. Sci. USA 1997, 94, 10895–10900. [Google Scholar] [CrossRef] [PubMed]
- Virchow, R. Die Krankhaften Geschwulste; A Hirschwald: Berlin, Germany, 1865; Volume 3. (In German) [Google Scholar]
- Stevens, L.C. Experimental production of testicular teratomas in mice. Proc. Natl. Acad. Sci. USA 1964, 52, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Pierce, G.B.; Dixon, F.J., Jr. Testicular teratomas. I. Demonstration of teratogenesis by metamorphosis of multipotential cells. Cancer 1959, 12, 573–583. [Google Scholar] [CrossRef]
- Sell, S.; Pierce, G.B. Maturation arrest of stem cell differentiation is a common pathway for the cellular origin of teratocarcinomas and epithelial cancers. Lab. Investig. 1994, 70, 6–22. [Google Scholar] [PubMed]
- Tu, S.M. Origin of Cancers: Clinical Perspectives and Implications of a Stem-Cell Theory of Cancer. In Cancer Treatment and Research; Rosen, S.T., Ed.; Springer: New York, NY, USA, 2010; Volume 154. [Google Scholar]
- Tu, S.M. Story of Hydra: Portrait of Cancer as a Stem-Cell Disease; Nova: New York, NY, USA, 2019; pp. 43–53. [Google Scholar]
- Tu, S.M.; Lin, S.H.; Logothetis, C.J. Stem-cell origin of metastasis and heterogeneity in solid tumours. Lancet Oncol. 2002, 3, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.M.; Moran, C.; Norton, W.; Zacharias, N.M. Stem cell theory of cancer: Origin of metastasis and sub-clonality. Semin. Diagn. Pathol. 2023, 40, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.M.; Zhang, M.; Wood, C.G.; Pisters, L.L. Stem cell theory of cancer: Origin of tumor heterogeneity and plasticity. Cancers 2021, 13, 4006. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.M.; Guo, C.C.; Chow, D.S.; Zacharias, N.M. Stem cell theory of cancer: Implications for drug resistance and chemosensitivity in cancer care. Cancers 2022, 14, 1548. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.M.; Estecio, M.; Lin, S.H.; Millward, N.Z. Stem cell theory of cancer: Rude awakening or bad dreams from cancer dormancy? Cancers 2022, 14, 655. [Google Scholar] [CrossRef] [PubMed]
- Simsek, T.; Kocabas, F.; Zheng, J.; Deberardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef]
- Hu, S.; Balakrishnan, A.; Bok, R.A.; Anderton, B.; Larson, P.E.Z.; Nelson, S.J.; Kurhanewicz, J.; Vigneron, D.B.; Goga, A. 13C-pyruvate imaging reveals alterations in glycolysis that precede c-myc-induced tumor formation and regression. Cell Metab. 2011, 14, 131–142. [Google Scholar] [CrossRef]
- San-Millan, I.; Julian, C.G.; Matarazzo, C.; Martinez, J.; Brooks, G.A. Is lactate an oncometabolite? Evidence supporting a role for lactate in the regulation of transcriptional activity of cancer-related genes in MCF7 breast cancer cells. Front. Oncol. 2020, 9, 1536. [Google Scholar] [CrossRef]
- Dang, C.V. PKM2 tyrosine phosphorylation and glutamine metabolism signal a different view of the Warburg effect. Sci. Signal. 2009, 2, 75. [Google Scholar] [CrossRef]
