
A Recipe for Successful Metastasis: Transition and Migratory Modes of Ovarian Cancer Cells
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Plasticity of Ovarian Cancer Cells
2.1. Epithelial-to-Mesenchymal Transition (EMT)
2.2. Mesenchymal-to-Amoeboid Transition (MAT)
2.3. Epithelial-to-Amoeboid Transition (EAT)
2.4. Redifferentiation of Cells
3. Migratory Modes of Ovarian Cancer Cells
3.1. Migration of Individual OC Cells
3.2. Collective Migration of OC Cells
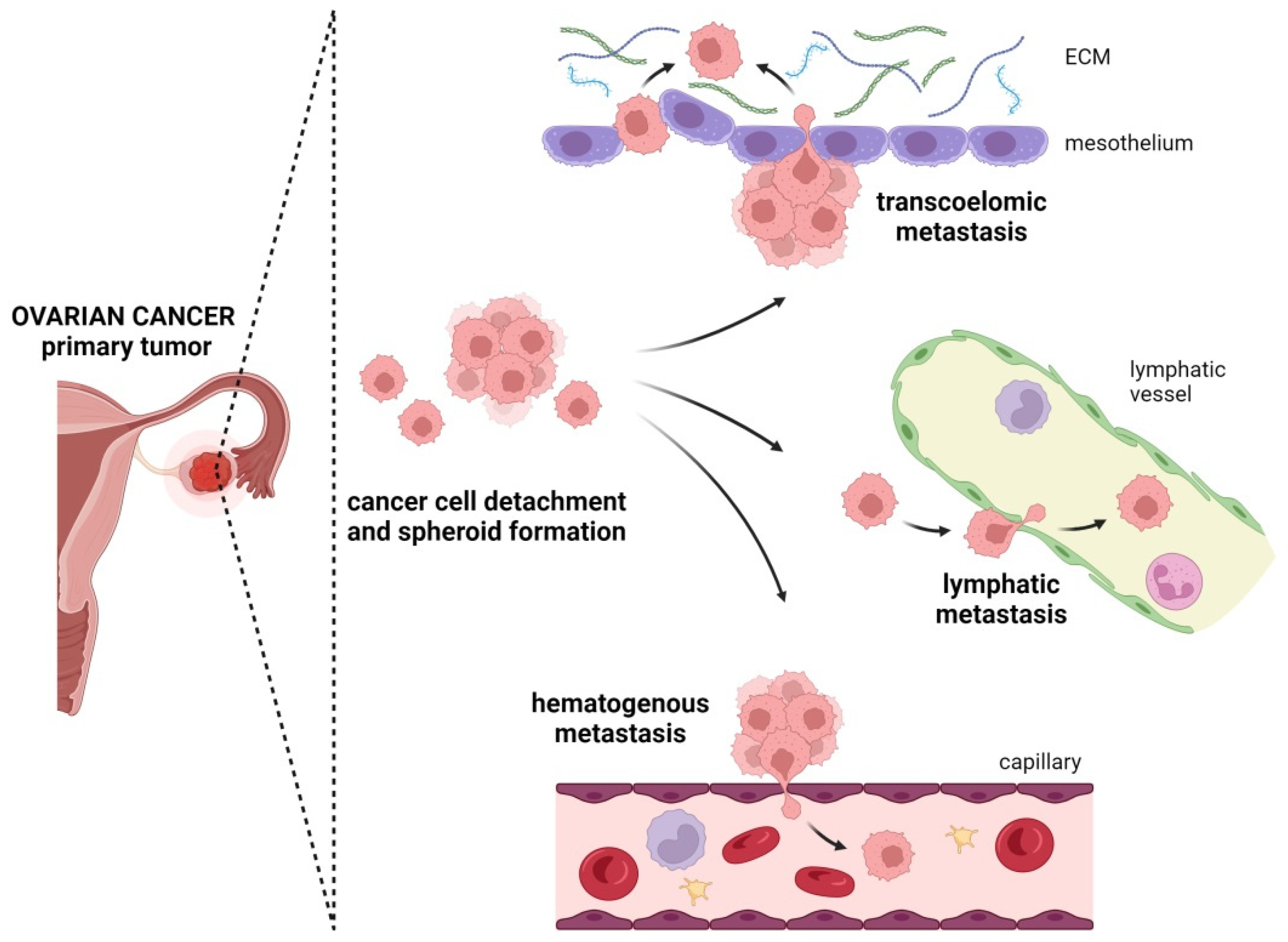
4. Metastatic Routes in Ovarian Cancer
4.1. Transcoelomic Metastasis
4.2. Lymphatic Metastasis
4.3. Hematogenous Metastasis
5. Therapeutic Strategies in OC
6. Clinical Implications of Different Metastatic Routes of OC
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Merino-Casallo, F.; Gomez-Benito, M.J.; Hervas-Raluy, S.; Garcia-Aznar, J.M. Unravelling Cell Migration: Defining Movement from the Cell Surface. Cell Adh. Migr. 2022, 16, 25–64. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018. CA. Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Chebouti, I.; Kuhlmann, J.D.; Buderath, P.; Weber, S.; Wimberger, P.; Bokeloh, Y.; Hauch, S.; Kimmig, R.; Kasimir-Bauer, S. ERCC1-Expressing Circulating Tumor Cells as a Potential Diagnostic Tool for Monitoring Response to Platinum-Based Chemotherapy and for Predicting Post-Therapeutic Outcome of Ovarian Cancer. Oncotarget 2017, 8, 24303–24313. [Google Scholar] [CrossRef] [PubMed]
- Chebouti, I.; Kasimir-Bauer, S.; Buderath, P.; Wimberger, P.; Hauch, S.; Kimmig, R.; Kuhlmann, J.D. EMT-like Circulating Tumor Cells in Ovarian Cancer Patients Are Enriched by Platinum-Based Chemotherapy. Oncotarget 2017, 8, 48820–48831. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Bai, X.; Feng, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Inhibition of PI3K/Akt/MTOR Signaling Pathway Alleviates Ovarian Cancer Chemoresistance through Reversing Epithelial-Mesenchymal Transition and Decreasing Cancer Stem Cell Marker Expression. BMC Cancer 2019, 19, 618. [Google Scholar] [CrossRef] [PubMed]
- Auer, K.; Bachmayr-Heyda, A.; Aust, S.; Sukhbaatar, N.; Reiner, A.T.; Grimm, C.; Horvat, R.; Zeillinger, R.; Pils, D. Peritoneal Tumor Spread in Serous Ovarian Cancer-Epithelial Mesenchymal Status and Outcome. Oncotarget 2015, 6, 17261–17275. [Google Scholar] [CrossRef] [PubMed]
- Penet, M.-F.; Krishnamachary, B.; Wildes, F.B.; Mironchik, Y.; Hung, C.-F.; Wu, T.; Bhujwalla, Z.M. Ascites Volumes and the Ovarian Cancer Microenvironment. Front. Oncol. 2018, 8, 595. [Google Scholar] [CrossRef] [PubMed]
- Haller, H.; Mamula, O.; Krasevic, M.; Rupcic, S.; Fischer, A.B.; Eminovic, S.; Manestar, M.; Perovic, D. Frequency and Distribution of Lymph Node Metastases in Epithelial Ovarian Cancer: Significance of Serous Histology. Int. J. Gynecol. Cancer 2011, 21, 245–250. [Google Scholar] [CrossRef]
- Bregenzer, M.E.; Horst, E.N.; Mehta, P.; Novak, C.M.; Repetto, T.; Snyder, C.S.; Mehta, G. Tumor Modeling Maintains Diverse Pathology in Vitro. Ann. Transl. Med. 2019, 7, S262. [Google Scholar] [CrossRef]
- Martins, F.C.; Couturier, D.L.; Paterson, A.; Karnezis, A.N.; Chow, C.; Nazeran, T.M.; Odunsi, A.; Gentry-Maharaj, A.; Vrvilo, A.; Hein, A.; et al. Clinical and Pathological Associations of PTEN Expression in Ovarian Cancer: A Multicentre Study from the Ovarian Tumour Tissue Analysis Consortium. Br. J. Cancer 2020, 123, 793–802. [Google Scholar] [CrossRef]
- Klymenko, Y.; Kim, O.; Stack, M.S. Complex Determinants of Epithelial: Mesenchymal Phenotypic Plasticity in Ovarian Cancer. Cancers 2017, 9, 104. [Google Scholar] [CrossRef]
- Siu, M.K.Y.; Jiang, Y.-X.; Wang, J.-J.; Leung, T.H.Y.; Ngu, S.F.; Cheung, A.N.Y.; Ngan, H.Y.S.; Chan, K.K.L. PDK1 Promotes Ovarian Cancer Metastasis by Modulating Tumor-Mesothelial Adhesion, Invasion, and Angiogenesis via A5β1 Integrin and JNK/IL-8 Signaling. Oncogenesis 2020, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Ji, G.; Le, X.; Luo, Z.; Wang, C.; Feng, M.; Xu, L.; Zhang, Y.; Lau, W.B.; Lau, B.; et al. An Integrated Analysis Identifies STAT4 as a Key Regulator of Ovarian Cancer Metastasis. Oncogene 2017, 36, 3384–3396. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Saha, P.; Samanta, A.; Bishayee, A. Emerging Concepts of Hybrid Epithelial-to-Mesenchymal Transition in Cancer Progression. Biomolecules 2020, 10, 1561. [Google Scholar] [CrossRef]
- McGrail, D.J.; Kieu, Q.M.N.; Dawson, M.R. The Malignancy of Metastatic Ovarian Cancer Cells Is Increased on Soft Matrices through a Mechanosensitive Rho-ROCK Pathway. J. Cell Sci. 2014, 127, 2621–2626. [Google Scholar] [CrossRef]
- Iwanicki, M.P.; Davidowitz, R.A.; Ng, M.R.; Besser, A.; Muranen, T.; Merritt, M.; Danuser, G.; Ince, T.A.; Brugge, J.S. Ovarian Cancer Spheroids Use Myosin-Generated Force to Clear the Mesothelium. Cancer Discov. 2011, 1, 144–157. [Google Scholar] [CrossRef]
