Pancreatic Cancer Treatment Targeting the HGF/c-MET Pathway: The MEK Inhibitor Trametinib

 , ,
, ,  and
and
Abstract
Simple Summary
Abstract
1. Pancreatic Cancer and Its Tumor Microenvironment
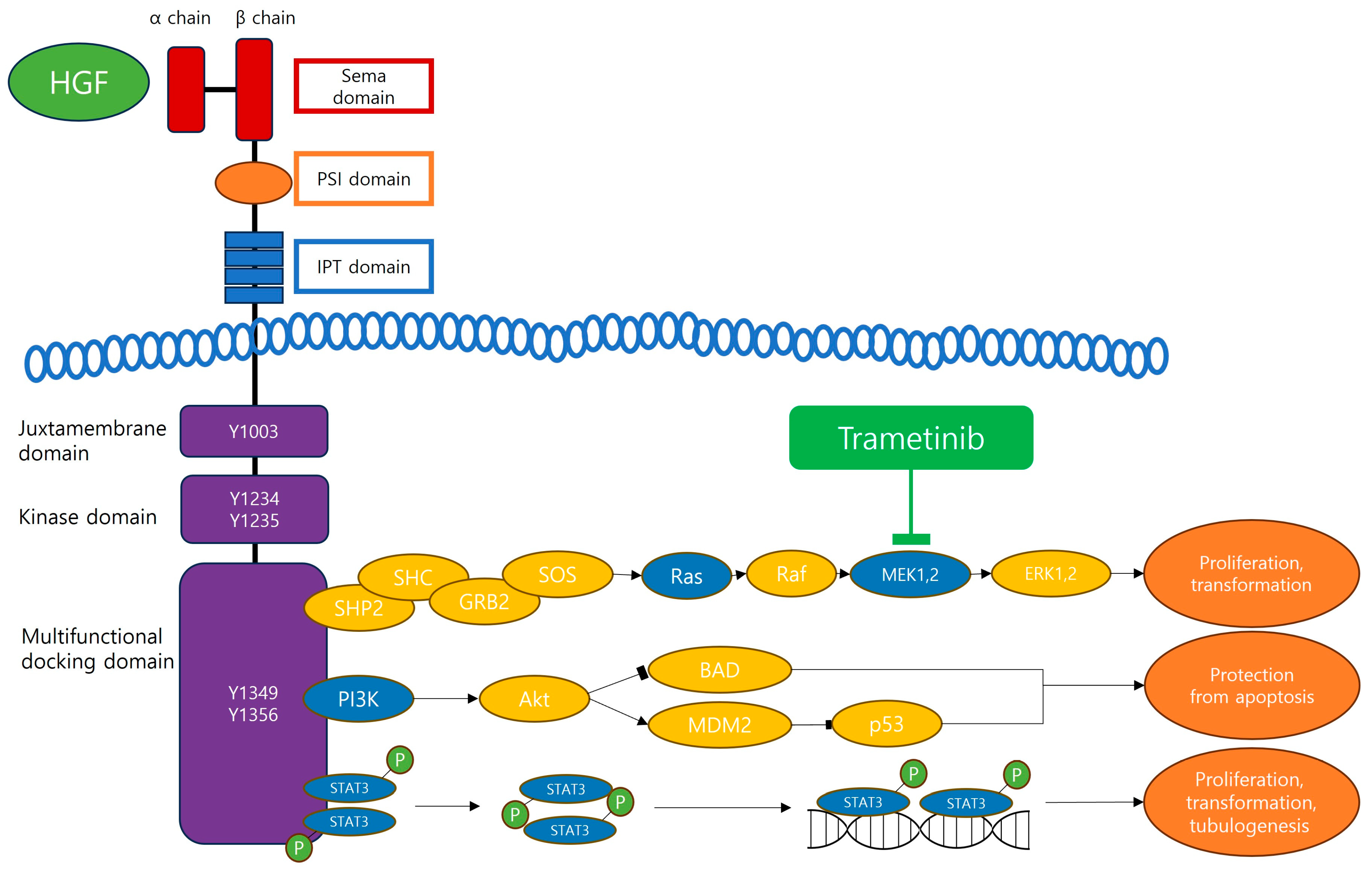
2. HGF/c-MET Pathway
3. HGF/c-MET Pathway in the Pancreatic Cancer
3.1. HGF/c-MET and MAPK/ERK (Ras, Ras/Raf/MEK/ERK) Pathway
3.2. HGF/c-MET and PI3K Pathway
3.3. HGF/c-MET and the Hypoxia, Angiogenesis, Metastasis of Pancreatic Cancer
3.4. HGF/c-MET and the Urokinase Plasminogen Activator (uPA) Positive Feed-Forward Loop
4. Trametinib, the MEK Inhibitor
5. Mechanisms of Trametinib in Pancreatic Cancer
5.1. Inhibition of the HGF/c-MET and MAPK/ERK Pathways
5.2. Alterations in the Tumor Microenvironment and Modulation of Immune Responses
6. Clinical Trials of Trametinib in Pancreatic Cancer
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| c-MET | Mesenchymal–epithelial transition factor |
| HGF | Hepatocyte growth factor |
| kDa | Kilodalton |
| MET | MAPK/ERK |
| PTK | Protein tyrosine kinase |
| GEFs | Guanine exchange factors |
| GTP | Guanosine triphosphate |
| PIP2 | Phosphatidylinositol-4,5-diphosphate |
| PIP3 | Phosphatidylinositol-3,4,5-triphosphate |
| Akt | protein kinase B |
| SHP2 | SH2 domain-containing tyrosine phosphatase |
| SHC | SH2 domain-containing transforming protein |
| GRB2 | Growth factor receptor-bound protein 2 |
| SOS | Son of sevenless |
| MEK | Mitogen-activated protein kinase kinase |
| ERK | Extracellular signal-regulated kinases |
| PI3K | Phosphoinositide 3-kinase |
| Akt | Protein kinase B |
| BAD | BCL-2 antagonist of cell death |
| MDM2 | Murine double minute 2 |
| STAT3 | Signal transducer and activator of transcription 3 |
| HIF-1 | Hypoxia-inducible factor-1 |
| HIF-1α | Hypoxia-inducible factor 1-alpha |
| mTOR | Mammalian target of rapamycin |
References
- Quaresma, M.; Coleman, M.P.; Rachet, B. 40-year trends in an index of survival for all cancers combined and survival adjusted for age and sex for each cancer in England and Wales, 1971–2011: A population-based study. Lancet 2015, 385, 1206–1218. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Aung, K.L.; Fischer, S.E.; Denroche, R.E.; Jang, G.H.; Dodd, A.; Creighton, S.; Southwood, B.; Liang, S.B.; Chadwick, D.; Zhang, A.; et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clin. Cancer Res. 2018, 24, 1344–1354. [Google Scholar] [CrossRef]
