
Drug Response of Patient-Derived Lung Cancer Cells Predicts Clinical Outcomes of Targeted Therapy

 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. PDC Establishment
2.2. Drug Sensitivity Screening Using PDCs
2.3. Cell Line and Reagents
2.4. Clinical Data Collection
2.5. Targeted Next-Generation Sequencing (NGS) of PDCs
2.6. Statistical Analysis
3. Results
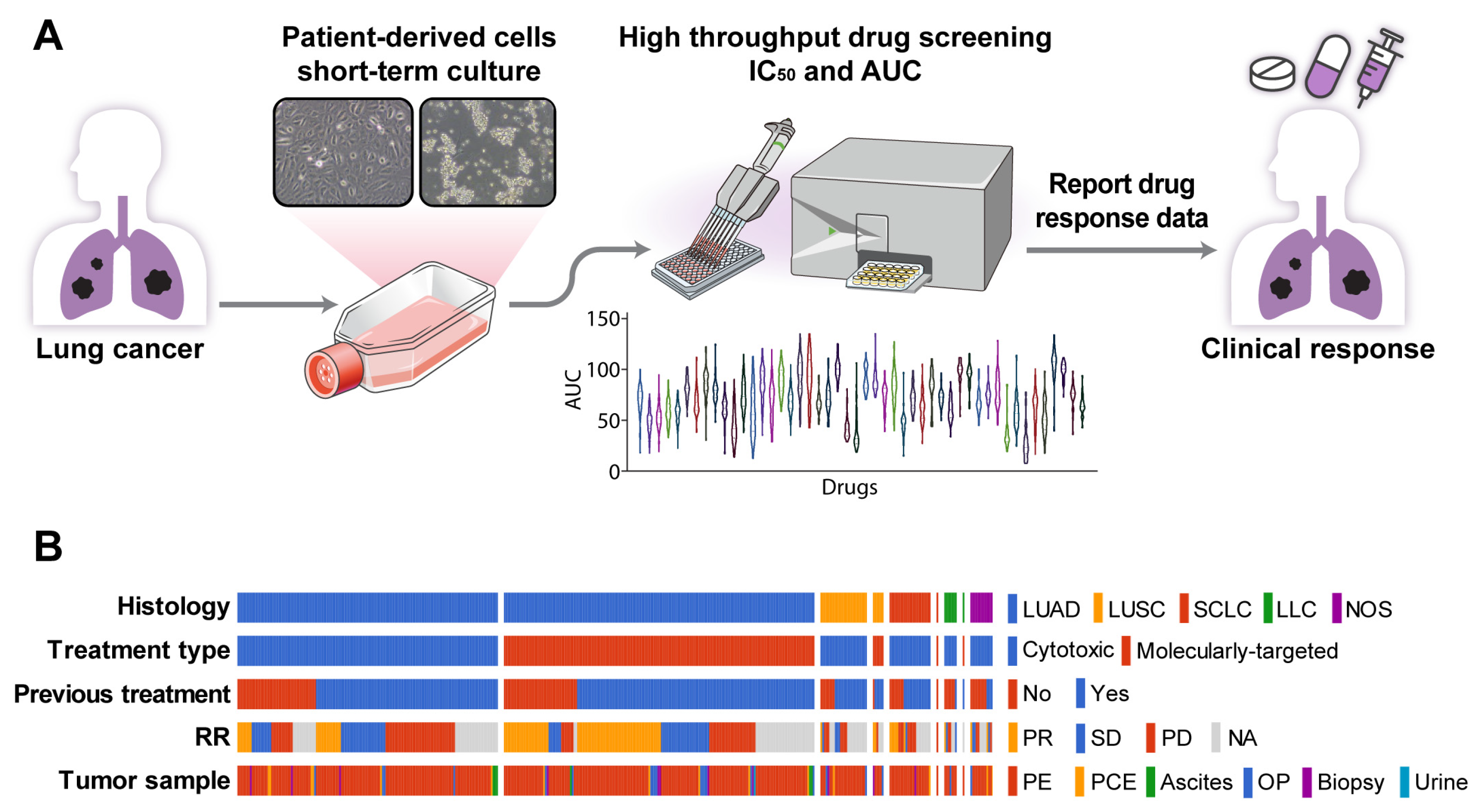
3.1. Establishment of PDC Drug Screening Platform
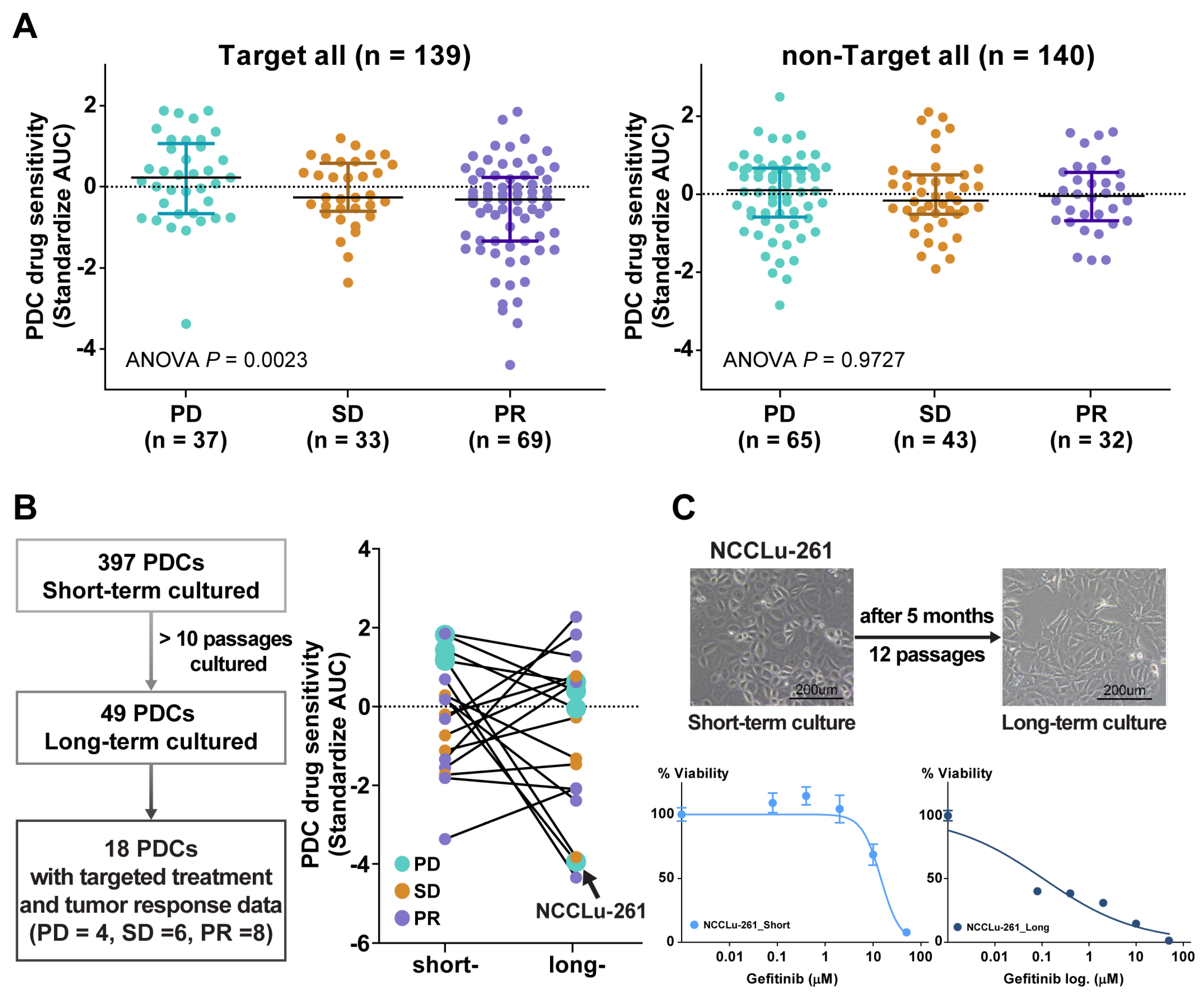
3.2. Association between PDC Drug Sensitivity and Clinical Tumor Response
3.3. PDC Drug Sensitivity Predicts Clinical Outcome in Treatment-Naive EGFR- or ALK-Positive NSCLC
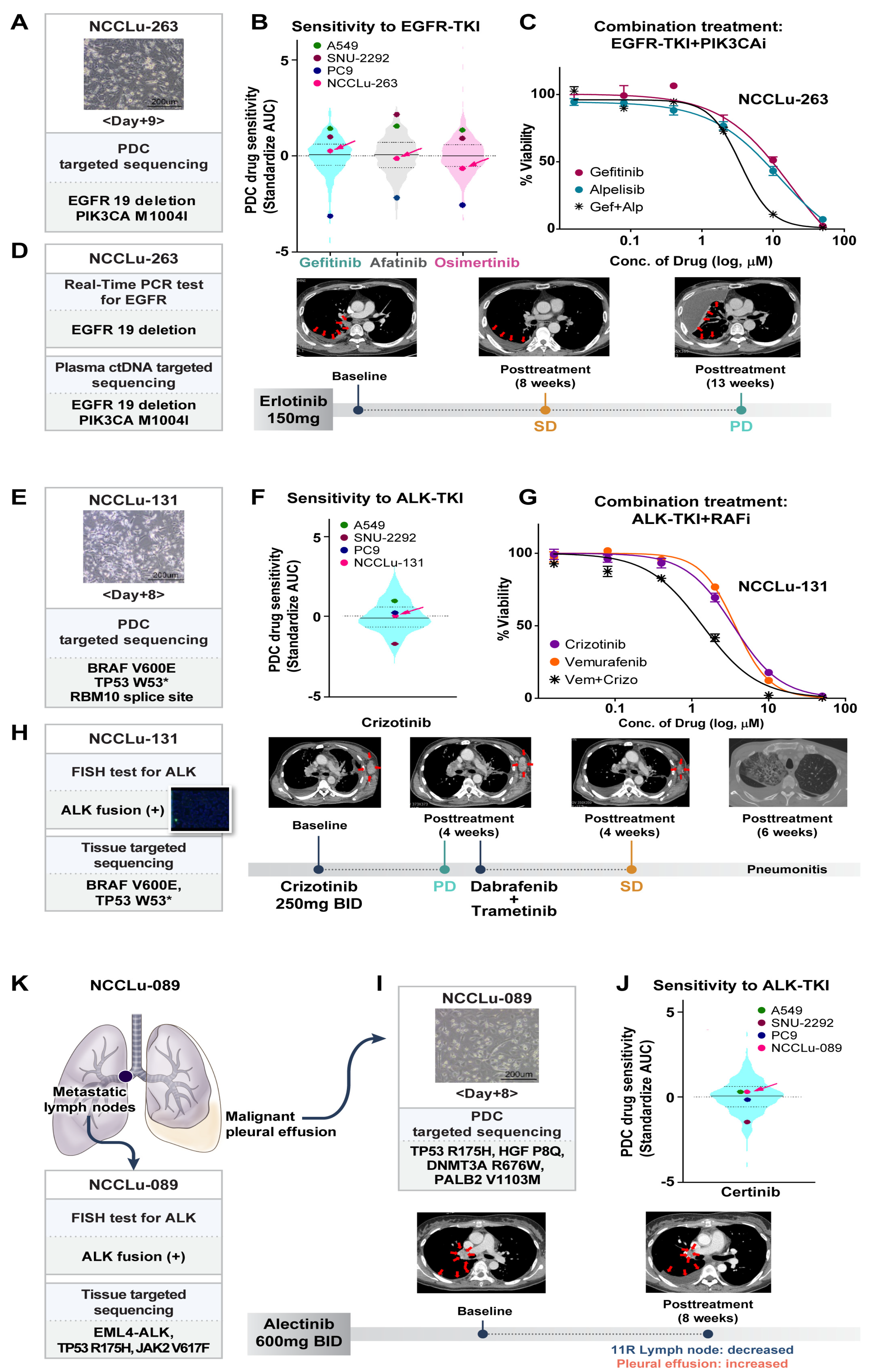
3.4. Causes of a Poor Response to EGFR- or ALK-TKIs
3.4.1. Concurrent Resistant Gene Alterations
3.4.2. Intra-Tumoral Heterogeneity
3.4.3. Inter-Lesional Tumor Heterogeneity
3.4.4. Histology
3.5. PDC-Based Prediction of an Unexpected Response to EGFR-Targeted Drugs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoeben, A.; Joosten, E.A.J.; van den Beuken-van Everdingen, M.H.J. Personalized Medicine: Recent Progress in Cancer Therapy. Cancers 2021, 13, 242. [Google Scholar] [CrossRef]
- Morgensztern, D.; Campo, M.J.; Dahlberg, S.E.; Doebele, R.C.; Garon, E.; Gerber, D.E.; Goldberg, S.B.; Hammerman, P.S.; Heist, R.S.; Hensing, T.; et al. Molecularly targeted therapies in non-small-cell lung cancer annual update 2014. J. Thorac. Oncol. 2015, 10, S1–S63. [Google Scholar] [CrossRef]
- Chan, B.A.; Hughes, B.G. Targeted therapy for non-small cell lung cancer: Current standards and the promise of the future. Transl. Lung Cancer Res. 2015, 4, 36–54. [Google Scholar]