- Jeong, K.J.; Park, S.Y.; Cho, K.H.; Sohn, J.S.; Lee, J.; Kim, Y.K.; Kang, J.; Park, C.G.; Han, J.W.; Lee, H.Y. The Rho/ROCK Pathway for Lysophosphatidic Acid-Induced Proteolytic Enzyme Expression and Ovarian Cancer Cell Invasion. Oncogene 2012, 31, 4279–4289. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Barai, A.; Thakur, B.; Das, A.; Patwardhan, S.R.; Monteiro, M.; Gaikwad, S.; Bukhari, A.B.; Mogha, P.; Majumder, A.; et al. Soft Drug-Resistant Ovarian Cancer Cells Migrate via Two Distinct Mechanisms Utilizing Myosin II-Based Contractility. Biochim. Biophys. Acta-Mol. Cell Res. 2018, 1865, 392–405. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Takahashi, T.; Shibuya, T.; Amita, M.; Henmi, N.; Takahashi, K.; Kurachi, H. Inhibition of the Rho/ROCK Pathway Enhances the Efficacy of Cisplatin through the Blockage of Hypoxia-Inducible Factor-1α in Human Ovarian Cancer Cells. Cancer Biol. Ther. 2012, 13, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, A.; Kikuchi, N.; Osada, R.; Wang, C.; Hayashi, A.; Nikaido, T.; Konishi, I. Overexpression of RhoA Enhances Peritoneal Dissemination: RhoA Suppression with Lovastatin May Be Useful for Ovarian Cancer. Cancer Sci. 2008, 99, 2532–2539. [Google Scholar] [CrossRef] [PubMed]
- Holle, A.W.; Govindan Kutty Devi, N.; Clar, K.; Fan, A.; Saif, T.; Kemkemer, R.; Spatz, J.P. Cancer Cells Invade Confined Microchannels via a Self-Directed Mesenchymal-to-Amoeboid Transition. Nano Lett. 2019, 19, 2280–2290. [Google Scholar] [CrossRef]
- Alexandrova, A.Y.; Chikina, A.S.; Svitkina, T.M. Actin Cytoskeleton in Mesenchymal-to-Amoeboid Transition of Cancer Cells. Int. Rev. Cell Mol. Biol. 2020, 356, 197–256. [Google Scholar]
- Hegerfeldt, Y.; Tusch, M.; Brocker, E.-B.; Friedl, P. Collective Cell Movement in Primary Melanoma Explants: Plasticity of Cell-Cell Interaction, $β$1-Integrin Function, and Migration Strategies. Cancer Res. 2002, 62, 2125–2130. [Google Scholar] [PubMed]
- Wolf, K.; Wu, Y.I.; Liu, Y.; Geiger, J.; Tam, E.; Overall, C.; Stack, M.S.; Friedl, P. Multi-Step Pericellular Proteolysis Controls the Transition from Individual to Collective Cancer Cell Invasion. Nat. Cell Biol. 2007, 9, 893–904. [Google Scholar] [CrossRef]
- Crosas-Molist, E.; Bertran, E.; Rodriguez-Hernandez, I.; Herraiz, C.; Cantelli, G.; Fabra, À.; Sanz-Moreno, V.; Fabregat, I. The NADPH Oxidase NOX4 Represses Epithelial to Amoeboid Transition and Efficient Tumour Dissemination. Oncogene 2017, 36, 3002–3014. [Google Scholar] [CrossRef]
- te Boekhorst, V.; Jiang, L.; Mählen, M.; Meerlo, M.; Dunkel, G.; Durst, F.C.; Yang, Y.; Levine, H.; Burgering, B.M.T.; Friedl, P. Calpain-2 Regulates Hypoxia/HIF-Induced Plasticity toward Amoeboid Cancer Cell Migration and Metastasis. Curr. Biol. 2022, 32, 412–427.e8. [Google Scholar] [CrossRef]
- Lehmann, S.; te Boekhorst, V.; Odenthal, J.; Bianchi, R.; van Helvert, S.; Ikenberg, K.; Ilina, O.; Stoma, S.; Xandry, J.; Jiang, L.; et al. Hypoxia Induces a HIF-1-Dependent Transition from Collective-to-Amoeboid Dissemination in Epithelial Cancer Cells. Curr. Biol. 2017, 27, 392–400. [Google Scholar] [CrossRef]
- Mohammadalipour, A.; Diaz, M.F.; Livingston, M.; Ewere, A.; Zhou, A.; Horton, P.D.; Olamigoke, L.T.; Lamar, J.M.; Hagan, J.P.; Lee, H.J.; et al. RhoA-ROCK Competes with YAP to Regulate Amoeboid Breast Cancer Cell Migration in Response to Lymphatic-like Flow. FASEB bioAdvances 2022, 4, 342–361. [Google Scholar] [CrossRef] [PubMed]
- Weigand, A.; Boos, A.M.; Tasbihi, K.; Beier, J.P.; Dalton, P.D.; Schrauder, M.; Horch, R.E.; Beckmann, M.W.; Strissel, P.L.; Strick, R. Selective Isolation and Characterization of Primary Cells from Normal Breast and Tumors Reveal Plasticity of Adipose Derived Stem Cells. Breast Cancer Res. 2016, 18, 32. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Jolly, M.K.; Lu, M.; Tsarfaty, I.; Ben-Jacob, E.; Onuchic, J.N. Modeling the Transitions between Collective and Solitary Migration Phenotypes in Cancer Metastasis. Sci. Rep. 2015, 5, 17379. [Google Scholar] [CrossRef]
- Thankamony, A.P.; Saxena, K.; Murali, R.; Jolly, M.K.; Nair, R. Cancer Stem Cell Plasticity—A Deadly Deal. Front. Mol. Biosci. 2020, 7, 79. [Google Scholar] [CrossRef]
- Shibamoto, S.; Hayakawa, M.; Takeuchi, K.; Hori, T.; Miyazawa, K.; Kitamura, N.; Johnson, K.R.; Wheelock, M.J.; Matsuyoshi, N.; Takeichi, M. Association of P120, a Tyrosine Kinase Substrate, with E-Cadherin/Catenin Complexes. J. Cell Biol. 1995, 128, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Yap, A.S.; Niessen, C.M.; Gumbiner, B.M. The Juxtamembrane Region of the Cadherin Cytoplasmic Tail Supports Lateral Clustering, Adhesive Strengthening, and Interaction with P120ctn. J. Cell Biol. 1998, 141, 779–789. [Google Scholar] [CrossRef]
- Engl, W.; Arasi, B.; Yap, L.L.; Thiery, J.P.; Viasnoff, V. Actin Dynamics Modulate Mechanosensitive Immobilization of E-Cadherin at Adherens Junctions. Nat. Cell Biol. 2014, 16, 584–591. [Google Scholar] [CrossRef]
- Yap, A.S.; Gomez, G.A.; Parton, R.G. Adherens Junctions Revisualized: Organizing Cadherins as Nanoassemblies. Dev. Cell 2015, 35, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Sipos, F.; Galamb, O. Epithelial-to-Mesenchymal and Mesenchymal-to-Epithelial Transitions in the Colon. World J. Gastroenterol. 2012, 18, 601–608. [Google Scholar] [CrossRef]
- Chen, J.; Wang, L.; Matyunina, L.V.; Hill, C.G.; McDonald, J.F. Overexpression of MiR-429 Induces Mesenchymal-to-Epithelial Transition (MET) in Metastatic Ovarian Cancer Cells. Gynecol. Oncol. 2011, 121, 200–205. [Google Scholar] [CrossRef]
- Brabletz, T. To Differentiate or Not—Routes towards Metastasis. Nat. Rev. Cancer 2012, 12, 425–436. [Google Scholar] [CrossRef]
