1. Introduction
Head and neck squamous cell carcinomas (HNSCC) are prevalent malignancies, accounting for about 900,000 new cases worldwide each year [
1,
2]. Many tumors are diagnosed in a locoregionally advanced stage, resulting in a 5-year survival rate of 40% to 60% despite multimodal treatment including surgery, radio- and chemotherapy [
3,
4]. Risk factors include tobacco smoking, alcohol consumption, and human papillomavirus (HPV) infection [
5]. Although HPV infections increase the risk for oropharyngeal cancer, they correlate with a better prognosis than HPV-negative disease, e.g., after radio-chemotherapy.
Cancer stem cells (CSCs) play a crucial role in therapy resistance and relapse in HNSCC, owing to their capacity for self-renewal and differentiation [
6,
7]. This is evident in the use of CSC markers such as CD44, CD98, and c-MET as prognostic biomarkers in HNSCC patients treated with radiotherapy [
8,
9,
10]. CSCs contribute to intra-tumor heterogeneity due to their high self-renewal and differentiation capacity and are associated with worse patient outcomes [
11,
12,
13]. With cellular barcoding, it was shown that single cells with high CD98 expression and ALDH activity in HNSCC cell lines contribute to clonogenic survival after irradiation [
14]. Moreover, it was demonstrated for various tumor entities that irradiation leads to an enrichment of CSCs with a radioresistant phenotype and elevated DNA repair capacity, controlled by metabolic and epigenetic mechanisms including DNA methylation, histone modification, and non-coding RNAs [
6,
15,
16,
17].
Notably, we demonstrated that the pharmacological inhibition of the histone methyltransferase (HMT) enhancer of zeste 2 polycomb repressive complex 2 (EZH2) using the chemical compound 3-deazaneplanocin A (DZNeP) resulted in the sensitization of radioresistant prostate cancer cells and reduced tumorigenicity [
18,
19]. Similarly, we identified epigenetic compounds that sensitized glioblastoma multiforme cell lines to photon and proton irradiation [
20].
The Cancer Genome Atlas (TCGA) network revealed a high frequency of mutations and focal deletions in genes such as the histone 3 lysine 36 methyltransferase 2D (
KMTD2, 5%) or the nuclear set domain gene (
NSD1/KMT3B, 4%) in HNSCC [
3]. Currently, there are several epigenetic targeting compounds under preclinical and clinical investigation for patients with HNSCC. Some of the compounds, such as the HDAC inhibitors vorinostat (SAHA) (phase II, NCT05608369), panobinostat (phase I, NCT00670553), and valproic acid (phase II, NCT01695122), are being tested in combination with radiotherapy (
www.clinicaltrials.gov).
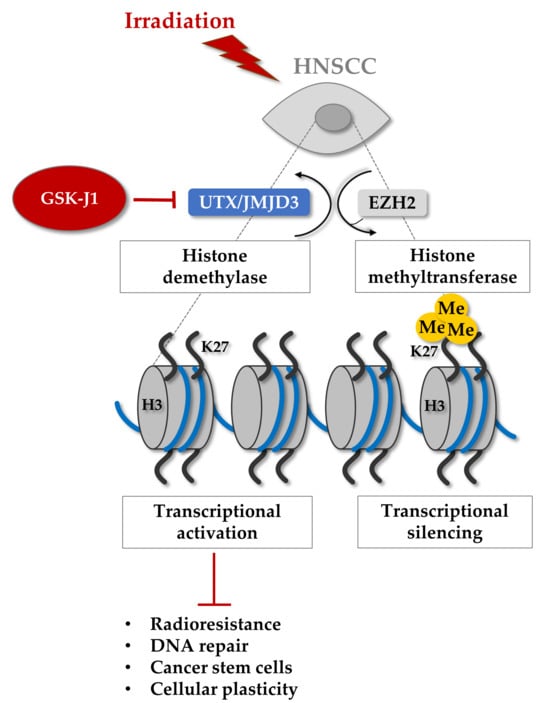
Therefore, we hypothesized that epigenetic targeting might be a promising strategy to sensitize HNSCC cells to irradiation by affecting CSC plasticity and DNA repair. In this study, we conducted a chemical library screen of epigenetic targeting agents to identify CSC-targeting and radiosensitizing compounds in HNSCC cell lines. We identified the histone demethylase inhibitor GSK-J1, characterized the underlying molecular mechanisms, and validated the prognostic potential of the target proteins UTX (KDM6A) and JMJD3 (KDM6B).