- Zhou, W.; Choi, M.; Margineantu, D.; Margaretha, L.; Hesson, J.; Cavanaugh, C.; Blau, C.A.; Horwitz, M.S.; Hockenbery, D.; Ware, C.; et al. HIF1α induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 2012, 31, 2103–2116. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.V.; Herst, P.M.; Tan, A.S. Metabolic flexibility and cell hierarchy in metastatic cancer. Mitochondrion 2010, 10, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Fischer, B.; Bavister, B.D. Oxygen tension in the oviduct and uterus of rhesus monkeys, hamsters and rabbits. J. Reprod. Fertil. 1993, 99, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Jeong, C.H.; Koo, S.Y.; Son, M.J.; Song, H.S.; Bae, S.K.; Raleigh, J.; Chung, H.Y.; Yoo, M.A.; Kim, K.W. Determination of hypoxic region by hypoxia marker in developing mouse embryos in vivo: A possible signal for vessel development. Dev. Dyn. 2002, 220, 175–186. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef]
- Pelicano, H.; Martin, D.S.; Xu, R.H.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef]
- Chu, Q.S.; Sanqha, R.; Spratlin, J.; Vos, L.J.; Mackey, J.R.; McEwan, A.J.B.; Venner, P.; Michelakis, E.D. A phase I open-labeled, ingle-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 603–610. [Google Scholar] [CrossRef]
- Raez, L.E.; Papadopoulos, K.; Ricart, A.D.; Chiorean, E.G.; DiPaola, R.S.; Stein, M.N.; Rocha Lima, C.M.; Schlesselman, J.J.; Tolba, K.; Langmuir, V.K.; et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 523–530. [Google Scholar] [CrossRef]
- Tu, S.M.; Singh, S.; Arnaoutakis, K.; Malapati, S.; Bhatti, S.A.; Joon, A.Y.; Atiq, O.T.; Posters, L.L. Stem cell theory of cancer: Implications for translational research from bedside to bench. Cancers 2022, 14, 3345. [Google Scholar] [CrossRef]
- Reitzer, L.J.; Wice, B.M.; Kennell, D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J. Biol. Chem. 1979, 254, 2669–2676. [Google Scholar] [CrossRef] [PubMed]
- Nutt, L.K. The Xenopus oocyte: A model for studying the metabolic regulation of cancer cell death. Sem. Cell Dev. Biol. 2012, 23, 412–418. [Google Scholar]
- Jin, J.; Byun, J.K.; Choi, Y.K.; Park, K.G. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp. Mol. Med. 2023, 55, 706–715. [Google Scholar] [PubMed]
- Lu, V.; Roy, I.J.; Torres, A., Jr.; Joly, J.H.; Ahsan, F.M.; Graham, N.A.; Teitell, M.A. Glutamine-dependent signaling controls pluripotent stem cell fate. Dev. Cell 2022, 57, 610–623. [Google Scholar] [PubMed]
- Vardhana, S.A.; Arnold, P.K.; Rosen, B.P.; Chen, Y.; Carey, B.W.; Huangfu, D.; Fontaine, C.C.; Thompson, C.B.; Finley, L.W.S. Glutamine independence is a selectable feature of pluripotent stem cells. Nat. Metab. 2019, 1, 676–687. [Google Scholar] [PubMed]
- Kim, C.S.; Ding, X.; Allmeroth, K.; Biggs, L.C.; Kolenc, O.I.; L’Hoest, N.; Chacon-Martinez, C.A.; Edlich-Muth, C.; Giavalisco, P.; Quinn, K.P.; et al. Glutamine metabolism controls stem cell fate reversibility and long-term maintenance in the hair follicle. Cell Metab. 2020, 32, 629–642. [Google Scholar] [PubMed]
- Liao, J.; Liu, P.P.; Hou, G.; Shao, J.; Yang, J.; Liu, K.; Lu, W.; Wen, S.; Hu, Y.; Huang, P. Regulation of stem-like cancer cells by glutamine through beta-catenin pathway mediated by redox signaling. Mol. Cancer 2017, 16, 51. [Google Scholar] [CrossRef]
- Pacifico, F.; Leonardi, A.; Crescenzi, E. Glutamine metabolism in cancer stem cells: A complex liaison in the tumor microenvironment. Int. J. Mol. Sci. 2023, 24, 2337. [Google Scholar] [CrossRef]