- Koensgen, D.; Freitag, C.; Klaman, I.; Dahl, E.; Mustea, A.; Chekerov, R.; Braicu, I.; Lichtenegger, W.; Sehouli, J. Expression and Localization of E-Cadherin in Epithelial Ovarian Cancer. Anticancer Res. 2010, 30, 2525–2530. [Google Scholar] [PubMed]
- de Toledo, M.; Anguille, C.; Roger, L.; Roux, P.; Gadea, G. Cooperative Anti-Invasive Effect of Cdc42/Rac1 Activation and ROCK Inhibition in SW620 Colorectal Cancer Cells with Elevated Blebbing Activity. PLoS ONE 2012, 7, e48344. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the Tumour Transition States Occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Gadea, G.; Ahn, J.; Paterson, H.; Marra, P.; Pinner, S.; Sahai, E.; Marshall, C.J. Rac Activation and Inactivation Control Plasticity of Tumor Cell Movement. Cell 2008, 135, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.-J.; Wong, M.K.; Tan, T.Z.; Kuay, K.T.; Ng, A.H.C.; Chung, V.Y.; Chu, Y.-S.; Matsumura, N.; Lai, H.-C.; Lee, Y.F.; et al. An EMT Spectrum Defines an Anoikis-Resistant and Spheroidogenic Intermediate Mesenchymal State That Is Sensitive to e-Cadherin Restoration by a Src-Kinase Inhibitor, Saracatinib (AZD0530). Cell Death Dis. 2013, 4, e915. [Google Scholar] [CrossRef] [PubMed]
- Rubtsova, S.N.; Zhitnyak, I.Y.; Gloushankova, N.A. Phenotypic Plasticity of Cancer Cells Based on Remodeling of the Actin Cytoskeleton and Adhesive Structures. Int. J. Mol. Sci. 2021, 22, 1821. [Google Scholar] [CrossRef] [PubMed]
- Spano, D.; Heck, C.; De Antonellis, P.; Christofori, G.; Zollo, M. Molecular Networks That Regulate Cancer Metastasis. Semin. Cancer Biol. 2012, 22, 234–249. [Google Scholar] [CrossRef] [PubMed]
- Paluch, E.; Piel, M.; Prost, J.; Bornens, M.; Sykes, C. Cortical Actomyosin Breakage Triggers Shape Oscillations in Cells and Cell Fragments. Biophys. J. 2005, 89, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Tsang, T.Y.; Wei, W.; Itamochi, H.; Tambouret, R.; Birrer, M.J. Integrated Genomic Analysis of Clear Cell Ovarian Cancers Identified PRKCI as a Potential Therapeutic Target. Oncotarget 2017, 8, 96482–96495. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-J.; Le Berre, M.; Lautenschlaeger, F.; Maiuri, P.; Callan-Jones, A.; Heuzé, M.; Takaki, T.; Voituriez, R.; Piel, M. Confinement and Low Adhesion Induce Fast Amoeboid Migration of Slow Mesenchymal Cells. Cell 2015, 160, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Lomakin, A.J.; Cattin, C.J.; Cuvelier, D.; Alraies, Z.; Molina, M.; Nader, G.P.F.; Srivastava, N.; Sáez, P.J.; Garcia-Arcos, J.M.; Zhitnyak, I.Y.; et al. The Nucleus Acts as a Ruler Tailoring Cell Responses to Spatial Constraints. Science 2020, 370, eaba2894. [Google Scholar] [CrossRef]
- Schick, J.; Raz, E. Blebs-Formation, Regulation, Positioning, and Role in Amoeboid Cell Migration. Front. Cell Dev. Biol. 2022, 10, 926394. [Google Scholar] [CrossRef]
- McKenzie, A.J.; Hicks, S.R.; Svec, K.V.; Naughton, H.; Edmunds, Z.L.; Howe, A.K. The Mechanical Microenvironment Regulates Ovarian Cancer Cell Morphology, Migration, and Spheroid Disaggregation. Sci. Rep. 2018, 8, 7228. [Google Scholar] [CrossRef]
- Madsen, C.D.; Sahai, E. Cancer Dissemination—Lessons from Leukocytes. Dev. Cell 2010, 19, 13–26. [Google Scholar] [CrossRef]
- Bronsert, P.; Enderle-Ammour, K.; Bader, M.; Timme, S.; Kuehs, M.; Csanadi, A.; Kayser, G.; Kohler, I.; Bausch, D.; Hoeppner, J.; et al. Cancer Cell Invasion and EMT Marker Expression: A Three-Dimensional Study of the Human Cancer-Host Interface. J. Pathol. 2014, 234, 410–422. [Google Scholar] [CrossRef]
- Ilina, O.; Campanello, L.; Gritsenko, P.G.; Vullings, M.; Wang, C.; Bult, P.; Losert, W.; Friedl, P. Intravital Microscopy of Collective Invasion Plasticity in Breast Cancer. Dis. Model. Mech. 2018, 11, dmm34330. [Google Scholar] [CrossRef] [PubMed]
- Hallou, A.; Jennings, J.; Kabla, A.J. Tumour Heterogeneity Promotes Collective Invasion and Cancer Metastatic Dissemination. R. Soc. Open Sci. 2017, 4, 161007. [Google Scholar] [CrossRef] [PubMed]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-Led Collective Invasion of Carcinoma Cells with Differing Roles for RhoGTPases in Leading and Following Cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Loret, N.; Denys, H.; Tummers, P.; Berx, G. The Role of Epithelial-to-Mesenchymal Plasticity in Ovarian Cancer Progression and Therapy Resistance. Cancers 2019, 11, 838. [Google Scholar] [CrossRef] [PubMed]
- Pagès, D.-L.; Dornier, E.; de Seze, J.; Gontran, E.; Maitra, A.; Maciejewski, A.; Wang, L.; Luan, R.; Cartry, J.; Canet-Jourdan, C.; et al. Cell Clusters Adopt a Collective Amoeboid Mode of Migration in Confined Nonadhesive Environments. Sci. Adv. 2022, 8, eabp8416. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Gilmour, D. Collective Cell Migration in Morphogenesis, Regeneration and Cancer. Nat. Rev. Mol. Cell Biol. 2009, 10, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E. Ovarian Cancer Development and Metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef]
- Kawanishi, K. Diverse Properties of the Mesothelial Cells in Health and Disease. Pleura Peritoneum 2016, 1, 79–89. [Google Scholar] [CrossRef]
- Cannistra, S.A.; Kansas, G.S.; Niloff, J.; DeFranzo, B.; Kim, Y.; Ottensmeier, C. Binding of Ovarian Cancer Cells to Peritoneal Mesothelium in Vitro Is Partly Mediated by CD44H. Cancer Res. 1993, 53, 3830–3838. [Google Scholar]
