
Breast Tumor Metastasis and Its Microenvironment: It Takes Both Seed and Soil to Grow a Tumor and Target It for Treatment

 , ,
, , 
 ,
,  , , , ,
, , , , 
Abstract
Simple Summary
Abstract
1. Introduction
2. Seed: Tumor Clonal Heterogeneity
2.1. Breast Cancer Subtypes
2.2. Clonal Evolution, Intratumor Heterogeneity and Metastasis
3. Soil: The Breast Cancer Metastasis Sites and the Immune Environment
3.1. Metastatic Sites of Breast Cancer
3.2. Metastasized Tumor-Immune Microenvironment
4. Soil: Tissue-Specific Properties at Metastatic Sites
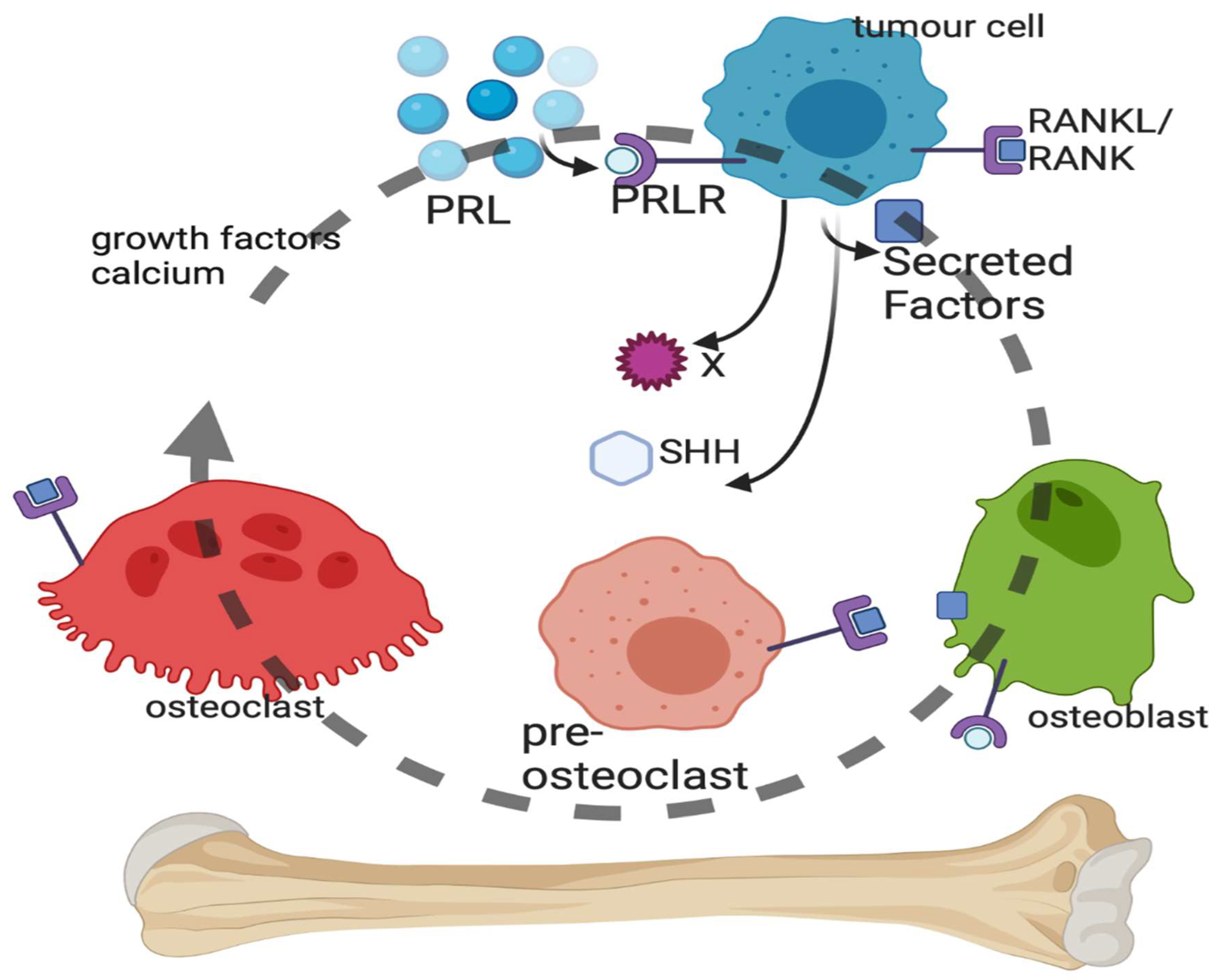
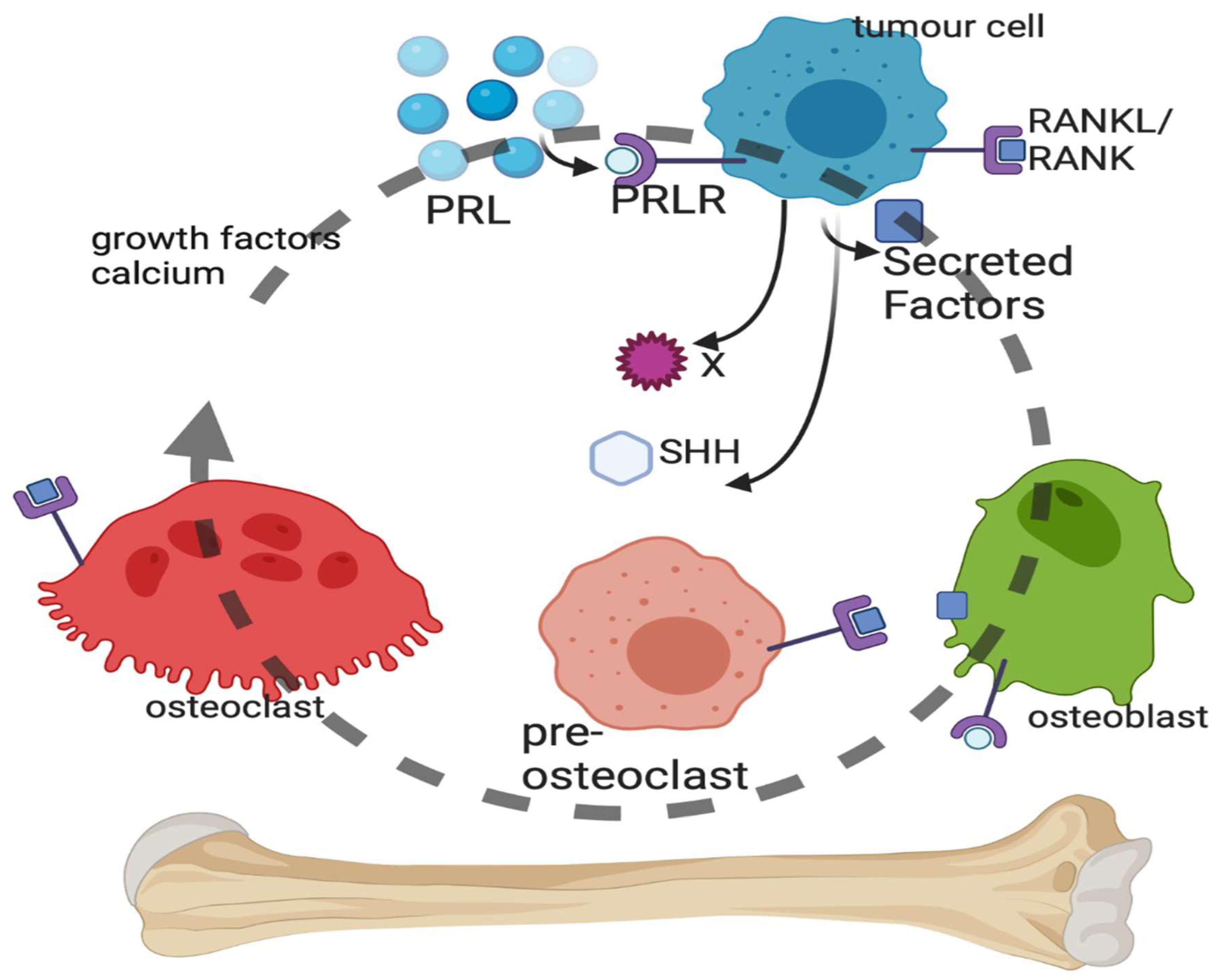
4.1. Bone Metastasis of Breast Cancer (Figure 2)
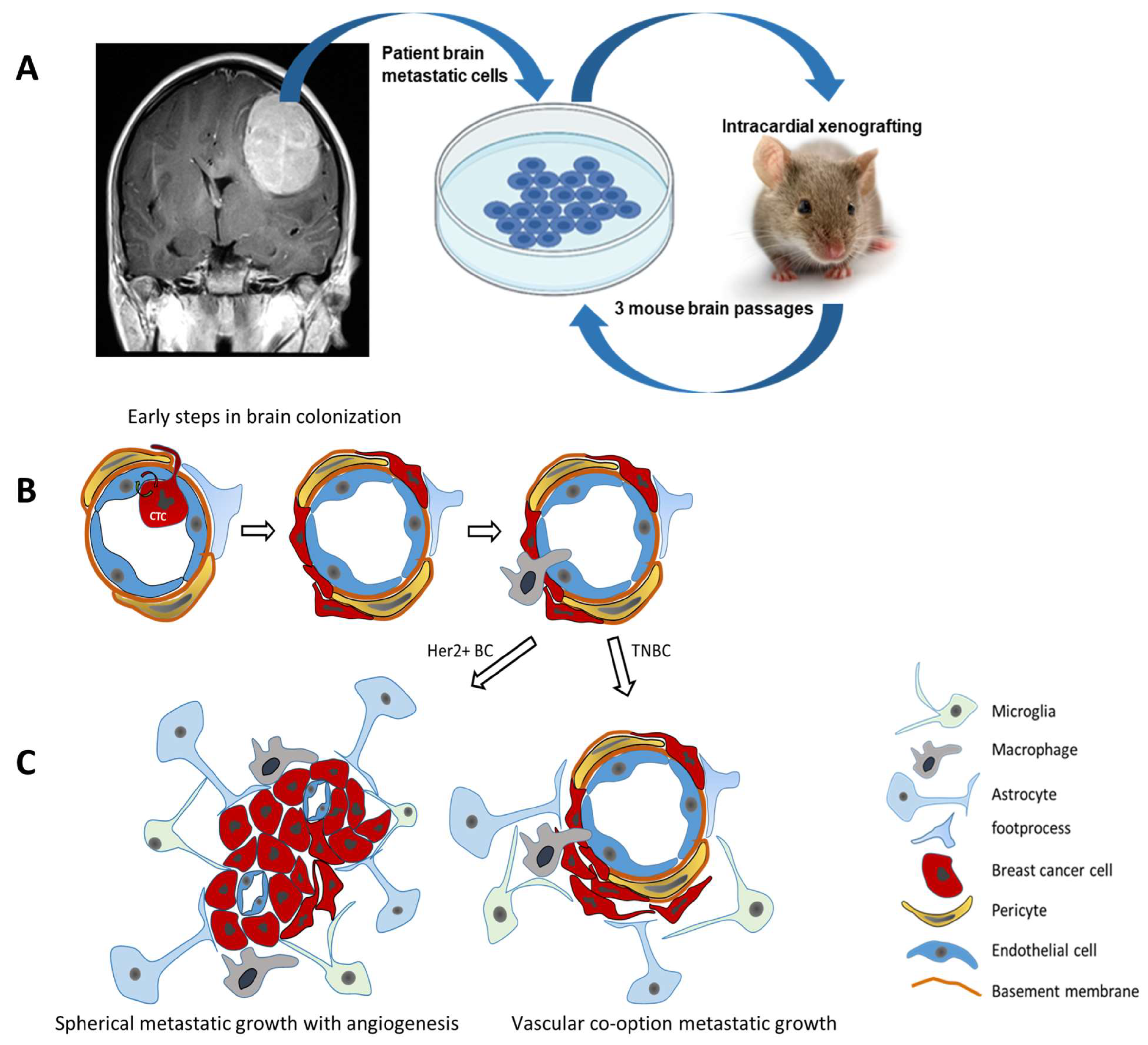