- Kalimuthu, S.N.; Wilson, G.W.; Grant, R.C.; Seto, M.; O’Kane, G.; Vajpeyi, R.; Notta, F.; Gallinger, S.; Chetty, R. Morphological classification of pancreatic ductal adenocarcinoma that predicts molecular subtypes and correlates with clinical outcome. Gut 2020, 69, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Chan-Seng-Yue, M.; Kim, J.C.; Wilson, G.W.; Ng, K.; Figueroa, E.F.; O’Kane, G.M.; Connor, A.A.; Denroche, R.E.; Grant, R.C.; McLeod, J.; et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 2020, 52, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Dijk, F.; Veenstra, V.L.; Soer, E.C.; Dings, M.P.G.; Zhao, L.; Halfwerk, J.B.; Hooijer, G.K.; Damhofer, H.; Marzano, M.; Steins, A.; et al. Unsupervised class discovery in pancreatic ductal adenocarcinoma reveals cell-intrinsic mesenchymal features and high concordance between existing classification systems. Sci. Rep. 2020, 10, 337. [Google Scholar] [CrossRef] [PubMed]
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.; et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013.e3. [Google Scholar] [CrossRef] [PubMed]
- Falcomata, C.; Barthel, S.; Widholz, S.A.; Schneeweis, C.; Montero, J.J.; Toska, A.; Mir, J.; Kaltenbacher, T.; Heetmeyer, J.; Swietlik, J.J.; et al. Selective multi-kinase inhibition sensitizes mesenchymal pancreatic cancer to immune checkpoint blockade by remodeling the tumor microenvironment. Nat. Cancer 2022, 3, 318–336. [Google Scholar] [CrossRef]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [PubMed]
- Pothula, S.P.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Pancreatic stellate cells: Aiding and abetting pancreatic cancer progression. Pancreatology 2020, 20, 409–418. [Google Scholar] [CrossRef]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Pothula, S.P.; Xu, Z.; Goldstein, D.; Biankin, A.V.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Hepatocyte growth factor inhibition: A novel therapeutic approach in pancreatic cancer. Br. J. Cancer 2016, 114, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Vonlaufen, A.; Joshi, S.; Qu, C.; Phillips, P.A.; Xu, Z.; Parker, N.R.; Toi, C.S.; Pirola, R.C.; Wilson, J.S.; Goldstein, D.; et al. Pancreatic stellate cells: Partners in crime with pancreatic cancer cells. Cancer Res. 2008, 68, 2085–2093. [Google Scholar] [CrossRef]
- Xu, Z.; Pang, T.C.Y.; Liu, A.C.; Pothula, S.P.; Mekapogu, A.R.; Perera, C.J.; Murakami, T.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; et al. Targeting the HGF/c-MET pathway in advanced pancreatic cancer: A key element of treatment that limits primary tumour growth and eliminates metastasis. Br. J. Cancer 2020, 122, 1486–1495. [Google Scholar] [CrossRef]
- Xu, Z.; Vonlaufen, A.; Phillips, P.A.; Fiala-Beer, E.; Zhang, X.; Yang, L.; Biankin, A.V.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am. J. Pathol. 2010, 177, 2585–2596. [Google Scholar] [CrossRef]
- Salgia, R. MET in Lung Cancer: Biomarker Selection Based on Scientific Rationale. Mol. Cancer Ther. 2017, 16, 555–565. [Google Scholar] [CrossRef]
- Fu, Y.T.; Zheng, H.B.; Zhou, L.; Zhang, D.Q.; Liu, X.L.; Sun, H. Valproic acid, targets papillary thyroid cancer through inhibition of c-Met signalling pathway. Am. J. Transl. Res. 2017, 9, 3138–3147. [Google Scholar]