- Mossmann, D.; Muller, C.; Park, S.; Ryback, B.; Colombi, M.; Ritter, N.; Weibenberger, D.; Dazert, E.; Coto-Llerena, M.; Nuciforo, S.; et al. Arginine reprograms metabolism in liver cancer via RBM39. Cell 2023, 186, 5068–5083. [Google Scholar]
- Zhao, T.; Goh, K.J.; Ng, H.H.; Vardy, L.A. A role for polyamine regulators in ESC self-renewal. Cell Cycle 2012, 11, 4517–4523. [Google Scholar] [CrossRef]
- Rana, A.B.; Horton, T.M.; Thakur, V.S.; Welford, S.M. The polyamine acetylation enzyme SAT1 drives mesenchymal features and therapeutic resistance in glioblastoma. Cancer Res. 2023, 83 (Suppl. 7), 3674. [Google Scholar] [CrossRef]
- Khan, A.; Gemble, L.D.; Upton, D.H.; Ung, C.; Yu, D.M.T.; Ehteda, A.; Pandher, R.; Mayoh, C.; Hebert, S.; Jabado, N.; et al. Dual targeting of polyamine synthesis and uptake in diffuse intrinsic pontine gliomas. Nat. Commun. 2021, 12, 971. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.C.; To, M.D.; Westcott, P.M.K.; Delrosario, R.; Kim, I.J.; Philips, M.; Tran, Q.; Bollam, S.R.; Goodarzi, H.; Bayani, N.; et al. Targeting KRAS4A splicing through the RBM39/DCAF15 pathway inhibits cancer stem cells. Nat. Commun. 2021, 12, 4288. [Google Scholar] [CrossRef] [PubMed]
- Sperber, H.; Mathieu, J.; Wang, Y.; Ferreccio, A.; Hesson, J.; Xu, Z.; Fischer, K.A.; Devi, A.; Detraux, D.; Gu, H.; et al. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat. Cell Biol. 2015, 17, 1523–1535. [Google Scholar]
- Cui, Y.; Zhang, L.; Wang, W.; Ma, S.; Liu, H.; Zang, X.; Zhang, Y.; Guan, F. Downregulation of nicotinamide N-methyltransferase inhibits migration and epithelial-mesenchymal transition of esophageal squamous cell carcinoma via Wnt/beta-catenin pathway. Mol. Cell Biochem. 2019, 460, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, Y.; Li, G.; Yu, H.; Xie, X. Down-regulation of nicotinamide N-methyltransferase induces apoptosis in human breast cancer cells via the mitochondria-mediated pathway. PLoS ONE 2014, 9, e89202. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD+ homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar]
- Roberti, A.; Fernandez, A.F.; Fraga, M.F. Nicotinamide N-methyltransferase: At the crossroads between cellular metabolism and epigenetic regulation. Mol. Metab. 2021, 45, 101165. [Google Scholar]
- Wang, W.; Yang, C.; Wang, T.; Deng, H. Complex roles of nicotinamide N-methyltransferase in cancer progression. Cell Death Dis. 2022, 13, 267. [Google Scholar] [CrossRef]
- Van Haren, M.J.; Gao, Y.; Buijs, N.; Campagna, R.; Sartini, D.; Emanuelli, M.; Mateuszuk, L.; Kij, A.; Chlopicki, S.; Martinez de Castilla, P.E.; et al. Esterase-sensitive prodrugs of a potent bisubstrate inhibitor of nicotinamide N-methyltransferase (NNMT) display cellular activity. Biomolecules 2021, 11, 1357. [Google Scholar] [CrossRef]
- Olney, J.W.; Farber, N.B.; Spitznagel, E.; Robins, L.N. Increasing brain tumor rates: Is there a link to aspartame? J. Neuropathol. Exp. Neurol. 1996, 55, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Soffritti, M.; Belpoggi, F.; Esposti, D.D.; Lambertini, L.; Tibaldi, E.; Rigano, A. First experimental demonstration of the multipotential carcinogenic effects of aspartame administered in the feed to Sprague-Dawley rats. Environ. Health Perspect. 2006, 114, 379–385. [Google Scholar] [CrossRef]