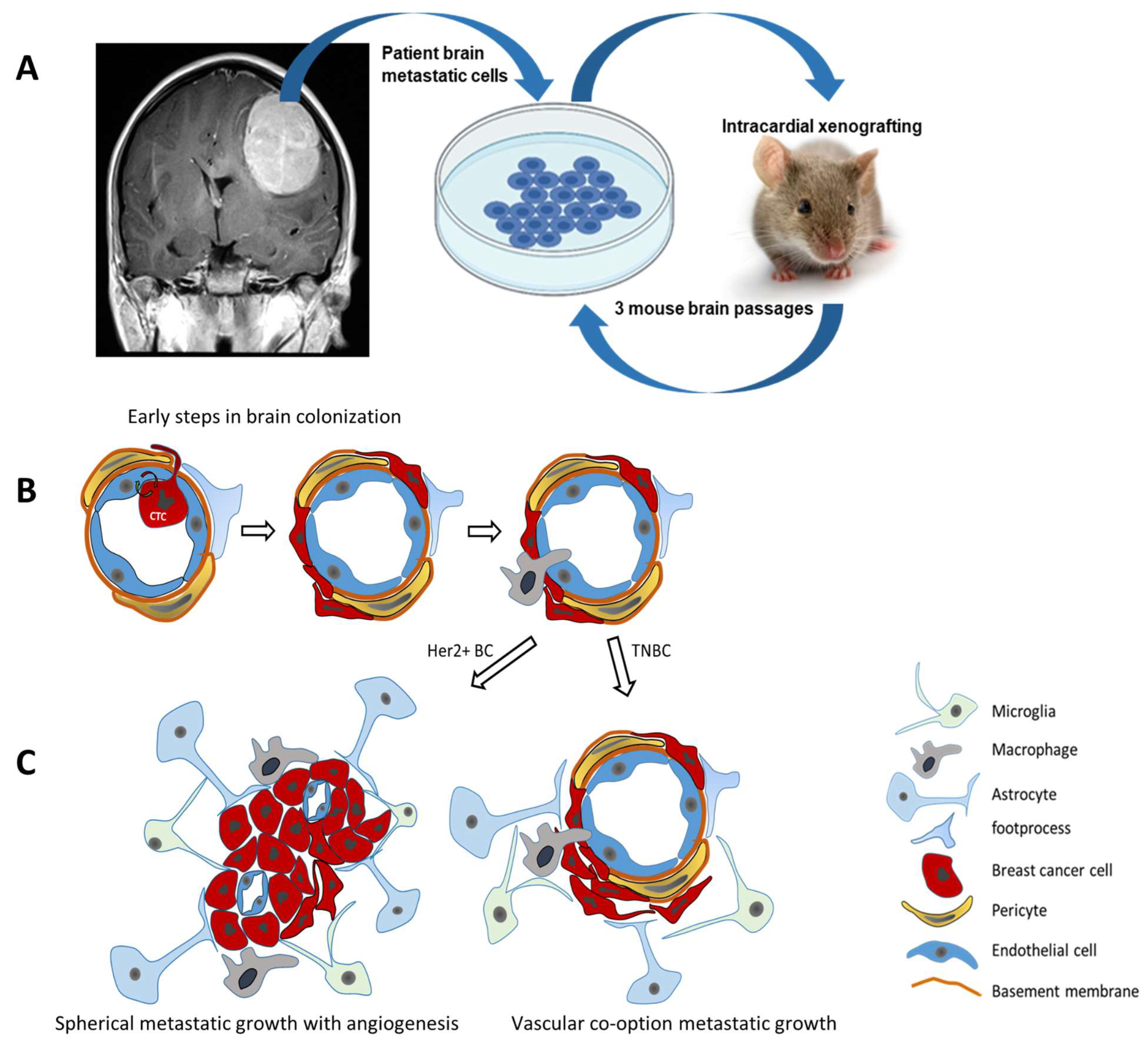
4.2. Brain Metastasis of Breast Cancer (Figure 3)
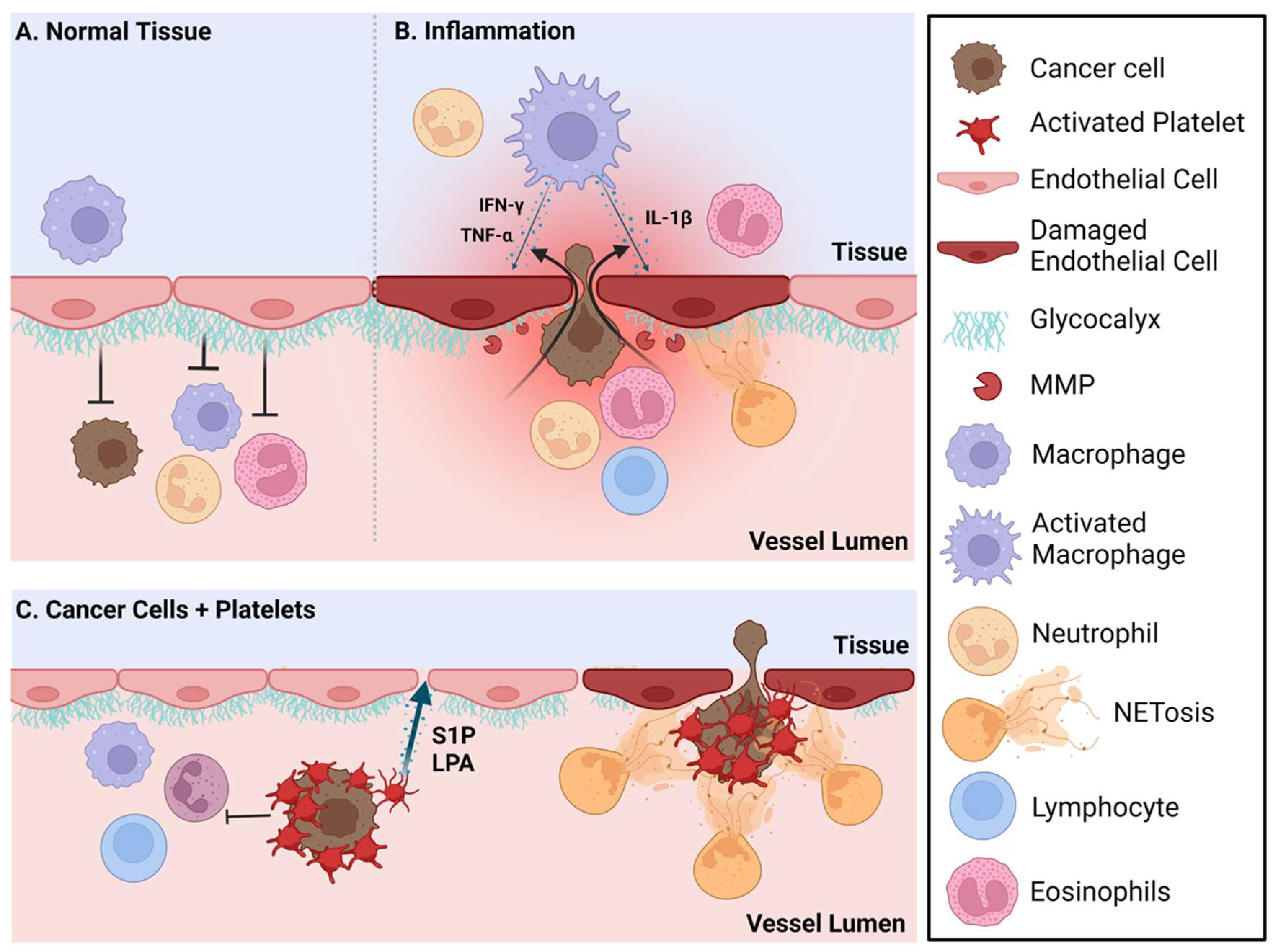
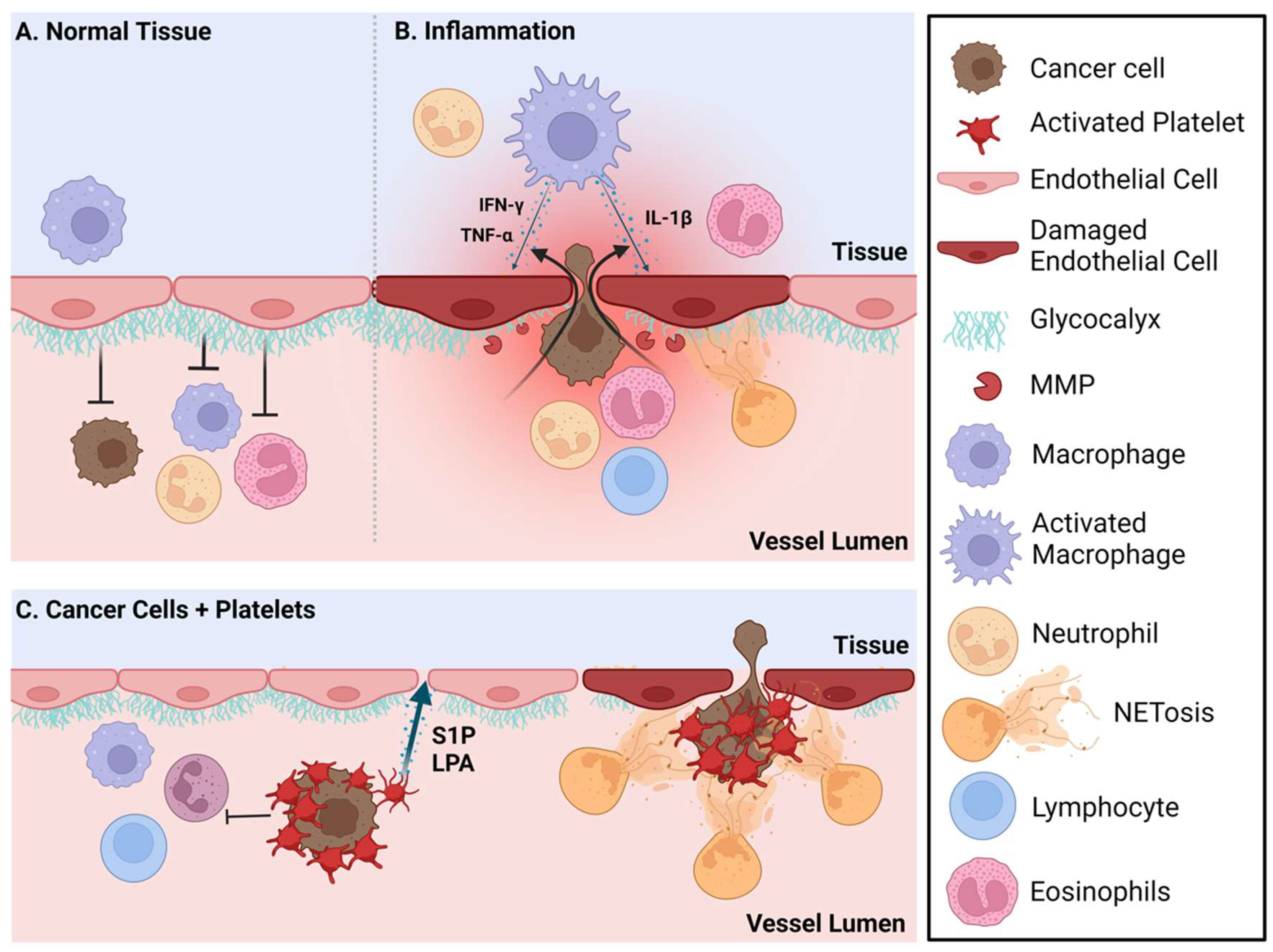
5. Treatment Opportunities for Targeted Versus Immunotherapy Approaches for Metastatic Breast Cancer
5.1. Seed: Targeted Treatments of Different Subtypes of Metastatic Breast Cancer Cells
5.2. Soil: Immunotherapy Targeting the Metastatic Breast Tumor through Alteration of Its Microenvironment
- (A)
- Immunomodulators: Upon exposure to a tumor-specific antigen, naïve T cells differentiate into effector cytotoxic CD8+ T lymphocytes (CTLs) that recognize and eliminate cancer cells through the secretion of cytokines and