- Rucki, A.A.; Xiao, Q.; Muth, S.; Chen, J.; Che, X.; Kleponis, J.; Sharma, R.; Anders, R.A.; Jaffee, E.M.; Zheng, L. Dual Inhibition of Hedgehog and c-Met Pathways for Pancreatic Cancer Treatment. Mol. Cancer Ther. 2017, 16, 2399–2409. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Xiong, X.; Kim, Y.; Okada, N.; Lu, F.; Zhang, H.; Sun, H. Acid sphingomyelinase is required for cell surface presentation of Met receptor tyrosine kinase in cancer cells. J. Cell Sci. 2016, 129, 4238–4251. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.T.; Jing, Y.Y.; Yu, G.F.; Chen, H.; Han, Z.P.; Yu, D.D.; Fan, Q.M.; Ye, F.; Li, R.; Gao, L.; et al. Hepatic stellate cell promoted hepatoma cell invasion via the HGF/c-Met signaling pathway regulated by p53. Cell Cycle 2016, 15, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, Z.; Zhang, M. Biomarker development in MET-targeted therapy. Oncotarget 2016, 7, 37370–37389. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, H.; Chen, J. Dysregulation of HGF/c-Met signal pathway and their targeting drugs in lung cancer. Zhongguo Fei Ai Za Zhi 2014, 17, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Su, X.; Li, Z.; Deng, L.; Liu, X.; Feng, X.; Peng, J. HGF/c-MET pathway in cancer: From molecular characterization to clinical evidence. Oncogene 2021, 40, 4625–4651. [Google Scholar] [CrossRef]
- Giordano, S.; Ponzetto, C.; Di Renzo, M.F.; Cooper, C.S.; Comoglio, P.M. Tyrosine kinase receptor indistinguishable from the c-met protein. Nature 1989, 339, 155–156. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef]
- Comoglio, P.M.; Trusolino, L.; Boccaccio, C. Known and novel roles of the MET oncogene in cancer: A coherent approach to targeted therapy. Nat. Rev. Cancer 2018, 18, 341–358. [Google Scholar] [CrossRef]
- Houldsworth, J.; Cordon-Cardo, C.; Ladanyi, M.; Kelsen, D.P.; Chaganti, R.S. Gene amplification in gastric and esophageal adenocarcinomas. Cancer Res. 1990, 50, 6417–6422. [Google Scholar]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef]
- Patel, M.B.; Pothula, S.P.; Xu, Z.; Lee, A.K.; Goldstein, D.; Pirola, R.C.; Apte, M.V.; Wilson, J.S. The role of the hepatocyte growth factor/c-MET pathway in pancreatic stellate cell-endothelial cell interactions: Antiangiogenic implications in pancreatic cancer. Carcinogenesis 2014, 35, 1891–1900. [Google Scholar] [CrossRef]
- Cecchi, F.; Rabe, D.C.; Bottaro, D.P. Targeting the HGF/Met signaling pathway in cancer therapy. Expert. Opin. Ther. Targets 2012, 16, 553–572. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Liebmann, C. Regulation of MAP kinase activity by peptide receptor signalling pathway: Paradigms of multiplicity. Cell Signal 2001, 13, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Rosario, M.; Birchmeier, W. How to make tubes: Signaling by the Met receptor tyrosine kinase. Trends Cell Biol. 2003, 13, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Mottet, D.; Dumont, V.; Deccache, Y.; Demazy, C.; Ninane, N.; Raes, M.; Michiels, C. Regulation of hypoxia-inducible factor-1alpha protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta pathway in HepG2 cells. J. Biol. Chem. 2003, 278, 31277–31285. [Google Scholar] [CrossRef]