- Fukuoka, M.; Wu, Y.; Thongprasert, S.; Sunpaweravong, P.; Leong, S.; Sriuranpong, V.; Chao, T.; Nakagawa, K.; Chu, D.; Saijo, N.; et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). J. Clin. Oncol. 2011, 29, 2866–2874. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.; Thongprasert, S.; Yang, C.; Chu, D.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Guo, Y.; Song, J.; Wang, Y.; Huang, L.; Sun, L.; Zhao, J.; Zhang, S.; Jing, W.; Ma, J.; Han, C. Concurrent Genetic Alterations and Other Biomarkers Predict Treatment Efficacy of EGFR-TKIs in EGFR-Mutant Non-Small Cell Lung Cancer: A Review. Front. Oncol. 2020, 10, 610923. [Google Scholar] [CrossRef]
- Janku, F. Tumor heterogeneity in the clinic: Is it a real problem? Ther. Adv. Med. Oncol. 2014, 6, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Du, C.; Lu, L.; Yuan, S.; Zhan, M.; You, P.; Du, H. Opportunities and challenges of patient-derived models in cancer research: Patient-derived xenografts, patient-derived organoid and patient-derived cells. World J. Surg. Oncol. 2022, 20, 37. [Google Scholar] [CrossRef]
- Huo, K.G.; D’Arcangelo, E.; Tsao, M.S. Patient-derived cell line, xenograft and organoid models in lung cancer therapy. Transl. Lung Cancer Res. 2020, 9, 2214–2232. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.Y.; Kim, S.Y.; Joo, J.Y.; Yun, D.H.; Jung, M.R.; Yun, J.D.; Heo, J.; Ahn, S.G.; Park, C.W.; et al. Patient-Derived Cells to Guide Targeted Therapy for Advanced Lung Adenocarcinoma. Sci. Rep. 2019, 9, 19909. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, S.Y.; Park, C.; Kim, N.K.; Jang, J.; Park, K.; Yi, J.H.; Hong, M.; Ahn, T.; Rath, O.; et al. Patient-derived cell models as preclinical tools for genome-directed targeted therapy. Oncotarget 2015, 6, 25619–25630. [Google Scholar] [CrossRef]