degrading enzymes through cell-to-cell contact. Ultimately, these effector T cells undergo apoptosis or further differentiate into tissue-resident memory T cells [175]. To prevent the prolonged activation of T cells, the immune system has evolved to develop an inhibitory mechanism to cause T cell dysfunction and exhaustion. This mechanism was initially described in a mouse model of chronic viral infection where T cell exhaustion was found to be due to antigen overstimulation [176,177,178]. In this context, T cell dysfunction or exhaustion was caused by increased expression of “checkpoint” inhibitory receptors such as PD-1, cytotoxic T lymphocyte antigen-4 (CTLA-4) and T cell immunoglobulin domain and mucin domain protein-3 (TIM-3) on the T cells [174]. Such inhibitory receptors are activated by the expression of their cognate ligands (e.g., PD-L1) on antigen presenting cells, such as dendritic cells and macrophages. In the microenvironments of solid tumors, such as BC, T cell exhaustion is frequently observed due to the increased expression of PD-L1 on the cancer cells and increased and sustained expression of inhibitory receptors on the TILs which could then lead to CTL exhaustion [179]. Perhaps the most convincing evidence was provided from experiments showing that blocking the PD-1 interaction with its ligand PD-L1, with a monoclonal antibody, reactivated the CTLs and suppressed the growth of tumors [180,181]. Based on this and similar confirmatory data, immune checkpoint blockade using monoclonal antibodies such nivolumab and avelumab have been approved for use in the clinic to treat melanomas, Hodgkin lymphoma, and lung and other cancers. More recently, to extend the effectiveness and duration of reactivity, some patients were treated with a combination of two immune checkpoint inhibitors: one to negate the PD-1/PD-L1 interaction and another to counteract the CTLA4/CD80 or/CD86 interactions. Clinical trials are now underway to test the effectiveness of these immune checkpoint inhibitors in other solid tumors including liver cancer, non-small cell lung cancer and some BCs [182] (Table 2).