- Soffritti, M.; Belpoggi, F.; Tibaldi, E.; Esposti, D.D.; Lauriola, M. Life-span exposure to low doses of aspartame beginning during prenatal life increases cancer effects in rats. Environ. Health Perspect. 2007, 115, 1293–1297. [Google Scholar] [CrossRef] [PubMed]
- Gnudi, F.; Panzacchi, S.; Tibaldi, E.; Iuliani, M.; Sgargi, D.; Bua, L.; Mandrioli, D. Hemolymphoreticular neoplasias from the Ramazzini Institute long-term mice and rat studies on aspartame. Ann. Glob. Health 2023, 89, 43. [Google Scholar] [CrossRef] [PubMed]
- Butchko, H.H.; Kotsonis, F.N. Acceptable daily intake vs actual intake: The aspartame example. J. Am. Coll. Nutr. 1991, 10, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Gezginci-Oktayoglu, S.; Ercin, M.; Sancar, S.; Celik, E.; Koyuturk, M.; Bolkent, S.; Bolkent, S. Aspartame induces cancer stem cell enrichment through p21, NICD and GLI1 in human PANC-1 pancreas adenocarcinoma cells. Food Chem. Toxicol. 2021, 153, 112264. [Google Scholar] [CrossRef]
- Pontel, L.B.; Rosado, I.V.; Burgos-Barragan, G.; Garaycoechea, J.I.; Yu, R.; Arends, M.J.; Chandrasekaran, G.; Broecker, V.; Wei, W.; Liu, L.; et al. Endogenous formaldehyde is a hematopoietic stem cell genotoxin and metabolic carcinogen. Mol. Cell 2015, 60, 177–188. [Google Scholar] [CrossRef]
- Pati, S.; Irfan, W.; Jameel, A.; Ahmed, S.; Shahid, R.K. Obesity and cancer: A current overview of epidemiology, pathogenesis, outcomes, and management. Cancers 2023, 15, 485. [Google Scholar] [CrossRef]
- Alvina, F.B.; Gouw, A.M.; Le, A. Cancer stem cell metabolism. The Heterogeneity of cancer metabolism. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2021; pp. 161–172. [Google Scholar]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.X. Lipid desaturation is a metabolic marker and therapeutic target of ovarian cancer stem cells. Cell Stem Cell 2017, 20, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Talib, W.H.; Mahmod, A.I.; Kamal, A.; Rashid, H.M.; Alashgar, A.M.D.; Khater, S.; Jamal, D.; Waly, M. Ketogenic diet in cancer prevention and therapy: Molecular targets and therapeutic opportunities. Curr. Issues Mol. Biol. 2021, 43, 558–589. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.T.; Lin, P.H.; Tolstikov, V.; Howard, L.; Chen, E.Y.; Bussberg, V.; Greenwood, B.; Narain, N.R.; Kiebish, M.A.; Freedland, S.J. Serum metabolomic analysis of men on a low-carbohydrate diet for biochemically recurrent prostate cancer reveals the potential role of ketogenesis to slow tumor growth: A secondary analysis of the CAPS2 diet trial. Prostate Cancer Prostatic Dis. 2022, 25, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Shah, U.A.; Iyengar, N.M. Plant-based and ketogenic diets as diverging paths to address cancer: A review. JAMA Oncol. 2022, 8, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.W.; Biton, M.; Haber, A.L.; Gunduz, N.; Eng, G.; Gaynor, L.T.; Tripathi, S.; Calibasi-Kocal, G.; Rickelt, S.; Butty, V.L.; et al. Ketone body signaling mediates intestinal stem cell homeostasis and adaptation to diet. Cell 2019, 178, 1115–1131. [Google Scholar] [CrossRef] [PubMed]