- Lee, J.K.; Liu, Z.; Sa, J.K.; Shin, S.; Wang, J.; Bordyuh, M.; Cho, H.J.; Elliott, O.; Chu, T.; Choi, S.W.; et al. Pharmacogenomic landscape of patient-derived tumor cells informs precision oncology therapy. Nat. Genet. 2018, 50, 1399–1411. [Google Scholar] [CrossRef]
- Sa, J.K.; Hwang, J.R.; Cho, Y.J.; Ryu, J.Y.; Choi, J.J.; Jeong, S.Y.; Kim, J.; Kim, M.S.; Paik, E.S.; Lee, Y.Y.; et al. Pharmacogenomic analysis of patient-derived tumor cells in gynecologic cancers. Genome Biol. 2019, 20, 253. [Google Scholar] [CrossRef] [PubMed]
- Sa, J.K.; Hong, J.Y.; Lee, I.K.; Kim, J.S.; Sim, M.H.; Kim, H.J.; An, J.Y.; Sohn, T.S.; Lee, J.H.; Bae, J.M.; et al. Comprehensive pharmacogenomic characterization of gastric cancer. Genome Med. 2020, 12, 17. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Bauer, T.M.; de Marinis, F.; Felip, E.; Goto, Y.; Liu, G.; Mazieres, J.; Kim, D.; Mok, T.; Polli, A.; et al. First-Line Lorlatinib or Crizotinib in Advanced ALK-Positive Lung Cancer. N. Engl. J. Med. 2020, 383, 2018–2029. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Yang, Y.; Hu, J.; Xu, H.; Zhang, S.; Wang, Y. EGFR exon 20 insertion variants A763_Y764insFQEA and D770delinsGY confer favorable sensitivity to currently approved EGFR-specific tyrosine kinase inhibitors. Front. Pharmacol. 2022, 13, 984503. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, L.; Zhu, Y.; Huang, C.; Qin, Y.; Liu, H.; Ren Heidenreich, L.; Shi, B.; Ren, H.; Chu, X.; et al. Coexistence of EGFR with KRAS, or BRAF, or PIK3CA somatic mutations in lung cancer: A comprehensive mutation profiling from 5125 Chinese cohorts. Br. J. Cancer 2014, 110, 2812–2820. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.; Woo, K.M.; Sima, C.S.; Plodkowski, A.; Hellmann, M.D.; Chaft, J.E.; Kris, M.G.; Arcila, M.E.; Ladanyi, M.; Drilon, A. Impact of Concurrent PIK3CA Mutations on Response to EGFR Tyrosine Kinase Inhibition in EGFR-Mutant Lung Cancers and on Prognosis in Oncogene-Driven Lung Adenocarcinomas. J. Thorac. Oncol. 