- (B)
- Adoptive cell transfer therapies: Cellular immunotherapy or the adoptive cell therapies refer to approaches that involve isolating the patient’s own T cells and either expanding them directly or genetically modifying them to enhance their anti-cancer effector functions prior to their expansion ex vivo. These activated T cells are then reinfused back into the patient with the idea that these cells are tumor reactive and will result in tumor regression. These treatments include TIL therapy and chimeric antigen receptor (CAR) T cell therapy.
- (C)
- Cancer vaccines: Vaccines for use as prophylactic measures to prevent tumor development have been developed against viral infections that cause malignancies such the human papilloma virus and the hepatitis B virus [174]. The role of other viruses such the human cytomegalovirus (HCMV) in the development of many malignancies including BC is an active area of research. Recent data indicate that evidence of an HCMV infection can be found in up to 90% of BC patients with expression of the HCMV viral proteins by BC cells [200]. On the other hand, the therapeutic cancer vaccines are still at various stages of development. For example, some prostate cancer cells exhibit overexpression of prostatic acid phosphatase which has led to the development of a vaccine to help the immune system detect and eliminate such prostate cancer cells. Another approach that is being actively considered is the creation of oncolytic viruses where a virus is used to cause forced expression of a toxic protein in cancer cells [174].
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, H.; Song, X.; Yang, Q. Metastatic Heterogeneity of Breast Cancer: Molecular Mechanism and Potential Therapeutic Targets. Semin. Cancer Biol. 2020, 60, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The Distribution of Secondary Growths in Cancer of the Breast. 1889. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [PubMed]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Herschkowitz, J.I.; Simin, K.; Weigman, V.J.; Mikaelian, I.; Usary, J.; Hu, Z.; Rasmussen, K.E.; Jones, L.P.; Assefnia, S.; Chandrasekharan, S.; et al. Identification of Conserved Gene Expression Features between Murine Mammary Carcinoma Models and Human Breast Tumors. Genome Biol. 2007, 8, R76. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and Molecular Characterization of the Claudin-Low Intrinsic Subtype of Breast Cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [PubMed]
- McGee, S.R.; Tibiche, C.; Trifiro, M.; Wang, E. Network Analysis Reveals A Signaling Regulatory Loop in the PIK3CA-Mutated Breast Cancer Predicting Survival Outcome. Genom. Proteom. Bioinform. 2017, 15, 121–129. [Google Scholar] [CrossRef]
- Li, J.; Lenferink, A.E.G.; Deng, Y.; Collins, C.; Cui, Q.; Purisima, E.O.; O’Connor-McCourt, M.D.; Wang, E. Identification of High-Quality Cancer Prognostic Markers and Metastasis Network Modules. Nat. Commun. 2010, 1, 34. [Google Scholar] [CrossRef]
- Wang, E.; Zaman, N.; Mcgee, S.; Milanese, J.-S.; Masoudi-Nejad, A.; O’Connor-McCourt, M. Predictive Genomics: A Cancer Hallmark Network Framework for Predicting Tumor Clinical Phenotypes Using Genome Sequencing Data. Semin. Cancer Biol. 2015, 30, 4–12. [Google Scholar] [CrossRef]
- Zaman, N.; Li, L.; Jaramillo, M.L.; Sun, Z.; Tibiche, C.; Banville, M.; Collins, C.; Trifiro, M.; Paliouras, M.; Nantel, A.; et al. Signaling Network Assessment of Mutations and Copy Number Variations Predict Breast Cancer Subtype-Specific Drug Targets. Cell Rep. 2013, 5, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Zou, J.; Zaman, N.; Beitel, L.K.; Trifiro, M.; Paliouras, M. Cancer Systems Biology in the Genome Sequencing Era: Part 1, Dissecting and Modeling of Tumor Clones and Their Networks. Semin. Cancer Biol. 2013, 23, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Zou, J.; Zaman, N.; Beitel, L.K.; Trifiro, M.; Paliouras, M. Cancer Systems Biology in the Genome Sequencing Era: Part 2, Evolutionary Dynamics of Tumor Clonal Networks and Drug Resistance. Semin. Cancer Biol. 2013, 23, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Wang, E. Understanding Genomic Alterations in Cancer Genomes Using an Integrative Network Approach. Cancer Lett. 2013, 340, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.A.; Carter, S.L.; Stojanov, P.; McKenna, A.; Stevenson, K.; Lawrence, M.S.; Sougnez, C.; Stewart, C.; Sivachenko, A.; Wang, L.; et al. Evolution and Impact of Subclonal Mutations in Chronic Lymphocytic Leukemia. Cell 2013, 152, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Loo, P.V.; Wedge, D.C.; Alexandrov, L.B.; Greenman, C.D.; Lau, K.W.; Raine, K.; Jones, D.; Marshall, J.; Ramakrishna, M.; et al. The Life History of 21 Breast Cancers. Cell 2012, 149, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Yates, L.R.; Gerstung, M.; Knappskog, S.; Desmedt, C.; Gundem, G.; Loo, P.V.; Aas, T.; Alexandrov, L.B.; Larsimont, D.; Davies, H.; et al. Subclonal Diversification of Primary Breast Cancer Revealed by Multiregion Sequencing. Nat. Med. 2015, 21, 751–759. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Siegel, M.B.; Kanchi, K.L.; Miller, C.A.; Ding, L.; Zhao, W.; He, X.; Parker, J.S.; Wendl, M.C.; Fulton, R.S.; et al. Tumor Evolution in Two Patients with Basal-like Breast Cancer: A Retrospective Genomics Study of Multiple Metastases. PLoS Med. 2016, 13, e1002174. [Google Scholar] [CrossRef]
- Bertucci, F.; Ng, C.K.Y.; Patsouris, A.; Droin, N.; Piscuoglio, S.; Carbuccia, N.; Soria, J.C.; Dien, A.T.; Adnani, Y.; Kamal, M.; et al. Genomic Characterization of Metastatic Breast Cancers. Nature 2019, 569, 560–564. [Google Scholar] [CrossRef]
- Angus, L.; Smid, M.; Wilting, S.M.; van Riet, J.; Hoeck, A.V.; Nguyen, L.; Nik-Zainal, S.; Steenbruggen, T.G.; Tjan-Heijnen, V.C.G.; Labots, M.; et al. The Genomic Landscape of Metastatic Breast Cancer Highlights Changes in Mutation and Signature Frequencies. Nat. Genet. 2019, 51, 1450–1458. [Google Scholar] [CrossRef]
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J.; et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N. Engl. J. Med. 2021, 384, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Milanese, J.-S.; Tibiche, C.; Zaman, N.; Zou, J.; Han, P.; Meng, Z.; Nantel, A.; Droit, A.; Wang, E. ETumorMetastasis: A Network-Based Algorithm Predicts Clinical Outcomes Using Whole-Exome Sequencing Data of Cancer Patients. Genom. Proteom. Bioinform. 2021, 19, 973–985. [Google Scholar] [CrossRef]
- Xu, X.; Zhou, Y.; Feng, X.; Li, X.; Asad, M.; Li, D.; Liao, B.; Li, J.; Cui, Q.; Wang, E. Germline Genomic Patterns Are Associated with Cancer Risk, Oncogenic Pathways, and Clinical Outcomes. Sci. Adv. 2020, 6, eaba4905. [Google Scholar] [CrossRef] [PubMed]
- Milanese, J.-S.; Tibiche, C.; Zou, J.; Meng, Z.; Nantel, A.; Drouin, S.; Marcotte, R.; Wang, E. Germline Variants Associated with Leukocyte Genes Predict Tumor Recurrence in Breast Cancer Patients. npj Precis. Oncol. 2019, 3, 28. [Google Scholar] [CrossRef] [PubMed]
- Piñeiro, R.; Martínez-Pena, I.; López-López, R. Circulating Tumor Cells in Breast Cancer Metastatic Disease. Adv. Exp. Med. Biol. 2020, 1220, 93–115. [Google Scholar] [CrossRef] [PubMed]
- Kotiyal, S.; Bhattacharya, S. Breast Cancer Stem Cells, EMT and Therapeutic Targets. Biochem. Biophys. Res. Commun. 2014, 453, 112–116. [Google Scholar] [CrossRef]
- Ibragimova, M.; Tsyganov, M.; Litviakov, N. Tumour Stem Cells in Breast Cancer. Int. J. Mol. Sci. 2022, 23, 5058. [Google Scholar] [CrossRef]
- Park, M.; Kim, D.; Ko, S.; Kim, A.; Mo, K.; Yoon, H. Breast Cancer Metastasis: Mechanisms and Therapeutic Implications. Int. J. Mol. Sci. 2022, 23, 6806. [Google Scholar] [CrossRef]
- Wang, H.; Guo, S.; Kim, S.-J.; Shao, F.; Ho, J.W.K.; Wong, K.U.; Miao, Z.; Hao, D.; Zhao, M.; Xu, J.; et al. Cisplatin Prevents Breast Cancer Metastasis through Blocking Early EMT and Retards Cancer Growth Together with Paclitaxel. Theranostics 2021, 11, 2442–2459. [Google Scholar] [CrossRef]
- Verstappe, J.; Berx, G. A Role for Partial Epithelial-to-Mesenchymal Transition in Enabling Stemness in Homeostasis and Cancer. Semin. Cancer Biol. 2023, 90, 15–28. [Google Scholar] [CrossRef]
- Grosse-Wilde, A.; d’Hérouël, A.F.; McIntosh, E.; Ertaylan, G.; Skupin, A.; Kuestner, R.E.; del Sol, A.; Walters, K.-A.; Huang, S. Stemness of the Hybrid Epithelial/Mesenchymal State in Breast Cancer and Its Association with Poor Survival. PLoS ONE 2015, 10, e0126522. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Mauri, F.; Song, Y.; de Cock, F.; Meeusen, B.; Swedlund, B.; Impens, F.; Haver, D.V.; Opitz, M.; Thery, M.; et al. Fat1 Deletion Promotes Hybrid EMT State, Tumour Stemness and Metastasis. Nature 2021, 589, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Mohme, M.; Riethdorf, S.; Pantel, K. Circulating and Disseminated Tumour Cells—Mechanisms of Immune Surveillance and Escape. Nat. Rev. Clin. Oncol. 2017, 14, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Lam, E.W.-F.; Sun, Y. Extracellular Vesicles in the Tumor Microenvironment: Old Stories, but New Tales. Mol. Cancer 2019, 18, 59. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Wang, X.; Gong, Z.; Yu, M.; Wu, H.; Zhang, D. Exosome-Mediated Metabolic Reprogramming: The Emerging Role in Tumor Microenvironment Remodeling and Its Influence on Cancer Progression. Signal Transduct. Target. Ther. 2020, 5, 242. [Google Scholar] [CrossRef] [PubMed]