- Popociciu, M.S.; Paduraru, L.; Yahya, G.; Metwally, K.; Cavalu, S. Emerging role of GLP-1 agonists in obesity: A comprehensive review of randomized controlled trials. Int. J. Mol. Sci. 2023, 24, 10449. [Google Scholar] [CrossRef]
- Turton, M.D.; O’Shea, D.; Gunn, I.; Beak, S.A.; Edwards, C.M.; Meeran, K.; Choi, S.J.; Taylor, G.M.; Heath, M.M.; Lambert, P.D.; et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature 1996, 379, 69–72. [Google Scholar] [CrossRef]
- Sandoval, D.A.; Bagnol, D.; Woods, S.C.; D’Alessio, D.A.; Seeley, R.J. Arcuate glucagon-like peptide 1 receptors regulate glucose homeostasis but not food intake. Diabetes 2008, 57, 2046–2054. [Google Scholar] [CrossRef]
- Wilding, J.P.H.; Batterham, R.L.; Calanna, S.; Davies, M.; Van Gaal, L.F.; Lingvay, I.; McGowan, B.M.; Rosenstock, J.; Tran, M.T.; Wadden, T.A.; et al. Once-Weekly Semaglutide in Adults with Overweight or Obesity. N. Engl. J. Med. 2021, 384, 989–1002. [Google Scholar] [CrossRef]
- Jastreboff, A.M.; Aronne, L.J.; Ahmad, N.N.; Wharton, S.; Connery, L.; Alves, B.; Kiyosue, A.; Zhang, S.; Liu, B.; Bunck, M.C.; et al. Tirzepatide Once Weekly for the Treatment of Obesity. N. Engl. J. Med. 2022, 387, 205–216. [Google Scholar] [CrossRef] [PubMed]
- The Diabetes Prevention Program Research Group. Long-term safety, tolerability, and weight loss associated with metformin in the Diabetes Prevention Program Outcomes Study. Diabetes Care 2012, 35, 731–737. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, M.; Wen, Z.; Lu, Z.; Cui, L.; Fu, C.; Xue, H.; Liu, Y.; Zhang, Y. GLP-1 receptor agonists: Beyond their pancreatic effects. Front. Endocrinol. 2021, 12, 2021. [Google Scholar] [CrossRef]
- Kershner, A.M.; Shin, H.; Hansen, T.J.; Kimble, J. Discovery of two GLP-1/Notch target genes that account for the role of GLP-1/Notch signaling in stem cell maintenance. Proc. Natl. Acad. Sci. USA 2014, 111, 3739–3744. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Mohammad, A.; Pazdernik, N.; Huang, H.; Bowman, B.; Tycksen, E.; Schdl, T. GLP-1 Notch-LAG-1 CSL control of the germline stem cell fate is mediated by transcriptional targets lst-1 and sygl-1. PLoS Genet. 2020, 16, 1008650. [Google Scholar] [CrossRef]
- Sforza, A.; Vigorelli, V.; Rurali, E.; Perrucci, G.L.; Gambini, E.; Arici, M.; Metallo, A.; Rinaldi, R.; Fiorina, P.; Barbuti, A.; et al. Liraglutide preserves CD34+ stem cells from dysfunction induced by high glucose exposure. Cardiovasc. Diabetol. 2022, 21, 51. [Google Scholar] [CrossRef]
- Sanz, C.; Vazquez, P.; Blazquez, C.; Barrio, P.A.; Alvarez Mdel, M.; Blazquez, E. Signaling and biological effects of glucagon-like peptide 1 on the differentiation of mesenchymal stem cells from human bone marrow. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E634–E643. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Joo, B.S.; Lee, C.H.; Kim, H.Y.; Ock, J.H.; Lee, Y.S. Effect of Glucagon-like Peptide-1 on the Differentiation of Adipose-derived Stem Cells into Osteoblasts and Adipocytes. J. Menopausal Med. 2015, 21, 93–103. [Google Scholar] [CrossRef]
- Stouras, I.; Vasileiou, M.; Kanatas, P.F.; Tziona, E.; Tsianava, C.; Theocharis, S. Metabolic profiles of cancer stem cells and normal stem cells and their therapeutic significance. Cells 2023, 12, 2686. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef]