2015, 10, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.G.; Chang, Y.L.; Yu, C.J.; Yang, P.C.; Shih, J.Y. The Role of PIK3CA Mutations among Lung Adenocarcinoma Patients with Primary and Acquired Resistance to EGFR Tyrosine Kinase Inhibition. Sci. Rep. 2016, 6, 35249. [Google Scholar] [CrossRef]
- Jin, Y.; Bao, H.; Le, X.; Fan, X.; Tang, M.; Shi, X.; Zhao, J.; Yan, J.; Xu, Y.; Quek, K.; et al. Distinct co-acquired alterations and genomic evolution during TKI treatment in non-small-cell lung cancer patients with or without acquired T790M mutation. Oncogene 2020, 39, 1846–1859. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Kim, J.; Kim, S.K.; Ha, S.; Mok, T.S.; Mitsudomi, T.; Cho, B.C. Lung cancer in never smokers: Change of a mindset in the molecular era. Lung Cancer 2011, 72, 9–15. [Google Scholar] [CrossRef]
- Le, X.; Desai, N.V.; Majid, A.; Karp, R.S.; Huberman, M.S.; Rangachari, D.; Kent, M.S.; Gangadharan, S.P.; Folch, E.; VanderLaan, P.A.; et al. De novo pulmonary small cell carcinomas and large cell neuroendocrine carcinomas harboring EGFR mutations: Lack of response to EGFR inhibitors. Lung Cancer 2015, 88, 70–73. [Google Scholar] [CrossRef]
- Lewis, W.E.; Hong, L.; Mott, F.E.; Simon, G.; Wu, C.; Rinsurongkawong, W.; Lee, J.J.; Lam, V.K.; Heymach, J.V.; Zhang, J.; et al. Efficacy of Targeted Inhibitors in Metastatic Lung Squamous Cell Carcinoma with EGFR or ALK Alterations. JTO Clin. Res. Rep. 2021, 2, 100237. [Google Scholar] [CrossRef]