2. Materials and Methods
2.1. Cell Culture
Within this study, the human head and neck squamous cell carcinoma (HNSCC) cell lines FaDu(DD) (RRID:CVCL_VP44, ATCC), SAS (RRID:CVCL_1675, Health Science Research Resources Bank, Osaka, Japan), Cal33 (RRID:CVCL_1108, Deutsche Sammlung von Mikroorganismen und Zellkulturen DSMZ GmbH), and UT-SCC-5 (RRID:CVCL_7858, University Turku, Finland) were used. Radioresistant (RR) sublines were generated from indicated parental cell lines via selection with at least 15 fractions of 4 Gy and analyzed together with age-matched controls [
17]. All HNSCC cell lines were cultured in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% L-Glutamine, 1% 2-(4-(2-Hydroxyethyl)-1-piperazinyl)-ethansulfonacid (HEPES), 1% minimum essential medium (MEM), 1% non-essential amino acid (NEAA), and 1% sodium pyruvate. The patient-derived xenograft (PDX) cell line DK19 and the immortalized human skin keratinocyte cell line HaCaT (RRID:CVCL_0038, DKFZ, Heidelberg, Germany) were used as controls and were cultured in DMEM supplemented with 1% NEAA, 1% HEPES, and 1% sodium pyruvate. All cell lines were used for experiments until passage 15, tested for mycoplasma infection and SNP-based cell authentication on a regular basis.
2.2. DNA Methylome Analysis
Genomic DNA was isolated in triplicate from each cell population with the Qiamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s recommendations. Approximately 1 microgram of DNA with a concentration of 25 ng/µL was used. Human DNA methylation bead array analysis with the EPIC array (v1_b4, Illumina, San Diego, CA, USA) was conducted at the Microarray Unit at the Genomics and Proteomics Core Facility (GPCF, DKFZ, Heidelberg, Germany). Raw data (IDAT format files) was imported and processed for differential methylation analysis with the R package RnBeads. Quality control was performed and data was normalized using the ‘wm.dasen’ method. Moreover, background normalization was performed using the ‘methylumi.noob’ method. The exploratory analysis module of RnBeads was activated to perform dimension reduction and statistical association tests to visualize and address association between methylation and experimental groups of interest. Beta values (β) estimating the methylation level, e.g., within gene bodies, promoter regions, and CpG islands, were calculated. Differential methylation analysis was conducted on site and at the regional level according to the sample groups specified in the analysis.
p-values on the site level were computed using the limma method; i.e., hierarchical linear models from the limma package were employed and fitted using an empirical Bayes approach on derived M-values. Differential methylation was defined based on a 10% mean methylation difference between groups of interest and a false discovery rate (FDR)-adjusted
p-value < 0.05 (
Supplementary Table S2).
2.3. Mass Spectrometry-Based Analysis of Cellular Metabolisms
Seeded cells were treated, washed with ice-cold PBS, and harvested in 500 μL methanol. Extracts were centrifuged, and supernatants plus internal standard mix were dried in a speed vac concentrator. To determine the concentrations of Krebs cycle metabolites (succinate, fumarate, malate, citrate, isocitrate, cis-aconitate, α-ketoglutarate), 2-hydroxyglutarate, pyruvate, lactate, glutamate, glutamine, aspartate, and asparagine, liquid chromatography–tandem mass spectrometry (LC-MS/MS) was used as described previously at the experimental LC-MS/MS unit (Institute for Clinical Chemistry and Laboratory Medicine, Dresden, Germany). Briefly, on the day of analysis, dried samples were resuspended in 99/1 mobile phases A and B. A QTRAP 5500 triple quadrupole mass spectrometer (AB Sciex, Darmstadt, Germany) was coupled to an ACQUITY UPLC system from the Waters Corporation (Waters Corp., Eschborn, Germany) using an ACQUITY UPLC® HSS T3 column (1.8 μm, 2.1 × 100 mm) with guard column (Waters Corp.) for separation. Mobile phases consisted of 0.2% formic acid in water (A) and 0.2% formic acid in acetonitrile. The MS was used in negative electrospray ionization and multiple-reaction monitoring scan mode. To quantify the metabolites, a comparison of the analyte peak areas to calibrator peak areas from the stable isotope labeled internal standards in samples was applied. Raw data were normalized to analyzed cell count and specified as pg per cell.