- Jiang, B.H.; Liu, L.Z. AKT signaling in regulating angiogenesis. Curr. Cancer Drug Targets 2008, 8, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Rue, E.A.; Iyer, N.V.; Pang, M.G.; Kearns, W.G. Assignment of the hypoxia-inducible factor 1alpha gene to a region of conserved synteny on mouse chromosome 12 and human chromosome 14q. Genomics 1996, 34, 437–439. [Google Scholar] [CrossRef]
- Hogenesch, J.B.; Chan, W.K.; Jackiw, V.H.; Brown, R.C.; Gu, Y.Z.; Pray-Grant, M.; Perdew, G.H.; Bradfield, C.A. Characterization of a subset of the basic-helix-loop-helix-PAS superfamily that interacts with components of the dioxin signaling pathway. J. Biol. Chem. 1997, 272, 8581–8593. [Google Scholar] [CrossRef]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef]
- Kitajima, Y.; Ide, T.; Ohtsuka, T.; Miyazaki, K. Induction of hepatocyte growth factor activator gene expression under hypoxia activates the hepatocyte growth factor/c-Met system via hypoxia inducible factor-1 in pancreatic cancer. Cancer Sci. 2008, 99, 1341–1347. [Google Scholar] [CrossRef]
- Ding, S.; Merkulova-Rainon, T.; Han, Z.C.; Tobelem, G. HGF receptor up-regulation contributes to the angiogenic phenotype of human endothelial cells and promotes angiogenesis in vitro. Blood 2003, 101, 4816–4822. [Google Scholar] [CrossRef]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef]
- Canadas, I.; Rojo, F.; Arumi-Uria, M.; Rovira, A.; Albanell, J.; Arriola, E. C-MET as a new therapeutic target for the development of novel anticancer drugs. Clin. Transl. Oncol. 2010, 12, 253–260. [Google Scholar] [CrossRef]
- Gibbons, A.V.; Lin, J.E.; Kim, G.W.; Marszalowicz, G.P.; Li, P.; Stoecker, B.A.; Blomain, E.S.; Rattan, S.; Snook, A.E.; Schulz, S.; et al. Intestinal GUCY2C prevents TGF-beta secretion coordinating desmoplasia and hyperproliferation in colorectal cancer. Cancer Res. 2013, 73, 6654–6666. [Google Scholar] [CrossRef] [PubMed]
- Kemik, O.; Purisa, S.; Kemik, A.S.; Tuzun, S. Increase in the circulating level of hepatocyte growth factor in pancreatic cancer patients. Bratisl. Lek. Listy 2009, 110, 627–629. [Google Scholar] [PubMed]
- Ueda, T.; Takeyama, Y.; Hori, Y.; Nishikawa, J.; Yamamoto, M.; Saitoh, Y. Hepatocyte growth factor in assessment of acute pancreatitis: Comparison with C-reactive protein and interleukin-6. J. Gastroenterol. 1997, 32, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.H.; Huang, C.; Qiu, Z.J.; Liu, J.; Zhang, Z.H.; Zhao, N.; Feng, Z.Z.; Lv, X.H. Expression and prognostic significance of CD151, c-Met, and integrin alpha3/alpha6 in pancreatic ductal adenocarcinoma. Dig. Dis. Sci. 2011, 56, 1090–1098. [Google Scholar] [CrossRef]