- Xiao, Y.; Yu, T.J.; Xu, Y.; Ding, R.; Wang, Y.P.; Jiang, Y.Z.; Shao, Z.M. Emerging therapies in cancer metabolism. Cell Metabol. 2023, 35, 1283–1303. [Google Scholar] [CrossRef]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed]
- Richards, K.A.; Liou, J.I.; Cryns, V.L.; Downs, T.M.; Abel, E.J.; Jarrard, D.F. Metformin use is associated with improved survival in patients with advanced prostate cancer on androgen deprivation therapy. J. Urol. 2018, 200, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Margel, D.; Urbach, D.R.; Lipscombe, L.L.; Bell, C.M.; Kulkarni, G.; Austin, P.C.; Flesher, N. Metformin use and all-cause and prostate cancer-specific mortality among men with diabetes. J. Clin. Oncol. 2013, 31, 3069–3075. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Zheng, Y.; Yao, Y.; Jia, R.; Ge, S.; Zhuang, A. Metformin and cancer hallmarks: Shedding new lights on therapeutic repurposing. J. Transl. Med. 2023, 21, 403. [Google Scholar] [CrossRef] [PubMed]
- Coyle, C.; Cafferty, F.H.; Vale, C.; Langley, R.E. Metformin as an adjuvant treatment for cancer: A systemic review and meta-analysis. Ann. Oncol. 2016, 27, 2184–2195. [Google Scholar] [CrossRef] [PubMed]
- Nishida, M.; Yamashita, N.; Ogawa, T.; Koseki, K.; Warabi, E.; Ohue, T.; Komatsu, M.; Matsushita, H.; Kakimi, K.; Kawakami, E.; et al. Mitochondrial reactive oxygen species trigger metformin-dependent antitumor immunity via activation of Nrf2/mTORC1/p62 axis in tumor-infiltrating CD8 T lymphocytes. J. Immunother. Cancer 2021, 9, e002954. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Yan, X.; Wintergerst, K.A.; Cai, L.; Keller, B.B.; Tan, Y. Nrf2: Redox and metabolic regulator of stem cell state and function. Trends Mol. Med. 2020, 26, 185–200. [Google Scholar] [CrossRef]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and act together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Del Barco, S.; Martin-Castillo, B.; Menendez, J.A. The anti-diabetic drug metformin suppresses self-renewal and proliferation of trastuzumab-resistant tumor-initiating breast cancer stem cells. Breast Cancer Res. Treat. 2011, 126, 355–364. [Google Scholar] [CrossRef]
- Cufi, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Martin-Castillo, B.; Joven, J.; Menendez, J.A. Metformin against TGFbeta-induced epithelial-to-mesenchymal transition (EMT): From cancer stem cell to age-associated fibrosis. Cell Cycle 2010, 9, 4461–4468. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Cufi, S.; Del Barco, S.; Martin-Castillo, B.; Menendez, J.A. Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-to-mesenchymal transition (EMT) status. Cell Cycle 2010, 9, 3807–3814. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Cufi, S.; Oliveras-Ferraros, C.; Martin-Castillo, B.; Joven, J.; Vellon, L.; Vazquez-Martin, A. Metformin and the ATM DNA damage response: Accelerating the onset of stress-induced senescence to boost protection against cancer. Aging 2011, 3, 1063–1077. [Google Scholar] [CrossRef]