2.4. Cell Irradiation
Cell irradiation was performed with the Maxishot Y.TU 320 machine (Yxlon International, Comet Group, Flamatt, The Switzerland) with 200 kV X-rays and a dose rate of 1.32 Gy/min at 20 mA. We used a collimator plate made of 7 mm beryllium and filtered the X-rays in the irradiation field (22 × 18 cm) with 0.5 mm copper. The absorbed dose was measured using a semiflex ionization chamber and the duplex dosimeter Unidos 11767 (PTW). Dose homogeneity was ensured through daily quality control and routine calibration.
2.5. Chemical Library Screen of Epigenetic Targeting Agents
For the chemical library screen, FaDu and Cal33 cells were seeded in 96-well plates for colony formation, sphere formation, and γH2AX assay. The following day, the Epigenetics Screening Library (Cat#11076, Cayman Chemical, Ann Arbor, MI, USA) was added at a final concentration of 5 μM. The chemical library contains 146 small molecules that are known to modulate the activity of different epigenetic ‘writer’, ‘reader’, and ‘eraser’ proteins, such as DNA and histone methyltransferases, DNA and histone demethylases, histone acetyltransferases, histone deacetylases, and acetylated histone binding proteins. After an additional 24 h, the plates were irradiated with 4 Gy. Colonies were counted on day 10, spheres on day 14, and γH2AX foci were counted 24 h after irradiation. The screen was performed in biological duplicates, each replicate consisting of one well per compound and 46 DMSO control wells. Compounds exhibiting a clonogenic plating efficiency of less than 5% in the unirradiated control were considered cytotoxic and excluded from further analysis. For each assay and compound, the values of both cell lines were grouped and compared to the mean DMSO value using a one-tailed t-test. Compounds with a statistically significant lower clonogenic/spherogenic survival fraction or higher number of γH2AX foci compared to the DMSO control were identified as hits, i.e., as having a radiosensitizing effect in the respective assay. Selected hits from the screen were further validated using a 3D colony formation assay in combination with single dose irradiation. A concentration equal to the 5% inhibitory concentration (IC5) was used after establishing the dose-response relationship by assessing cell viability using the CellTiterGlo (Promega, Madison, WI, USA).
2.6. Histone Modification Assay
The nuclear extraction of cells grown in culture was performed with the EpiQuik™ Nuclear Extraction Kit I (#OP-0002) according to the manufacturer’s recommendations. Five to 10 µg nuclear extracts were analyzed with the EpiQuik Histone Demethylase (H3K4 Specific) Activity/Inhibition Assay Kit (#P-3074-48) and EpiQuik Histone Demethylase (H3K9 Specific) Activity/Inhibition Fast Assay Kit (#P-3077-48) to screen for histone demethylase activity in the different cells lines after irradiation as well as with the Epigenase™ JMJD2 Demethylase Activity/Inhibition Assay Kit (Fluorometric) (#P-3081-48), Epigenase JARID Demethylase Activity/Inhibition Assay Kit (Fluorometric) (#P-3083-48), and Epigenase JMJD3/UTX Demethylase Activity/Inhibition Assay Kit (Fluorometric) (#P-3085-48). The plates were scanned with the Tecan Infinite.