- Park, J.K.; Kim, M.A.; Ryu, J.K.; Yoon, Y.B.; Kim, S.W.; Han, H.S.; Kang, G.H.; Kim, H.; Hwang, J.H.; Kim, Y.T. Postoperative prognostic predictors of pancreatic ductal adenocarcinoma: Clinical analysis and immunoprofile on tissue microarrays. Ann. Surg. Oncol. 2012, 19, 2664–2672. [Google Scholar] [CrossRef] [PubMed]
- Hage, C.; Rausch, V.; Giese, N.; Giese, T.; Schonsiegel, F.; Labsch, S.; Nwaeburu, C.; Mattern, J.; Gladkich, J.; Herr, I. The novel c-Met inhibitor cabozantinib overcomes gemcitabine resistance and stem cell signaling in pancreatic cancer. Cell Death Dis. 2013, 4, e627. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.W.; Somcio, R.J.; Fan, F.; Liu, W.; Johnson, M.; Lesslie, D.P.; Evans, D.B.; Gallick, G.E.; Ellis, L.M. Regulatory role of c-Met in insulin-like growth factor-I receptor-mediated migration and invasion of human pancreatic carcinoma cells. Mol. Cancer Ther. 2006, 5, 1676–1682. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.S.; Gaziova, I.; Harrigal, L.; Guerra, Y.A.; Qiu, S.; Sastry, S.K.; Arumugam, T.; Logsdon, C.D.; Elferink, L.A. Met receptor tyrosine kinase signaling induces secretion of the angiogenic chemokine interleukin-8/CXCL8 in pancreatic cancer. PLoS ONE 2012, 7, e40420. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.W.; Liu, W.; Fan, F.; Camp, E.R.; Yang, A.; Somcio, R.J.; Bucana, C.D.; Callahan, J.; Parry, G.C.; Evans, D.B.; et al. Targeting of urokinase plasminogen activator receptor in human pancreatic carcinoma cells inhibits c-Met- and insulin-like growth factor-I receptor-mediated migration and invasion and orthotopic tumor growth in mice. Cancer Res. 2005, 65, 7775–7781. [Google Scholar] [CrossRef]
- Buckley, B.J.; Aboelela, A.; Minaei, E.; Jiang, L.X.; Xu, Z.; Ali, U.; Fildes, K.; Cheung, C.Y.; Cook, S.M.; Johnson, D.C.; et al. 6-Substituted Hexamethylene Amiloride (HMA) Derivatives as Potent and Selective Inhibitors of the Human Urokinase Plasminogen Activator for Use in Cancer. J. Med. Chem. 2018, 61, 8299–8320. [Google Scholar] [CrossRef]
- Zeiser, R. Trametinib. Recent. Results Cancer Res. 2014, 201, 241–248. [Google Scholar] [CrossRef]
- Wright, C.J.; McCormack, P.L. Trametinib: First global approval. Drugs 2013, 73, 1245–1254. [Google Scholar] [CrossRef]
- Brannon, A., 3rd; Drouillard, D.; Steele, N.; Schoettle, S.; Abel, E.V.; Crawford, H.C.; Pasca di Magliano, M. Beta 1 integrin signaling mediates pancreatic ductal adenocarcinoma resistance to MEK inhibition. Sci. Rep. 2020, 10, 11133. [Google Scholar] [CrossRef]
- Lugowska, I.; Kosela-Paterczyk, H.; Kozak, K.; Rutkowski, P. Trametinib: A MEK inhibitor for management of metastatic melanoma. Onco Targets Ther. 2015, 8, 2251–2259. [Google Scholar] [CrossRef]