- Oliveras-Ferraros, C.; Cufi, S.; Torres-Garcia, V.Z.; Del Barco, S.; Martin-Castillo, B.; Menendez, J.A. MicroRNA expression profile of breast cancer epithelial cells treated with the anti-diabetic drug metformin: Induction of the tumor suppressor miRNA let-7a and suppression of the TGF beta-induced oncomiRNA-181a. Cell Cycle 2011, 10, 1144–1151. [Google Scholar] [CrossRef]
- Alimova, I.N.; Liu, B.; Fan, Z.; Edgerton, S.M.; Dillon, T.; Lind, S.E.; Thor, A.D. Metformin inhibits breast cancer cell growth, colony formation, and induces cell cycle arrest in vitro. Cell Cycle 2009, 8, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Menendez, J.A. The antidiabetic drug metformin suppresses HER2 (erbB2) oncoprotein overexpression via inhibition of the mTOR effector p70S6K1 in human breast carcinoma cells. Cell Cycle 2009, 8, 88–96. [Google Scholar] [CrossRef]
- Zhuang, Y.; Miskimins, W.K. Cell cycle arrest in metformin treated breast cancer cells involves activation of AMPK, downregulation of cyclin D1, and requires p27kip1 or p21Cip1. J. Mol. Signal. 2008, 3, 18. [Google Scholar] [CrossRef] [PubMed]
- Sahra, I.B.; Laurent, K.; Loubat, A.; Giorgetti-Peraldi, S.; Colosetti, P.; Auberger, P.; Tanti, J.F.; Marchand-Brustel, Y.L.; Bost, F. The antidiabetic drug metformin exerts an anti-tumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 2008, 27, 3576–3586. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Gestwicki, J.E.; Murphy, L.O.; Klionsky, D.J. Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov. 2007, 6, 304–312. [Google Scholar] [CrossRef]
- Wu, J.B.; Shao, C.; Li, X.; Li, Q.; Hu, P.; Shi, C.; Li, Y.; Chen, Y.T.; Yin, F.; Liao, C.P.; et al. Monoamine oxidase A mediates prostate tumorigenesis and cancer metastasis. J. Clin. Investig. 2014, 124, 2891–2908. [Google Scholar] [CrossRef]
- Gross, M.E.; Agus, D.B.; Dorff, T.B.; Pinski, J.K.; Quinn, D.I.; Castellanos, O.; Gilmore, P.; Shih, J.C. Phase 2 trial of monoamine oxidase inhibitor phenelzine in biochemical recurrent prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 61–68. [Google Scholar] [CrossRef]
- Cheng, T.S.; Chen, W.C.; Lin, Y.Y.; Tsai, C.H.; Liao, C.I.; Shyu, H.Y.; Ko, C.J.; Tzeng, S.F.; Huang, C.Y.; Yang, P.C.; et al. Curcumin-targeting pericellular serine protease matriptase role in suppression of prostate cancer cell invasion, tumor growth and metastasis. Cancer Prev. Res. 2013, 6, 495–505. [Google Scholar] [CrossRef]
- Park, W.; Amin, A.R.; Chen, Z.G.; Shin, D.M. New perspectives of curcumin in cancer prevention. Cancer Prev. Res. 2013, 6, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Lord, S.R.; Harris, A.L. Is it still worth pursuing the repurposing of metformin as a cancer therapeutic? Br. J. Cancer 2023, 128, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.M.; Bilen, M.A.; Tannir, N.M. The scientific method: Pillar and pitfall of cancer research. Cancer Med. 2014, 3, 1035–1037. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, L.; Li, X.F. Targeting hypoxia: Hypoxia-activated prodrugs in cancer therapy. Front. Oncol. 2021, 11, 700407. [Google Scholar] [CrossRef]
- Williamson, S.K.; Crowley, J.J.; Lara, P.N., Jr.; McCoy, J.; Lau, D.H.; Tucker, R.W.; Mills, G.M.; Gandara, D.R.; Southwest Oncology Group Trial S0003. Phase III trial of paclitaxel plus carboplatin with or without tirapazamine in advanced non-small-cell lung cancer: Southwest Oncology Group Trial S0003. J. Clin. Oncol. 2005, 23, 9097–9104. [Google Scholar] [CrossRef] [PubMed]