2.7. Cell Proliferation Assay
Cells were plated in 96-well plates with cell counts between 3000 to 8000 cells per well depending on the cell line; 24 h after plating, cells were treated with increasing inhibitor concentrations and cell survival after 24 h was analyzed using the Luminescent Cell Viability Assay (CellTiterGlo, Promega) according to the manufacturer’s recommendation. The luminescence signal was measured with the Tecan microplate reader, and the half maximal inhibitory concentration (IC50) was calculated with GraphPad Prism software (version 8).
2.8. 3D Colony Forming Assay
The clonogenic survival assay was performed on Matrigel (Fisher Scientific, Waltham, MA, USA)-based 3D cultures after 1 h pre-treatment with the defined 5% inhibitory concentration (IC
5) of every compound with 2 Gy, 4 Gy, 6 Gy and 8 Gy irradiation. For this purpose, 1000 cells per well were seeded into 96-well culture plates coated with 50 µL of 1% low-melting agarose (Sigma Aldrich). A 100 µL quantity of cell suspension was mixed with Matrigel (1:20) and one hour before irradiation overlayed with 50 µL medium inhibitor containing media. We used the 5% inhibitory concentration (IC
5) of GSK-J1 with 4.5 µM and the solvent dimethyl sulfoxide (DMSO) as control. The colonies were scanned with the Celigo S image Cytometer (Nexcelom Biosience, Lawrence, MA, USA) after 6 to 8 days and counted with a threshold of diameter > 50 µm. The plating efficiency and surviving fraction were calculated according to the following formulas:
The obtained surviving fraction (SF) after a defined radiation dose (D) was fitted with a weighted, stratified, linear regression according to the linear–quadratic (LQ) formula: SF(D) = e−αD−βD^2 to determine the linear parameter α and the quadratic parameter β for different treatment conditions to perform group comparisons with the Statistical Package for the Social Sciences (SPSS) v23 software (IBM®, Chicago, IL, USA). To indicate the GSK-J1 inhibitor effect within the combined treatment together with irradiation, we calculated the dose modifying factor (DMF) as the ratio of doses with monotherapy versus a combination that causes the same level of biological effect.
2.9. Sphere Formation Assay
To determine self-renewal properties, cells were grown under non-adherent conditions in growth factor-defined human mammary epithelial cell growth medium (MEBM) consisting of B27 (Sigma-Aldrich, St. Louis, MO, USA), epidermal growth factor (Peqlab, Erlangen, Germany; 20 ng/mL), fibroblast growth factor (20 ng/mL), insulin (4 µg/mL), 1% penicillin-streptomycin, and glutamine. Cells were added into 24-well ultra-low attachment plates, with 5000 cells in one milliliter per well. One hour before irradiation, the cells were treated with an IC5 concentration of GSK-J1 (4.5 µM) and cultured for 14 days. Spheres were scanned with the Celigo S image Cytometer (Nexcelom Bioscience, Lawrence, Massachusetts, USA) and counted with a diameter >100 µm.
2.10. Generation of Color-Coded Cell Lines
To generate HNSCC cell lines stably expressing blue fluorescent protein (BFP), tandem-dimer Tomato (tdTomato), or enhanced green fluorescent protein (eGFP), HEK293 cells were used for lentivirus production. The cells were transfected, 24 h after plating, with calcium phosphate containing 2 μg psPAX2, 1 μg pMD2.G, and 2.5 μg pWPXL plasmid (Addgene, Watertown, Massachusetts, USA). The original green fluorescent protein (GFP) insert was replaced with either mTagBFP (blue fluorescent protein) or tdTomato. The viruses were collected at 24, 48, and 96 h after transfection, pooled, and passed through a 45 μm filter (Sarstedt). Between 100,000 and 200,000 HNSCC cells per well were plated in a 6-well plate one day before transduction individually defined per cell line. To obtain pure BFP and tdTomato-expressing cell populations, cells with the highest 5% intensity were isolated with fluorescence-activated cell sorting (FACS, BD FACSAria™ III; BD Biosciences, Franklin Lakes, NJ, USA). The stable expression of the fluorescent proteins was examined via flow cytometry and revealed a purity of 98.6% for Cal33-BFP, 97.7% for Cal33-tdTomato, 51.4% for FaDu-BFP, and 96.8% for FaDu-tdTomato cells.