- Infante, J.R.; Fecher, L.A.; Falchook, G.S.; Nallapareddy, S.; Gordon, M.S.; Becerra, C.; DeMarini, D.J.; Cox, D.S.; Xu, Y.; Morris, S.R.; et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.S.; Lewis, K.D.; Infante, J.R.; Gordon, M.S.; Vogelzang, N.J.; DeMarini, D.J.; Sun, P.; Moy, C.; Szabo, S.A.; Roadcap, L.T.; et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 782–789. [Google Scholar] [CrossRef]
- Caunt, C.J.; Sale, M.J.; Smith, P.D.; Cook, S.J. MEK1 and MEK2 inhibitors and cancer therapy: The long and winding road. Nat. Rev. Cancer 2015, 15, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Brauswetter, D.; Gurbi, B.; Varga, A.; Varkondi, E.; Schwab, R.; Banhegyi, G.; Fabian, O.; Keri, G.; Valyi-Nagy, I.; Petak, I. Molecular subtype specific efficacy of MEK inhibitors in pancreatic cancers. PLoS ONE 2017, 12, e0185687. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.S.; McDonald, P.C.; Nemirovsky, O.; Awrey, S.; Chafe, S.C.; Schaeffer, D.F.; Li, J.; Renouf, D.J.; Stanger, B.Z.; Dedhar, S. Overcoming Adaptive Resistance to KRAS and MEK Inhibitors by Co-targeting mTORC1/2 Complexes in Pancreatic Cancer. Cell Rep. Med. 2020, 1, 100131. [Google Scholar] [CrossRef]
- Aung, K.L.; McWhirter, E.; Welch, S.; Wang, L.; Lovell, S.; Stayner, L.A.; Ali, S.; Malpage, A.; Makepeace, B.; Ramachandran, M.; et al. A phase II trial of GSK2256098 and trametinib in patients with advanced pancreatic ductal adenocarcinoma. J. Gastrointest. Oncol. 2022, 13, 3216–3226. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Cao, Y.; Liu, W.; Ju, X.; Zhao, X.; Jiang, L.; Ye, Y.; Jin, G.; Zhang, H. Stereotactic body radiotherapy plus pembrolizumab and trametinib versus stereotactic body radiotherapy plus gemcitabine for locally recurrent pancreatic cancer after surgical resection: An open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2022, 23, e105–e115. [Google Scholar] [CrossRef]
- Infante, J.R.; Somer, B.G.; Park, J.O.; Li, C.P.; Scheulen, M.E.; Kasubhai, S.M.; Oh, D.Y.; Liu, Y.; Redhu, S.; Steplewski, K.; et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur. J. Cancer 2014, 50, 2072–2081. [Google Scholar] [CrossRef]
- Tai, D. Study of Trametinib and Ruxolitinib in Colorectal Cancer and Pancreatic Adenocarcinoma. Available online: https://clinicaltrials.gov/study/NCT04303403 (accessed on 19 February 2024).
- Vasquez, S. Combined MEK, STAT3 and PD-1 Inhibition in Metastatic Pancreatic Ductal Adenocarcinoma. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT05440942 (accessed on 20 February 2024).
- Sharry, S. Trametinib and Hydroxychloroquine in Treating Patients with Pancreatic Cancer (THREAD). Available online: https://clinicaltrials.gov/study/NCT03825289 (accessed on 19 February 2024).