- Rischin, D.; Peters, L.J.; O’Sullivan, B.; Giralt, J.; Fisher, R.; Yuen, K.; Trotti, A.; Bernier, J.; Bourhis, J.; Ringash, J.; et al. Tirapazamine, cisplatin, and radiation versus cisplatin and radiation for advanced squamous cell carcinoma of the head and neck (Trog 02.02, Headstart): A phase III trial of the Trans-Tasman Radiation Oncology Group. J. Clin. Oncol. 2010, 28, 2989–2995. [Google Scholar] [CrossRef]
- Fallah, J.; Brave, M.H.; Weinstock, C.; Mehta, G.U.; Bradford, D.; Gittleman, H.; Bloomquist, E.W.; Charlab, R.; Hamed, S.S.; Miller, C.P.; et al. FDA approval summary: Belzutifan for von Hippel-Lindau disease-associated tumors. Clin. Cancer Res. 2022, 28, 4843–4848. [Google Scholar] [CrossRef]
- Albiges, L.; Rini, B.I.; Peltola, K.; De Velasco Oria, G.A.; Burotto, M.; Suarez Rodriguez, C.; Ghatalia, P.; Iacovelli, R.; Lam, E.T.; Verzoni, E.; et al. Belzutifan versus everolimus in participants with previously treated advanced clear cell renal cell carcinoma: Randomized open-label phase III LITESPARK-005 study. Ann. Oncol. 2023, 34 (Suppl. 2), S1329–S1330. [Google Scholar] [CrossRef]
| Metabolism | Treatments | Mechanisms/Targets | References |
|---|---|---|---|
| Glucose | Dichloroacetate | Pyruvate dehydrogenase | [32] |
| 2-deoxyglucose | Hexokinase 2 | [33] | |
| Glutamine | L-asparaginase | Beta-catenin | [41] |
| Arginine | Indisulam | RBM39 | [47] |
| S-adenosyl-L-methionine | GYZ-319 | NAD+/NNMT | [48,49,50,51,52,53,54,55] |
| Lipids | Ketogenic diet | Ketone bodies | [67] |
| Semaglutide | GLP-1RA | [73] | |
| Tirzepatide | GIP and GLP-1RA | [74] | |
| Glycolysis | Metformin | Nrf | [91] |
| EMT | [94,95,96] | ||
| miRNA let-7 | [97] | ||
| HER2 | [98,99] | ||
| Cyclin D1 | [100,101] | ||
| AMPK, mTOR | [102] | ||
| Monoamines | MAOAi | AKT/FOXO1/TWIST1 | [103,104] |
| Curcumin | COX-2, NFkB | [105,106] | |
| Hypoxia | Tirapazamine | Hypoxia | [109,110,111] |
| Belzutifan | HIF1-alpha | [112,113] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tu, S.-M.; Chen, J.Z.; Singh, S.R.; Maraboyina, S.; Gokden, N.; Hsu, P.-C.; Langford, T. Stem Cell Theory of Cancer: Clinical Implications for Cellular Metabolism and Anti-Cancer Metabolomics. Cancers 2024, 16, 624. https://doi.org/10.3390/cancers16030624
Tu S-M, Chen JZ, Singh SR, Maraboyina S, Gokden N, Hsu P-C, Langford T. Stem Cell Theory of Cancer: Clinical Implications for Cellular Metabolism and Anti-Cancer Metabolomics. Cancers. 2024; 16(3):624. https://doi.org/10.3390/cancers16030624
Chicago/Turabian StyleTu, Shi-Ming, Jim Z. Chen, Sunny R. Singh, Sanjay Maraboyina, Neriman Gokden, Ping-Ching Hsu, and Timothy Langford. 2024. "Stem Cell Theory of Cancer: Clinical Implications for Cellular Metabolism and Anti-Cancer Metabolomics" Cancers 16, no. 3: 624. https://doi.org/10.3390/cancers16030624
APA StyleTu, S. -M., Chen, J. Z., Singh, S. R., Maraboyina, S., Gokden, N., Hsu, P. -C., & Langford, T. (2024). Stem Cell Theory of Cancer: Clinical Implications for Cellular Metabolism and Anti-Cancer Metabolomics. Cancers, 16(3), 624. https://doi.org/10.3390/cancers16030624