2.11. siRNA-Mediated Knockdown
To test the radiobiological effects of the genes KDM6A and KDM6B in HNSCC cells, 250,000 cells per well were plated in 6-well plates and treated 24 h later with 0.4 µL siRNA (40 pmol, Eurofins, Luxembourg City, Luxembourg) mixed with 9 µL of Lipofectamine RNAiMAX (Thermo Fisher, Waltham, MA, USA) in OptiMEM media (Thermo Fisher). Untreated, Lipofectamine-treated (MOCK control) and scrambled siRNA (Scrambled#3: ‘CCC UUC AAC UCG GGA GCA ATT’) transfected cells were used as controls. Three specific siRNA sequences to target KDM6A (KDM6A-2852: ‘GGU UUA CUA AGU UCA GAC AAT’, KDM6A-738: ‘GGU ACU ACA GUU UAC AGU CTG’, KDM6A-4523: ‘GCU CAU UAC UGUAGC AUU UGT’) and KDM6B (KDM6B-3120: ‘GGU GCU AGA ATA GAU CAG CCG’, KDM6B-3657: ‘GCU GCG CUC ACU UAF UFA GGG’, KDM6B-1715: ‘GCA GUC GGA AAC CGU UCU UGG’) were used for each transcript. Treated cells were seeded into 96-well plates with 1500 cells in each well, plated for clonogenic survival assay, and irradiated for 24 h after knock-down. The knock-down efficiency was validated on protein level via Western blot.
2.12. Western Blot
Cell cultures were washed with PBS and lysed with radioimmunoprecipitation assay (RIPA) buffer with cell scraper on ice. After centrifugation, protein extracts were measured with the bicinchoninic acid (BCA) assay. Bovine serum albumin (BSA) was used as reference to calculate the protein concentration. Equally protein concentrations between different samples were applied. Before loading the NuPAGE 1–12% gels (Invitrogen), samples were mixed with 4× SDS sample puffer including dithiothreitol (DDT) for 5 min at 95 °C. After protein separation according to their kDa size within the gel, they were transferred to an 0.2 µm nitrocellulose membrane (Amersham Protran, GE Healthcare, Chicago, IL, USA). The semi-dry transfer was performed within 1× NuPage transfer buffer. Afterwards, the membrane was blocked with 5% bovine serum albumin (BSA) in 1× PBS containing 0.1% Triton-X100. To analyze specific proteins, the membranes were incubated with following primary antibodies overnight: anti-UTX (Cell Signaling, Danvers, MA, USA, #33510S, 1:1000), anti-JMJD3 (Cell Signaling, #3457S 1:1000), anti-Histone H3 (clone: D1H2, Cell Signaling, #4499T, 1:2000), anti-H3K27me3 (Active Motif, Carlsbad, CA, USA, #039158, 1:5000), and, as loading control, anti-β-actin (#8H10D10). After washing, the membranes were incubated with secondary anti-mouse or anti-rabbit antibody (Cell Signaling, #3700, 1:1000; GE Healthcare, #NA931, 1:10,000 or GE Healthcare, #NA934, 1:10,000). For protein detection, SuperSignal™ West Dura Extended Duration Substrate (Thermo Scientific, Waltham, MA, USA) was added. The chemiluminescence signals were imaged with Fusion FX EDGE (Vilber, Marne-la-vallée, France) and automatically analyzed with ImageJ software (win-64, Fiji.app, accessed on 28 of April 2019).