- Duffy, A. PaTcH Study: A Phase 2 Study of Trametinib and Hydroxychloroquine in Patients with Metastatic Refractory Pancreatic Cancer (PaTcH). Available online: https://clinicaltrials.gov/study/NCT05518110 (accessed on 3 March 2024).


{kind=link}
{kind=link}
| ClinicalTrials.gov ID | Study Design | Intervention | Diagnosis and Stage | Number of Patients Treated with Trametinib | Median Progression Free Survival (95% Confidence Interval) | Median Overall Survival (95% Confidence Interval) | Publication Year | PMID |
|---|---|---|---|---|---|---|---|---|
| NCT04303403 | Phase Ib trial | Combination treatment of trametinib and ruxolitinib | Advanced RAS mutant colorectal cancer and pancreatic adenocarcinoma | NA | NA | NA | NA | |
| NCT05440942 | Phase I trial | Combination treatment of trametinib, retifanlimab, and ruxolitinib | Metastatic pancreatic ductal adenocarcinoma | NA | NA | NA | NA | |
| NCT03825289 | Phase I trial | Combination treatment of trametininb and hydroxychloroquine | Metastatic or locally advanced, unresectable pancreatic carcinoma | NA | NA | NA | NA | |
| NCT02428270 | Phase II trial of investigational drugs GSK2256098 and trametinib | GSK2256098 and Trametinib | Advanced PDAC patients whose disease progressed after first-line palliative chemotherapy | 16 | 1.6 months (1.5–1.8 months) | 3.6 months (2.7 months-not reached) | 2022 | 36636049 |
| NCT02704156 | Phase II trial with an open-label, randomized control design | Stereotactic body radiation therapy plus pembrolizumab and trametinib | Locally recurrent pancreatic cancer after surgical resection | 85 | 8.2 months (6.9–9.5 months) | 14.9 months (12.7–17.1 months) | 2022 | 35240087 |
| NCT05518110 | Phase II trial | Combination treatment of trametinib and hydroxychloroquine | Metastatic pancreatic cancer which has previously progressed on at least one line of systemic therapy | NA | NA | NA | NA | |
| NCT01231581 | Phase II trial with randomized, double-blind, placebo-controlled design | Combination treatment of trametinib and gemcitabine | Untreated metastatic adenocarcinoma of the pancreas | 80 | 8.4 months (7.8–9.9 months) | 16.1 weeks (14.0–23.4) | 2014 | 24915778 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Lee, T.S.; Lee, M.H.; Cho, I.R.; Ryu, J.K.; Kim, Y.-T.; Lee, S.H.; Paik, W.H. Pancreatic Cancer Treatment Targeting the HGF/c-MET Pathway: The MEK Inhibitor Trametinib. Cancers 2024, 16, 1056. https://doi.org/10.3390/cancers16051056
Kim J, Lee TS, Lee MH, Cho IR, Ryu JK, Kim Y-T, Lee SH, Paik WH. Pancreatic Cancer Treatment Targeting the HGF/c-MET Pathway: The MEK Inhibitor Trametinib. Cancers. 2024; 16(5):1056. https://doi.org/10.3390/cancers16051056
Chicago/Turabian StyleKim, Junyeol, Tae Seung Lee, Myeong Hwan Lee, In Rae Cho, Ji Kon Ryu, Yong-Tae Kim, Sang Hyub Lee, and Woo Hyun Paik. 2024. "Pancreatic Cancer Treatment Targeting the HGF/c-MET Pathway: The MEK Inhibitor Trametinib" Cancers 16, no. 5: 1056. https://doi.org/10.3390/cancers16051056
APA StyleKim, J., Lee, T. S., Lee, M. H., Cho, I. R., Ryu, J. K., Kim, Y.-T., Lee, S. H., & Paik, W. H. (2024). Pancreatic Cancer Treatment Targeting the HGF/c-MET Pathway: The MEK Inhibitor Trametinib. Cancers, 16(5), 1056. https://doi.org/10.3390/cancers16051056