- Lam, V.K.; Tran, H.T.; Banks, K.C.; Lanman, R.B.; Rinsurongkawong, W.; Peled, N.; Lewis, J.; Lee, J.J.; Roth, J.; Roarty, E.B.; et al. Targeted Tissue and Cell-Free Tumor DNA Sequencing of Advanced Lung Squamous-Cell Carcinoma Reveals Clinically Significant Prevalence of Actionable Alterations. Clin. Lung Cancer 2019, 20, 30–36.e3. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed]
- Oh, I.; Ban, H.; Kim, K.; Kim, Y. Retreatment of gefitinib in patients with non-small-cell lung cancer who previously controlled to gefitinib: A single-arm, open-label, phase II study. Lung Cancer 2012, 77, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.; Katakami, N.; Yoshioka, H.; Fujita, S.; Kunimasa, K.; Nanjo, S.; Otsuka, K.; Kaji, R.; Tomii, K.; Iwasaku, M.; et al. Erlotinib after gefitinib failure in relapsed non-small cell lung cancer: Clinical benefit with optimal patient selection. Lung Cancer 2011, 74, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, P.E.N.S.; Gergis, C.; Viray, H.; Varkaris, A.; Fujii, M.; Rangachari, D.; VanderLaan, P.A.; Kobayashi, I.S.; Kobayashi, S.; Costa, D.B. EGFR-A763_Y764insFQEA Is a Unique Exon 20 Insertion Mutation That Displays Sensitivity to Approved and In-Development Lung Cancer EGFR Tyrosine Kinase Inhibitors. JTO Clin. Res. Rep. 2020, 1, 100051. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Park, E.; Yun, C.; Sng, N.J.; Lucena Araujo, A.R.; Yeo, W.; Huberman, M.S.; Cohen, D.W.; Nakayama, S.; Ishioka, K.; et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci. Transl. Med. 2013, 5, 216ra177. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | No. | Univariate Analysis | Multivariate Analysis | |||
|---|---|---|---|---|---|---|
| HR (95% CI) | p † | HR (95% CI) | p † | |||
| Age | ≥65 vs. <65 years | 28 vs. 31 | 0.81 (0.43–1.52) | 0.507 | ||
| Gender | Male vs. Female | 23 vs. 36 | 1.70 (0.87–3.32) | 0.119 | ||
| ECOG PS | 0–1 vs. 2–3 | 41 vs. 18 | 0.41 (0.21–0.81) | 0.010 | 0.56 (0.28–1.13) | 0.106 |