- Misra, S.; Hascall, V.C.; Markwald, R.R.; Ghatak, S. Interactions between Hyaluronan and Its Receptors (CD44, RHAMM) Regulate the Activities of Inflammation and Cancer. Front. Immunol. 2015, 6, 201. [Google Scholar] [CrossRef]
- Desjardins, M.; Xie, J.; Gurler, H.; Muralidhar, G.G.; Sacks, J.D.; Burdette, J.E.; Barbolina, M. V Versican Regulates Metastasis of Epithelial Ovarian Carcinoma Cells and Spheroids. J. Ovarian Res. 2014, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.L.; Reinke, L.M.; Damerow, M.S.; Perez, D.; Chodosh, L.A.; Yang, J.; Cheng, C. CD44 Splice Isoform Switching in Human and Mouse Epithelium Is Essential for Epithelial-Mesenchymal Transition and Breast Cancer Progression. J. Clin. Investig. 2011, 121, 1064–1074. [Google Scholar] [CrossRef]
- Coelho, R.; Ricardo, S.; Amaral, A.L.; Huang, Y.-L.; Nunes, M.; Neves, J.P.; Mendes, N.; López, M.N.; Bartosch, C.; Ferreira, V.; et al. Regulation of Invasion and Peritoneal Dissemination of Ovarian Cancer by Mesothelin Manipulation. Oncogenesis 2020, 9, 61. [Google Scholar] [CrossRef]
- Stoeck, A.; Schlich, S.; Issa, Y.; Gschwend, V.; Wenger, T.; Herr, I.; Marmé, A.; Bourbie, S.; Altevogt, P.; Gutwein, P. L1 on Ovarian Carcinoma Cells Is a Binding Partner for Neuropilin-1 on Mesothelial Cells. Cancer Lett. 2006, 239, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Gurler Main, H.; Xie, J.; Muralidhar, G.G.; Elfituri, O.; Xu, H.; Kajdacsy-Balla, A.A.; Barbolina, M. V Emergent Role of the Fractalkine Axis in Dissemination of Peritoneal Metastasis from Epithelial Ovarian Carcinoma. Oncogene 2017, 36, 3025–3036. [Google Scholar] [CrossRef] [PubMed]
- Kenny, H.A.; Chiang, C.-Y.; White, E.A.; Schryver, E.M.; Habis, M.; Romero, I.L.; Ladanyi, A.; Penicka, C.V.; George, J.; Matlin, K.; et al. Mesothelial Cells Promote Early Ovarian Cancer Metastasis through Fibronectin Secretion. J. Clin. Investig. 2014, 124, 4614–4628. [Google Scholar] [CrossRef]
- Carroll, M.J.; Fogg, K.C.; Patel, H.A.; Krause, H.B.; Mancha, A.-S.; Patankar, M.S.; Weisman, P.S.; Barroilhet, L.; Kreeger, P.K. Alternatively-Activated Macrophages Upregulate Mesothelial Expression of P-Selectin to Enhance Adhesion of Ovarian Cancer Cells. Cancer Res. 2018, 78, 3560–3573. [Google Scholar] [CrossRef]
- Gharpure, K.M.; Lara, O.D.; Wen, Y.; Pradeep, S.; LaFargue, C.; Ivan, C.; Rupaimoole, R.; Hu, W.; Mangala, L.S.; Wu, S.Y.; et al. ADH1B Promotes Mesothelial Clearance and Ovarian Cancer Infiltration. Oncotarget 2018, 9, 25115–25126. [Google Scholar] [CrossRef] [PubMed]
- Roy, L.; Bobbs, A.; Sattler, R.; Kurkewich, J.L.; Dausinas, P.B.; Nallathamby, P.; Cowden Dahl, K.D. CD133 Promotes Adhesion to the Ovarian Cancer Metastatic Niche. Cancer Growth Metastasis 2018, 11, 117906441876788. [Google Scholar] [CrossRef] [PubMed]
- Sodek, K.L.; Ringuette, M.J.; Brown, T.J. MT1-MMP Is the Critical Determinant of Matrix Degradation and Invasion by Ovarian Cancer Cells. Br. J. Cancer 2007, 97, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Fisher, J.; Fishman, D.A. MMP-1-PAR1 Axis Mediates LPA-Induced Epithelial Ovarian Cancer (EOC) Invasion. Gynecol. Oncol. 2011, 120, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Kenny, H.A.; Kaur, S.; Coussens, L.M.; Lengyel, E. The Initial Steps of Ovarian Cancer Cell Metastasis Are Mediated by MMP-2 Cleavage of Vitronectin and Fibronectin. J. Clin. Investig. 2008, 118, 1367–1379. [Google Scholar] [CrossRef]
- Jackson-Jones, L.H.; Smith, P.; Portman, J.R.; Magalhaes, M.S.; Mylonas, K.J.; Vermeren, M.M.; Nixon, M.; Henderson, B.E.P.; Dobie, R.; Vermeren, S.; et al. Stromal Cells Covering Omental Fat-Associated Lymphoid Clusters Trigger Formation of Neutrophil Aggregates to Capture Peritoneal Contaminants. Immunity 2020, 52, 700–715.e6. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes Promote Ovarian Cancer Metastasis and Provide Energy for Rapid Tumor Growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef]
- Horowitz, N.S.; Miller, A.; Rungruang, B.; Richard, S.D.; Rodriguez, N.; Bookman, M.A.; Hamilton, C.A.; Krivak, T.C.; Maxwell, G.L. Does Aggressive Surgery Improve Outcomes? Interaction between Preoperative Disease Burden and Complex Surgery in Patients with Advanced-Stage Ovarian Cancer: An Analysis of GOG 182. J. Clin. Oncol. 2015, 33, 937–943. [Google Scholar] [CrossRef]
- Arie, A.B.; McNally, L.; Kapp, D.S.; Teng, N.N.H. The Omentum and Omentectomy in Epithelial Ovarian Cancer: A Reappraisal: Part II--The Role of Omentectomy in the Staging and Treatment of Apparent Early Stage Epithelial Ovarian Cancer. Gynecol. Oncol. 2013, 131, 784–790. [Google Scholar] [CrossRef]
- Bilbao, M.; Aikins, J.K.; Ostrovsky, O. Is Routine Omentectomy of Grossly Normal Omentum Helpful in Surgery for Ovarian Cancer? A Look at the Tumor Microenvironment and Its Clinical Implications. Gynecol. Oncol. 2021, 161, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Angelico, G.; Piermattei, A.; Inzani, F.; Valente, M.; Arciuolo, D.; Spadola, S.; Mulè, A.; Zorzato, P.; Fagotti, A.; et al. Pathological Chemotherapy Response Score in Patients Affected by High Grade Serous Ovarian Carcinoma: The Prognostic Role of Omental and Ovarian Residual Disease. Front. Oncol. 2019, 9, 778. [Google Scholar] [CrossRef]
- Hao, Z.; Yu, Y.; Yang, S. The Impact of Omentectomy on Cause-Specific Survival of Stage I-IIIA Epithelial Ovarian Cancer: A PSM-IPTW Analysis Based on the SEER Database. Front. Surg. 2022, 9, 1052788. [Google Scholar] [CrossRef]