2.13. Flow Cytometry
Cells were harvested with Accutase (Sigma-Aldrich) and stained either with the Aldefluor assay (Stem cell technologies) according to the manufacture’s recommendation or in PBS buffer with 5% FBS, 1% HEPES and 0.5 mM EDTA with anti-human CD44 (1:100, clone: IM7, #48-0441-80, Thermo Fisher) and anti-CD133 (1:50, clone: TMP4, #130133186, eBioscience). To exclude dead cells from the analysis, cells were additionally stained with the viability dye 7-Aminoactionomycin D (1:1000, Thermo Fisher). The samples were acquired with the BD FACSCelesta and analyzed with FlowJo software (version 10.9.0). The ALDH+ population was gated based on background fluorescence from the negative control, the diethylaminobenzaldehyde (DEAB) treated cells. The other CSC markers were gated according to the isotype controls (IgG-eFluor 450, eBioscience, #48-403182; rat anti-mouse IgG1-PE, eBioscience, #130-113-200).
2.14. RNA Sequencing
RNA was isolated after GSK-J1 treatment cells using the RNeasy mini kit (Qiagen; #74104) according to the manufacturer’s recommendation, in triplicate. The concentrations were measured with the NanoDrop ND-1000 (Peqlab). RNA sequencing was performed at the Genomics and Proteomics Core Facility (GPCF, DKFZ, Heidelberg, Germany). Libraries were prepared using the Illumina TruSeq Stranded Total RNA Library Prep Kit following the manufacturer’s instructions. Libraries concentration and quality control were performed using Qubit (Invitrogen) and Tapestation (Agilent). The samples were sequenced in a 2 × 100 bp paired-end setting on an Illumina HiSeq 4000 system according to the manufacturer’s protocol. The RNAseq raw data was demultiplexed, aligned, and mapped to genes to generate a raw count table following the DKFZ/ODCF RNAseq workflow (version 1.3.0-2). Down-stream data processing and statistical analysis were performed using DeSeq2 (version 1.30.1) in R 4.0.3 [
21] excluding genes with low count number (<one count) from further analysis. Raw feature counts were transformed by the variance-stabilizing transformation (vst) and subsequent visualization with the ggplot2 R package (version 3.3.3,
https://ggplot2.tidyverse.org, accessed on 4 January 2021). Functional enrichment and pathway analysis were carried out, with the genes having a Log2 fold change >2 and adjusted
p-value < 0.05 in gProfiler (
https://biit.cs.ut.ee/gprofiler/gost).
2.15. HNSCC Xenograft Model
Cal33 and FaDu xenografts were generated via subcutaneous transplantation of small pieces of tumors generated from a cryopreserved stock into the right hind leg of anesthetized NMRI (nu/nu) mice. Cisplatin (Calbiochem, Germany, 3 mg/kg b.w.) was dissolved in sodium chloride (0.9%) and administrated i.p. at the first day of treatment and then once weekly directly before irradiation as described by Koi et al. [
22]. Control animals were injected with the same volume of sodium chloride. Local tumor irradiations were given with 18 and 12 Gy in 10 fractions for FaDu and Cal33, respectively. Tumors were excised 24 h after the last irradiation fraction for histological evaluation. The experimental procedure and the workflows within the animal facility followed the German animal welfare regulations and the ARRIVE guidelines and were approved by the institutional ethics committee (no: DD24-5131/207/34).
2.16. Immunfluorescence Staining
Tumor tissues were sectioned into 7-µm-thick slices for immunofluorescence staining. Slides were fixed with methanol and acetone and washed with PBS. Unspecific binding was prevented via incubation in DAKO protein block (Agilent Dako, Santa Clara, California, USA) for 60 min at room temperature (RT). Primary antibodies against tri-methyl-histone H3 (#9733S, Cell Signaling), Histone H3 (#4499S, Cell Signaling), JMJD3 (#PA535012, Thermo Fisher Scientific) and UTX (#33510S, Cell Signaling) were diluted in 0.5% BSA in 1× PBS and incubated overnight at 4 °C in a humidified chamber. The samples were washed with 3% BSA in 1× PBS. Diluted Alexa Fluor 488 conjugated secondary antibodies were added and incubated for one hour at room temperature in the dark. Cell nuclei were stained with DAPI, and the samples were mounted with Mowiol mounting medium. Finally, the slides were scanned with a 20× magnification using a Zeiss Axio Scan.Z1 slide scanner (Zeiss, Jena, Germany).