| Smoking | Never vs. Ever | 30 vs. 19 | 0.73 (0.38–1.38) | 0.327 | ||
| Histology | ADC vs. non-ADC | 56 vs. 3 | 0.20 (0.06–0.70) | 0.012 | 0.43 (0.11–1.69) | 0.227 |
| Operation | No vs. Yes | 42 vs. 12 | 2.71 (1.25–5.91) | 0.012 | ||
| BM metastasis | No vs. Yes | 41 vs. 18 | 0.51 (0.26–1.02) | 0.056 | 0.87 (0.40–1.88) | 0.722 |
| TP53 | WT vs. MT | 14 vs. 11 | 0.71 (0.29–1.72) | 0.451 | ||
| PDC response | Yes vs. No | 40 vs. 19 | 0.31 (0.16–0.60) | 0.001 | 0.38 (0.18–0.77) | 0.007 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Lee, Y.; Song, B.R.; Sim, H.; Kang, E.H.; Hwang, M.; Yu, N.; Hong, S.; Park, C.; Ahn, B.-C.; et al. Drug Response of Patient-Derived Lung Cancer Cells Predicts Clinical Outcomes of Targeted Therapy. Cancers 2024, 16, 778. https://doi.org/10.3390/cancers16040778
Kim S, Lee Y, Song BR, Sim H, Kang EH, Hwang M, Yu N, Hong S, Park C, Ahn B-C, et al. Drug Response of Patient-Derived Lung Cancer Cells Predicts Clinical Outcomes of Targeted Therapy. Cancers. 2024; 16(4):778. https://doi.org/10.3390/cancers16040778
Chicago/Turabian StyleKim, Sunshin, Youngjoo Lee, Bo Ram Song, Hanna Sim, Eun Hye Kang, Mihwa Hwang, Namhee Yu, Sehwa Hong, Charny Park, Beung-Chul Ahn, and et al. 2024. "Drug Response of Patient-Derived Lung Cancer Cells Predicts Clinical Outcomes of Targeted Therapy" Cancers 16, no. 4: 778. https://doi.org/10.3390/cancers16040778
APA StyleKim, S., Lee, Y., Song, B. R., Sim, H., Kang, E. H., Hwang, M., Yu, N., Hong, S., Park, C., Ahn, B.-C., Lim, E. J., Hwang, K. H., Park, S.-Y., Choi, J.-H., Lee, G. K., & Han, J.-Y. (2024). Drug Response of Patient-Derived Lung Cancer Cells Predicts Clinical Outcomes of Targeted Therapy. Cancers, 16(4), 778. https://doi.org/10.3390/cancers16040778