- McNally, L.; Teng, N.N.H.; Kapp, D.S.; Karam, A. Does Omentectomy in Epithelial Ovarian Cancer Affect Survival? An Analysis of the Surveillance, Epidemiology, and End Results Database. Int. J. Gynecol. Cancer 2015, 25, 607–615. [Google Scholar] [CrossRef]
- Meza-Perez, S.; Randall, T.D. Immunological Functions of the Omentum. Trends Immunol. 2017, 38, 526–536. [Google Scholar] [CrossRef]
- Chkourko Gusky, H.; Diedrich, J.; MacDougald, O.A.; Podgorski, I. Omentum and Bone Marrow: How Adipocyte-rich Organs Create Tumour Microenvironments Conducive for Metastatic Progression. Obes. Rev. 2016, 17, 1015–1029. [Google Scholar] [CrossRef]
- Suh, D.H.; Kim, H.S.; Kim, B.; Song, Y.S. Metabolic Orchestration between Cancer Cells and Tumor Microenvironment as a Co-Evolutionary Source of Chemoresistance in Ovarian Cancer: A Therapeutic Implication. Biochem. Pharmacol. 2014, 92, 43–54. [Google Scholar] [CrossRef]
- Motohara, T.; Masuda, K.; Morotti, M.; Zheng, Y.; El-Sahhar, S.; Chong, K.Y.; Wietek, N.; Alsaadi, A.; Carrami, E.M.; Hu, Z.; et al. An Evolving Story of the Metastatic Voyage of Ovarian Cancer Cells: Cellular and Molecular Orchestration of the Adipose-Rich Metastatic Microenvironment. Oncogene 2019, 38, 2885–2898. [Google Scholar] [CrossRef]
- Krishnan, V.; Tallapragada, S.; Schaar, B.; Kamat, K.; Chanana, A.M.; Zhang, Y.; Patel, S.; Parkash, V.; Rinker-Schaeffer, C.; Folkins, A.K.; et al. Omental Macrophages Secrete Chemokine Ligands That Promote Ovarian Cancer Colonization of the Omentum via CCR1. Commun. Biol. 2020, 3, 524. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A.; Lu, Z.; Jennings, N.B.; Etemadmoghadam, D.; Capalbo, L.; Jacamo, R.O.; Barbosa-Morais, N.; Le, X.-F.; Vivas-Mejia, P.; Lopez-Berestein, G.; et al. SIK2 Is a Centrosome Kinase Required for Bipolar Mitotic Spindle Formation That Provides a Potential Target for Therapy in Ovarian Cancer. Cancer Cell 2010, 18, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Alfraidi, A.; Zhang, S.; Santiago-O’Farrill, J.M.; Yerramreddy Reddy, V.K.; Alsaadi, A.; Ahmed, A.A.; Yang, H.; Liu, J.; Mao, W.; et al. A Novel Compound ARN-3236 Inhibits Salt-Inducible Kinase 2 and Sensitizes Ovarian Cancer Cell Lines and Xenografts to Paclitaxel. Clin. Cancer Res. 2017, 23, 1945–1954. [Google Scholar] [CrossRef] [PubMed]
- Miranda, F.; Ahmed, A.A. How to Make Ovarian Cancer Cells “Sick-Too”. Cell Cycle 2017, 16, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Etzerodt, A.; Moulin, M.; Doktor, T.K.; Delfini, M.; Mossadegh-Keller, N.; Bajenoff, M.; Sieweke, M.H.; Moestrup, S.K.; Auphan-Anezin, N.; Lawrence, T. Tissue-Resident Macrophages in Omentum Promote Metastatic Spread of Ovarian Cancer. J. Exp. Med. 2020, 217, 1869. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Ko, S.Y.; Mohamed, M.S.; Kenny, H.A.; Lengyel, E.; Naora, H. Neutrophils Facilitate Ovarian Cancer Premetastatic Niche Formation in the Omentum. J. Exp. Med. 2019, 216, 176–194. [Google Scholar] [CrossRef] [PubMed]
- Teijeira, A.; Garasa, S.; Ochoa, M.C.; Villalba, M.; Olivera, I.; Cirella, A.; Eguren-Santamaria, I.; Berraondo, P.; Schalper, K.A.; de Andrea, C.E.; et al. IL8, Neutrophils, and NETs in a Collusion against Cancer Immunity and Immunotherapy. Clin. Cancer Res. 2021, 27, 2383–2393. [Google Scholar] [CrossRef] [PubMed]
- Masucci, M.T.; Minopoli, M.; Del Vecchio, S.; Carriero, M.V. The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Front. Immunol. 2020, 11, 1749. [Google Scholar] [CrossRef]
- Khan, S.M.; Funk, H.M.; Thiolloy, S.; Lotan, T.L.; Hickson, J.; Prins, G.S.; Drew, A.F.; Rinker-Schaeffer, C.W. In Vitro Metastatic Colonization of Human Ovarian Cancer Cells to the Omentum. Clin. Exp. Metastasis 2010, 27, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.; Klink, M. The Role of Tumor-Associated Macrophages in the Progression and Chemoresistance of Ovarian Cancer. Cells 2020, 9, 1299. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Geng, X.; Li, Y. Milky Spots: Omental Functional Units and Hotbeds for Peritoneal Cancer Metastasis. Tumour Biol. 2016, 37, 5715–5726. [Google Scholar] [CrossRef]
- Han, Q.; Huang, B.; Huang, Z.; Cai, J.; Gong, L.; Zhang, Y.; Jiang, J.; Dong, W.; Wang, Z. Tumor Cell-fibroblast Heterotypic Aggregates in Malignant Ascites of Patients with Ovarian Cancer. Int. J. Mol. Med. 2019, 44, 2245–2255. [Google Scholar] [CrossRef]
- Yin, M.; Shen, J.; Yu, S.; Fei, J.; Zhu, X.; Zhao, J.; Zhai, L.; Sadhukhan, A.; Zhou, J. Tumor-Associated Macrophages (TAMs): A Critical Activator In Ovarian Cancer Metastasis. Onco. Targets. Ther. 2019, 12, 8687–8699. [Google Scholar] [CrossRef]
- Kim, S.; Kim, B.; Song, Y.S. Ascites Modulates Cancer Cell Behavior, Contributing to Tumor Heterogeneity in Ovarian Cancer. Cancer Sci. 2016, 107, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Rickard, B.P.; Conrad, C.; Sorrin, A.J.; Ruhi, M.K.; Reader, J.C.; Huang, S.A.; Franco, W.; Scarcelli, G.; Polacheck, W.J.; Roque, D.M.; et al. Malignant Ascites in Ovarian Cancer: Cellular, Acellular, and Biophysical Determinants of Molecular Characteristics and Therapy Response. Cancers 2021, 13, 4318. [Google Scholar] [CrossRef]