2.17. In Silico Analysis of HNSCC Patient Data
KDM6A/B expression was investigated in tumor specimens of HNSCC patients that had received adjuvant radio/chemotherapy (RCTx). This independent explorative, retrospective study was conducted by the German Cancer Consortium Radiation Oncology Group (DKTK-ROG) and published by Lohaus et al. [
23]. Ethical approval for clinical and biological data was obtained from the Ethics Committees of all DKTK partner sites. Briefly, 221 patients with locally advanced HNSCC received post-operative radio/chemotherapy (RCTx) based on cisplatin (81.6%) or mitomycin C (18.4%) between 2005 and 2011. For 187 out of 221 patients, tissue was available for gene expression profiling with the Affymetrix HTA2.0 platform. Patients were stratified according to a median threshold defined with the maxstat function (log-rank method) with the endpoints loco-regional control (LRC) to illustrate radiotherapy-specific local relapse, overall survival (OS), and distant metastasis (DM). Kaplan–Meier survival curves, log–rank statistics and hazard ratio were calculated with the GraphPad Prism software (version 8). The validation was performed on available data from the retrospective HNSCC cohort (
n = 137) of the DKTK-ROG that received primary RCTx [
24], including univariable and multivariable Cox regression analysis (
Supplementary Table S1). The parameter age, tumor localization, HPV16 DNA, HPV16 RNA, P16 protein, p53 protein,
EZH2,
HIF1A,
KDM5B, and
KDM6A were included for multivariate analysis. Clinical and research data management was managed with the open-source software RadPlanBio (v1.0.0.14, 10.5281/zenodo.8298621,cacess date: 29 August 2023), a radiation/dose/plan, image/biomarker, and outcome platform (RPB).
2.18. Statistics
Radiobiological experiments were performed at least in three technical replicates, partially containing additional technical replicates depending on the readout. Statistical analyses to test for significant differences between experimental groups were performed using either a two-tailed t-test or ANOVA, depending on the experimental design, using SPSS (v23, IBM®, Chicago, IL, USA) or GraphPad Prism software (version 8, LLC Company, Boston, MA, USA). A one-tailed t-test was used for the Epigenetics Library Screen. A p-value < 0.05 was considered statistically significant and marked with *. Data are illustrated as violin plots including individual values or mean values including error bars as standard deviation (SD) or standard error of the mean (SEM) as indicated.
4. Discussion
Tumor heterogeneity and cellular plasticity have a significant impact on tumor progression, therapy resistance, and metastatic dissemination. This heterogeneity may arise from CSCs that can dynamically respond to environmental changes, therapeutic interventions, or immune cell attacks [
26,
27]. CSCs play a critical role in influencing tumor radiosensitivity, and achieving lasting tumor control necessitates the complete elimination of all CSCs [
6,
28]. However, an increased cellular plasticity observed during radiotherapy has been documented, which hinders the effective elimination of CSCs in various tumor types, including HNSCC [
15,
17,
18,
29]. CSC plasticity may result from epigenetic reprogramming [
26] and involves histone modifications as well as DNA methylation [
30,
31,
32,
33].
In our study, we observed profound epigenetic and metabolic alterations in radioresistant HNSCC cells and ALDH
+ CSCs. These findings led us to hypothesize that modulating the epigenetic landscape through the inhibition of epigenetic regulatory proteins could serve as a promising therapeutic strategy for radiosensitizing of HNSCC. To identify potential compounds, we conducted a high-throughput chemical library screening including various clinically relevant epigenetic inhibitors, which revealed six compounds with high radiosensitizing and CSC-targeting potential. One of these compounds is the histone H3K27 methyltransferase EZH2 inhibitor DZNeP, which we previously identified as a candidate that targets ALDH
+ CSCs in prostate cancer [
18]. The most promising new candidate we identified for HNSCC is the histone demethylase inhibitor GSK-J1, which targets UTX (
KDM6A) and JMJD3 (
KDM6B).
UTX is required for embryonic development and lineage-specific differentiation [
34]. In HNSCC, UTX is frequently mutated and has been described as a metabolically regulated, X-linked tumor suppressor [
35,
36,
37]. Moreover, the activity of the oxygen-sensitive UTX (
KDM6A) is reduced under hypoxic conditions, whereas its paralog JMJD3 (
KDM6B) remains unaffected [
38]. Therefore, GSK-J1 may influence cancer cell radiosensitivity by influencing the oxidative stress response of cancer cells [
39]. Another proposed inducer of JMJD3 (
KDM6B) transcription is the canonical NOTCH signaling [
40]. Down-stream of JMJD3, genes involved in response to cellular and environmental stresses, differentiation, proliferation, senescence, and apoptosis are positively regulated. Both UTX (
KDM6A) and JMJD3 (
KDM6B) are Jumonji motif-containing demethylases and dependent on α-ketoglutarate and Fe
2+ for efficient enzymatic activity. The identified compound GSK-J1 is a competitive inhibitor and prevents the binding of both cofactors to JMJD3 and UTX.
Chemosensitizing effects, reduced cell proliferation, and apoptosis induction have been previously shown for GSK-J1 in HNSCC cells. Zhang et al. combined GSK-J1 with TCP, an inhibitor of the H3K4me1/2-specific HDM LSD1 (
KDM1A), and found synergistic effects [
41]. Kruidenier et al. assessed the target specificity of GSK-J1 with an AlphaScreen assay and found inhibitory potency against JARID1B (
KDM5B) and JARID1B (
KDM5C) [
42]. Additionally, GSK-J4, a pro-drug and ethyl ester derivative of GSK-J1, which penetrates cells more effectively and is hydrolyzed intracellularly to GSK-J1, demonstrated radiosensitizing effects in patient-derived xenograft models of diffuse intrinsic pontine glioma (DIPG). The authors noted a reduced expression of DNA repair genes and changes in DNA accessibility through ATAC sequencing after GSK-J4 treatment. This led to an increased number of sustained DNA double-strand breaks after irradiation, as demonstrated by the γH2AX and 53BP1 foci assay, as well as reduced clonogenic survival and increased survival of tumor-bearing mice treated with GSK-J4 in combination with radiotherapy [
43]. Our study in HNSCC cell lines revealed similar findings, showing radiosensitizing effects of GSK-J1 in 3D clonogenic survival and γH2AX assays. Furthermore, we discovered that GSK-J1 treatment prior to irradiation prevents irradiation-induced plasticity, as indicated by reduced sphere forming potential and a decreased CSC marker expression.
Limitation: The epigenetic inhibitor screen was carried out within 2D culture models. Those model systems have the disadvantage that they do not represent cell-cell and cell-ECM interactions. Therefore, we applied Matrigel-based 3D culture and sphere formation assays in liquid cultures because those model systems mimic cell interactions, cell morphology, microenvironment, oxygen, and nutrient gradients as existing in human tumors. The strengths of the performed epigenetic inhibitor screening is the read-out with three different biological endpoints for radiosensitivity, self-renewal and DNA repair to identify compounds targeting several pathways involved in radioresistance. The screening approach could be improved in the future through the addition of different compound concentrations and treatment time points. Moreover, a validation of our findings in murine models would be necessary before clinical translation.
Outlook: The identification of UTX (
KDM6A) as putative therapeutic target and/or potential prognostic biomarker for HNSCC patients undergoing radiotherapy draws our attention to the currently upcoming discussion about the importance of X- and Y-linked tumor suppressor genes and/or oncogenes influencing sex-specific cancer incidence and prognosis [
44]. UTX (
KDM6A) is located on the X chromosome and carries sex-specific mutations associated with an increased cancer incidence in males [
45,
46]. Within our study, we used the HNSCC cell lines Cal33 and FaDu, which originate from male patients, whereas SAS was derived from a female HNSCC patient. These sex differences may influence the sensitivity to GSK-J1 treatment. Further projects investigating these findings are needed to implement them within clinical workflows and improve cancer therapy efficiency through sex-specific prognosis and treatment.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