- den Ouden, J.E.; Zaman, G.J.R.; Dylus, J.; van Doornmalen, A.M.; Mulder, W.R.; Grobben, Y.; van Riel, W.E.; de Hullu, J.A.; Buijsman, R.C.; van Altena, A.M. Chemotherapy Sensitivity Testing on Ovarian Cancer Cells Isolated from Malignant Ascites. Oncotarget 2020, 11, 4570–4581. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Yang, Z.; Xu, S.; Li, X.; Yang, X.; Jin, P.; Liu, Y.; Zhou, X.; Zhang, T.; Gong, C.; et al. Heterotypic CAF-Tumor Spheroids Promote Early Peritoneal Metastatis of Ovarian Cancer. J. Exp. Med. 2019, 216, 688–703. [Google Scholar] [CrossRef] [PubMed]
- Al Habyan, S.; Kalos, C.; Szymborski, J.; McCaffrey, L. Multicellular Detachment Generates Metastatic Spheroids during Intra-Abdominal Dissemination in Epithelial Ovarian Cancer. Oncogene 2018, 37, 5127–5135. [Google Scholar] [CrossRef] [PubMed]
- Latifi, A.; Luwor, R.B.; Bilandzic, M.; Nazaretian, S.; Stenvers, K.; Pyman, J.; Zhu, H.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K.; et al. Isolation and Characterization of Tumor Cells from the Ascites of Ovarian Cancer Patients: Molecular Phenotype of Chemoresistant Ovarian Tumors. PLoS ONE 2012, 7, e46858. [Google Scholar] [CrossRef]
- Nguyen, D.X.; Bos, P.D.; Massagué, J. Metastasis: From Dissemination to Organ-Specific Colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef]
- Takeshima, N.; Hirai, Y.; Umayahara, K.; Fujiwara, K.; Takizawa, K.; Hasumi, K. Lymph Node Metastasis in Ovarian Cancer: Difference between Serous and Non-Serous Primary Tumors. Gynecol. Oncol. 2005, 99, 427–431. [Google Scholar] [CrossRef]
- Schoppmann, S.; Fenzl, A.; Nagy, K.; Unger, S.; Bayer, G.; Geleff, S.; Gnant, M.; Horvat, R.; Jakesz, R.; Birner, P. VEGF-C Expressing Tumor-Associated Macrophages in Lymph Node Positive Breast Cancer: Impact on Lymphangiogenesis and Survival. Surgery 2006, 139, 839–846. [Google Scholar] [CrossRef]
- Wang, X.; Deavers, M.; Patenia, R.; Bassett, R.L.; Mueller, P.; Ma, Q.; Wang, E.; Freedman, R.S. Monocyte/Macrophage and T-Cell Infiltrates in Peritoneum of Patients with Ovarian Cancer or Benign Pelvic Disease. J. Transl. Med. 2006, 4, 30. [Google Scholar] [CrossRef]
- Harter, P.; Heitz, F.; Ataseven, B.; Schneider, S.; Baert, T.; Prader, S.; du Bois, A. How to Manage Lymph Nodes in Ovarian Cancer. Cancer 2019, 125, 4573–4577. [Google Scholar] [CrossRef] [PubMed]
- Erdem, B.; Yüksel, I.T.; Peker, N.; Ulukent, S.C.; Aşıcıoğlu, O.; Özaydin, I.Y.; Ülker, V.; Akbayir, O. Evaluation of Factors Affecting Lymph Node Metastasis in Clinical Stage I–II Epithelial Ovarian Cancer. Oncol. Res. Treat. 2018, 41, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.E.; Werner, B.; Hacker, N.F.; Warton, K. The Untapped Potential of Ascites in Ovarian Cancer Research and Treatment. Br. J. Cancer 2020, 123, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Pradeep, S.; Kim, S.W.; Wu, S.Y.; Nishimura, M.; Chaluvally-Raghavan, P.; Miyake, T.; Pecot, C.V.; Kim, S.-J.; Choi, H.J.; Bischoff, F.Z.; et al. Hematogenous Metastasis of Ovarian Cancer: Rethinking Mode of Spread. Cancer Cell 2014, 26, 77–91. [Google Scholar] [CrossRef]
- Coffman, L.G.; Burgos-Ojeda, D.; Wu, R.; Cho, K.; Bai, S.; Buckanovich, R.J. New Models of Hematogenous Ovarian Cancer Metastasis Demonstrate Preferential Spread to the Ovary and a Requirement for the Ovary for Abdominal Dissemination. Transl. Res. 2016, 175, 92–102.e2. [Google Scholar] [CrossRef] [PubMed]
- Figueras, A.; Alsina-Sanchís, E.; Lahiguera, Á.; Abreu, M.; Muinelo-Romay, L.; Moreno-Bueno, G.; Casanovas, O.; Graupera, M.; Matias-Guiu, X.; Vidal, A.; et al. A Role for CXCR4 in Peritoneal and Hematogenous Ovarian Cancer Dissemination. Mol. Cancer Ther. 2018, 17, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Block, M.S.; Vierkant, R.A.; Rambau, P.F.; Winham, S.J.; Wagner, P.; Traficante, N.; Tołoczko, A.; Tiezzi, D.G.; Taran, F.A.; Sinn, P.; et al. MyD88 and TLR4 Expression in Epithelial Ovarian Cancer. Mayo Clin. Proc. 2018, 93, 307–320. [Google Scholar] [CrossRef]
- Kolostova, K.; Pinkas, M.; Jakabova, A.; Pospisilova, E.; Svobodova, P.; Spicka, J.; Cegan, M.; Matkowski, R.; Bobek, V. Molecular Characterization of Circulating Tumor Cells in Ovarian Cancer. Am. J. Cancer Res. 2016, 6, 973–980. [Google Scholar]
- Obermayr, E.; Maritschnegg, E.; Agreiter, C.; Pecha, N.; Speiser, P.; Helmy-Bader, S.; Danzinger, S.; Krainer, M.; Singer, C.; Zeillinger, R. Efficient Leukocyte Depletion by a Novel Microfluidic Platform Enables the Molecular Detection and Characterization of Circulating Tumor Cells. Oncotarget 2018, 9, 812–823. [Google Scholar] [CrossRef]
- Lou, E.; Vogel, R.I.; Teoh, D.; Hoostal, S.; Grad, A.; Gerber, M.; Monu, M.; Łukaszewski, T.; Deshpande, J.; Linden, M.A.; et al. Assessment of Circulating Tumor Cells as a Predictive Biomarker of Histology in Women With Suspected Ovarian Cancer. Lab. Med. 2018, 49, 134–139. [Google Scholar] [CrossRef]
- Szczerba, A.; Śliwa, A.; Pieta, P.P.; Jankowska, A. The Role of Circulating Tumor Cells in Ovarian Cancer Dissemination. Cancers 2022, 14, 6030. [Google Scholar] [CrossRef]
- Asante, D.-B.; Calapre, L.; Ziman, M.; Meniawy, T.M.; Gray, E.S. Liquid Biopsy in Ovarian Cancer Using Circulating Tumor DNA and Cells: Ready for Prime Time? Cancer Lett. 2020, 468, 59–71. [Google Scholar] [CrossRef]
- Banys-Paluchowski, M.; Fehm, T.; Neubauer, H.; Paluchowski, P.; Krawczyk, N.; Meier-Stiegen, F.; Wallach, C.; Kaczerowsky, A.; Gebauer, G. Clinical Relevance of Circulating Tumor Cells in Ovarian, Fallopian Tube and Peritoneal Cancer. Arch. Gynecol. Obstet. 2020, 301, 1027–1035. [Google Scholar] [CrossRef]
- Yousefi, M.; Dehghani, S.; Nosrati, R.; Ghanei, M.; Salmaninejad, A.; Rajaie, S.; Hasanzadeh, M.; Pasdar, A. Current Insights into the Metastasis of Epithelial Ovarian Cancer—Hopes and Hurdles. Cell Oncol. 2020, 43, 515–538. [Google Scholar] [CrossRef]
- Zeng, L.; Liang, X.; Liu, Q.; Yang, Z. The Predictive Value of Circulating Tumor Cells in Ovarian Cancer: A Meta Analysis. Int. J. Gynecol. Cancer 2017, 27, 1109–1117. [Google Scholar] [CrossRef]
- Lee, M.; Kim, E.J.; Cho, Y.; Kim, S.; Chung, H.H.; Park, N.H.; Song, Y.-S. Predictive Value of Circulating Tumor Cells (CTCs) Captured by Microfluidic Device in Patients with Epithelial Ovarian Cancer. Gynecol. Oncol. 2017, 145, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Poveda, A.; Kaye, S.B.; McCormack, R.; Wang, S.; Parekh, T.; Ricci, D.; Lebedinsky, C.A.; Tercero, J.C.; Zintl, P.; Monk, B.J. Circulating Tumor Cells Predict Progression Free Survival and Overall Survival in Patients with Relapsed/Recurrent Advanced Ovarian Cancer. Gynecol. Oncol. 2011, 122, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Obermayr, E.; Castillo-Tong, D.C.; Pils, D.; Speiser, P.; Braicu, I.; Van Gorp, T.; Mahner, S.; Sehouli, J.; Vergote, I.; Zeillinger, R. Molecular Characterization of Circulating Tumor Cells in Patients with Ovarian Cancer Improves Their Prognostic Significance—A Study of the OVCAD Consortium. Gynecol. Oncol. 2013, 128, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Pearl, M.L.; Zhao, Q.; Yang, J.; Dong, H.; Tulley, S.; Zhang, Q.; Golightly, M.; Zucker, S.; Chen, W.-T. Prognostic Analysis of Invasive Circulating Tumor Cells (ICTCs) in Epithelial Ovarian Cancer. Gynecol. Oncol. 2014, 134, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Pearl, M.L.; Dong, H.; Tulley, S.; Zhao, Q.; Golightly, M.; Zucker, S.; Chen, W.-T. Treatment Monitoring of Patients with Epithelial Ovarian Cancer Using Invasive Circulating Tumor Cells (ICTCs). Gynecol. Oncol. 2015, 137, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Amintas, S.; Bedel, A.; Moreau-Gaudry, F.; Boutin, J.; Buscail, L.; Merlio, J.-P.; Vendrely, V.; Dabernat, S.; Buscail, E. Circulating Tumor Cell Clusters: United We Stand Divided We Fall. Int. J. Mol. Sci. 2020, 21, 2653. [Google Scholar] [CrossRef]
- Kenda Suster, N.; Virant-Klun, I. Presence and Role of Stem Cells in Ovarian Cancer. World J. Stem Cells 2019, 11, 383–397. [Google Scholar] [CrossRef]
- Bapat, S.A.; Mali, A.M.; Koppikar, C.B.; Kurrey, N.K. Stem and Progenitor-Like Cells Contribute to the Aggressive Behavior of Human Epithelial Ovarian Cancer. Cancer Res. 2005, 65, 3025–3029. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.-C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.-M.; Nephew, K.P. Identification and Characterization of Ovarian Cancer-Initiating Cells from Primary Human Tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Kadife, E.; Raza, A.; Short, M.; Jubinsky, P.T.; Kannourakis, G. Ovarian Cancer, Cancer Stem Cells and Current Treatment Strategies: A Potential Role of Magmas in the Current Treatment Methods. Cells 2020, 9, 719. [Google Scholar] [CrossRef] [PubMed]
- Parte, S.; Bhartiya, D.; Telang, J.; Daithankar, V.; Salvi, V.; Zaveri, K.; Hinduja, I. Retraction: Detection, Characterization, and Spontaneous Differentiation In Vitro of Very Small Embryonic-Like Putative Stem Cells in Adult Mammalian Ovary. Stem Cells Dev. 2011, 20, 1451–1464. [Google Scholar] [CrossRef]
- Iżycka, N.; Zaborowski, M.P.; Ciecierski, Ł.; Jaz, K.; Szubert, S.; Miedziarek, C.; Rezler, M.; Piątek-Bajan, K.; Synakiewicz, A.; Jankowska, A.; et al. Cancer Stem Cell Markers—Clinical Relevance and Prognostic Value in High-Grade Serous Ovarian Cancer (HGSOC) Based on The Cancer Genome Atlas Analysis. Int. J. Mol. Sci. 2023, 24, 12746. [Google Scholar] [CrossRef] [PubMed]
- Bharathan, R. Surgical Management of Ovarian Cancer. In Ovarian Cancer—From Pathogenesis to Treatment; InTech: London, UK, 2018. [Google Scholar]
- Huang, C.-Y.; Cheng, M.; Lee, N.-R.; Huang, H.-Y.; Lee, W.-L.; Chang, W.-H.; Wang, P.-H. Comparing Paclitaxel–Carboplatin with Paclitaxel–Cisplatin as the Front-Line Chemotherapy for Patients with FIGO IIIC Serous-Type Tubo-Ovarian Cancer. Int. J. Environ. Res. Public Health 2020, 17, 2213. [Google Scholar] [CrossRef]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review, 1975–2017; National Cancer Institute: Bethesda, MD, USA, 2020. [Google Scholar]
- Arend, R.C.; Jackson-Fisher, A.; Jacobs, I.A.; Chou, J.; Monk, B.J. Ovarian Cancer: New Strategies and Emerging Targets for the Treatment of Patients with Advanced Disease. Cancer Biol. Ther. 2021, 22, 89–105. [Google Scholar] [CrossRef]
- Varaganti, P.; Buddolla, V.; Lakshmi, B.A.; Kim, Y.-J. Recent Advances in Using Folate Receptor 1 (FOLR1) for Cancer Diagnosis and Treatment, with an Emphasis on Cancers That Affect Women. Life Sci. 2023, 326, 121802. [Google Scholar] [CrossRef]
- Cheung, A.; Bax, H.J.; Josephs, D.H.; Ilieva, K.M.; Pellizzari, G.; Opzoomer, J.; Bloomfield, J.; Fittall, M.; Grigoriadis, A.; Figini, M.; et al. Targeting Folate Receptor Alpha for Cancer Treatment. Oncotarget 2016, 7, 52553–52574. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, L.; Guntupalli, S.R. Treatment of Epithelial Ovarian Cancer. BMJ 2020, 371, m3773. [Google Scholar] [CrossRef]
- Pignata, S.; C Cecere, S.; Du Bois, A.; Harter, P.; Heitz, F. Treatment of Recurrent Ovarian Cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, viii51–viii56. [Google Scholar] [CrossRef] [PubMed]
- Gandalovičová, A.; Rosel, D.; Fernandes, M.; Veselý, P.; Heneberg, P.; Čermák, V.; Petruželka, L.; Kumar, S.; Sanz-Moreno, V.; Brábek, J. Migrastatics—Anti-Metastatic and Anti-Invasion Drugs: Promises and Challenges. Trends Cancer 2017, 3, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Salbreux, G.; Charras, G.; Paluch, E. Actin Cortex Mechanics and Cellular Morphogenesis. Trends Cell Biol. 2012, 22, 536–545. [Google Scholar] [CrossRef]
- Hong, L.; Kenney, S.R.; Phillips, G.K.; Simpson, D.; Schroeder, C.E.; Nöth, J.; Romero, E.; Swanson, S.; Waller, A.; Strouse, J.J.; et al. Characterization of a Cdc42 Protein Inhibitor and Its Use as a Molecular Probe. J. Biol. Chem. 2013, 288, 8531–8543. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Kenney, S.R.; Muller, C.Y.; Adams, S.; Rutledge, T.; Romero, E.; Murray-Krezan, C.; Prekeris, R.; Sklar, L.A.; Hudson, L.G.; et al. R-Ketorolac Targets Cdc42 and Rac1 and Alters Ovarian Cancer Cell Behaviors Critical for Invasion and Metastasis. Mol. Cancer Ther. 2015, 14, 2215–2227. [Google Scholar] [CrossRef]
- Guo, Y.; Kenney, S.R.; Cook, L.; Adams, S.F.; Rutledge, T.; Romero, E.; Oprea, T.I.; Sklar, L.A.; Bedrick, E.; Wiggins, C.L.; et al. A Novel Pharmacologic Activity of Ketorolac for Therapeutic Benefit in Ovarian Cancer Patients. Clin. Cancer Res. 2015, 21, 5064–5072. [Google Scholar] [CrossRef]
- Akhshi, T.K.; Wernike, D.; Piekny, A. Microtubules and Actin Crosstalk in Cell Migration and Division. Cytoskeleton 2014, 71, 1–23. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Microtubules, Microtubule-Interfering Agents and Apoptosis. Apoptosis 2003, 8, 413–450. [Google Scholar] [CrossRef]
- De Geest, K.; Blessing, J.A.; Morris, R.T.; Yamada, S.D.; Monk, B.J.; Zweizig, S.L.; Matei, D.; Muller, C.Y.; Richards, W.E. Phase II Clinical Trial of Ixabepilone in Patients with Recurrent or Persistent Platinum- and Taxane-Resistant Ovarian or Primary Peritoneal Cancer: A Gynecologic Oncology Group Study. J. Clin. Oncol. 2010, 28, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Bozkaya, Y.; Doğan, M.; Umut Erdem, G.; Tulunay, G.; Uncu, H.; Arık, Z.; Demirci, U.; Yazıcı, O.; Zengin, N. Effectiveness of Low-Dose Oral Etoposide Treatment in Patients with Recurrent and Platinum-Resistant Epithelial Ovarian Cancer. J. Obstet. Gynaecol. 2017, 37, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Kokkinos, M.I.; Wafai, R.; Wong, M.K.; Newgreen, D.F.; Thompson, E.W.; Waltham, M. Vimentin and Epithelial-Mesenchymal Transition in Human Breast Cancer--Observations in Vitro and in Vivo. Cells. Tissues. Organs 2007, 185, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Satelli, A.; Li, S. Vimentin in Cancer and Its Potential as a Molecular Target for Cancer Therapy. Cell Mol. Life Sci. 2011, 68, 3033–3046. [Google Scholar] [CrossRef] [PubMed]
- Karantza, V. Keratins in Health and Cancer: More than Mere Epithelial Cell Markers. Oncogene 2011, 30, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Momenimovahed, Z.; Tiznobaik, A.; Taheri, S.; Salehiniya, H. Ovarian Cancer in the World: Epidemiology and Risk Factors. Int. J. Womens. Health 2019, 11, 287–299. [Google Scholar] [CrossRef]
- Yang, C.P.H.; Horwitz, S.B. Taxol®: The First Microtubule Stabilizing Agent Taxol. Int. J. Mol. Sci. 2017, 18, 1733. [Google Scholar] [CrossRef]
- Results, T.; Piccart, M.J.; Bertelsen, K.; James, K.; Cassidy, J.; Simonsen, E.; Stuart, G.; Kaye, S.; Vergote, I.; Grimshaw, R.; et al. Randomized Intergroup Trial of Cisplatin–Paclitaxel Versus Cisplatin–Cyclophosphamide in Women With Advanced Epithelial Ovarian Cancer: Three-Year Results. JNCI J. Natl. Cancer Inst. 2000, 92, 699–708. [Google Scholar]
- Stehn, J.R.; Haass, N.K.; Bonello, T.; Desouza, M.; Kottyan, G.; Treutlein, H.; Zeng, J.; Nascimento, P.R.B.B.; Sequeira, V.B.; Butler, T.L.; et al. A Novel Class of Anticancer Compounds Targets the Actin Cytoskeleton in Tumor Cells. Cancer Res. 2013, 73, 5169–5182. [Google Scholar] [CrossRef]
- Nürnberg, A.; Kollmannsperger, A.; Grosse, R. Pharmacological Inhibition of Actin Assembly to Target Tumor Cell Motility. Rev. Physiol. Biochem. Pharmacol. 2013, 166, 23–42. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Śliwa, A.; Szczerba, A.; Pięta, P.P.; Białas, P.; Lorek, J.; Nowak-Markwitz, E.; Jankowska, A. A Recipe for Successful Metastasis: Transition and Migratory Modes of Ovarian Cancer Cells. Cancers 2024, 16, 783. https://doi.org/10.3390/cancers16040783
Śliwa A, Szczerba A, Pięta PP, Białas P, Lorek J, Nowak-Markwitz E, Jankowska A. A Recipe for Successful Metastasis: Transition and Migratory Modes of Ovarian Cancer Cells. Cancers. 2024; 16(4):783. https://doi.org/10.3390/cancers16040783
Chicago/Turabian StyleŚliwa, Aleksandra, Anna Szczerba, Paweł Piotr Pięta, Piotr Białas, Jakub Lorek, Ewa Nowak-Markwitz, and Anna Jankowska. 2024. "A Recipe for Successful Metastasis: Transition and Migratory Modes of Ovarian Cancer Cells" Cancers 16, no. 4: 783. https://doi.org/10.3390/cancers16040783