- Crompot, E.; Damme, M.V.; Pieters, K.; Vermeersch, M.; Perez-Morga, D.; Mineur, P.; Maerevoet, M.; Meuleman, N.; Bron, D.; Lagneaux, L.; et al. Extracellular Vesicles of Bone Marrow Stromal Cells Rescue Chronic Lymphocytic Leukemia B Cells from Apoptosis, Enhance Their Migration and Induce Gene Expression Modifications. Haematologica 2017, 102, 1594–1604. [Google Scholar] [CrossRef]
- Ye, F.; Liang, Y.; Wang, Y.; Yang, R.L.; Luo, D.; Li, Y.; Jin, Y.; Han, D.; Chen, B.; Zhao, W.; et al. Cancer-Associated Fibroblasts Facilitate Breast Cancer Progression through Exosomal CircTBPL1-Mediated Intercellular Communication. Cell Death Dis. 2023, 14, 471. [Google Scholar] [CrossRef]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.F.; Li, S.; Chin, A.R.; et al. Breast-Cancer-Secreted MiR-122 Reprograms Glucose Metabolism in Premetastatic Niche to Promote Metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef]
- Yan, W.; Wu, X.; Zhou, W.; Fong, M.Y.; Cao, M.; Liu, J.; Liu, X.; Chen, C.-H.; Fadare, O.; Pizzo, D.P.; et al. Cancer-Cell-Secreted Exosomal MiR-105 Promotes Tumour Growth through the MYC-Dependent Metabolic Reprogramming of Stromal Cells. Nat. Cell Biol. 2018, 20, 597–609. [Google Scholar] [CrossRef]
- Ma, C.; He, D.; Tian, P.; Wang, Y.; He, Y.; Wu, Q.; Jia, Z.; Zhang, X.; Zhang, P.; Ying, H.; et al. MiR-182 Targeting Reprograms Tumor-Associated Macrophages and Limits Breast Cancer Progression. Proc. Natl. Acad. Sci. USA 2022, 119, e2114006119. [Google Scholar] [CrossRef]
- Gray, J.I.; Farber, D.L. Tissue-Resident Immune Cells in Humans. Annu. Rev. Immunol. 2022, 40, 195–220. [Google Scholar] [CrossRef] [PubMed]
- Altan-Bonnet, G.; Mukherjee, R. Cytokine-Mediated Communication: A Quantitative Appraisal of Immune Complexity. Nat. Rev. Immunol. 2019, 19, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Rudensky, A.Y. Hallmarks of Tissue-Resident Lymphocytes. Cell 2016, 164, 1198–1211. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.N.; Mackay, L.K. Tissue-Resident Memory T Cells: Local Specialists in Immune Defence. Nat. Rev. Immunol. 2016, 16, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Koscsó, B.; Rajani, G.M.; Stevanovic, K.; Berres, M.-L.; Hashimoto, D.; Mortha, A.; Leboeuf, M.; Li, X.-M.; Mucida, D.; et al. Crosstalk between Muscularis Macrophages and Enteric Neurons Regulates Gastrointestinal Motility. Cell 2014, 158, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Mortha, A.; Chudnovskiy, A.; Hashimoto, D.; Bogunovic, M.; Spencer, S.P.; Belkaid, Y.; Merad, M. Microbiota-Dependent Crosstalk between Macrophages and ILC3 Promotes Intestinal Homeostasis. Science 2014, 343, 1249288. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Ruhland, M.K.; Loza, A.J.; Capietto, A.-H.; Luo, X.; Knolhoff, B.L.; Flanagan, K.C.; Belt, B.A.; Alspach, E.; Leahy, K.; Luo, J.; et al. Stromal Senescence Establishes an Immunosuppressive Microenvironment that Drives Tumorigenesis. Nat. Commun. 2016, 7, 11762. [Google Scholar] [CrossRef]
- Fane, M.E.; Chhabra, Y.; Alicea, G.M.; Maranto, D.A.; Douglass, S.M.; Webster, M.R.; Rebecca, V.W.; Marino, G.E.; Almeida, F.; Ecker, B.L.; et al. Stromal Changes in the Aged Lung Induce an Emergence from Melanoma Dormancy. Nature 2022, 606, 396–405. [Google Scholar] [CrossRef]
- Yang, Z.; Tang, X.; Hasing, M.E.; Pang, X.; Ghosh, S.; McMullen, T.P.W.; Brindley, D.N.; Hemmings, D.G. Human Cytomegalovirus Seropositivity and Viral DNA in Breast Tumors Are Associated with Poor Patient Prognosis. Cancers 2022, 14, 1148. [Google Scholar] [CrossRef]
- Yang, Z.; Tang, X.; Meng, G.; Benesch, M.G.K.; Mackova, M.; Belon, A.P.; Serrano-Lomelin, J.; Goping, I.S.; Brindley, D.N.; Hemmings, D.G. Latent Cytomegalovirus Infection in Female Mice Increases Breast Cancer Metastasis. Cancers 2019, 11, 447. [Google Scholar] [CrossRef] [PubMed]
- Pooladanda, V.; Thatikonda, S.; Muvvala, S.P.; Godugu, C. Acute Respiratory Distress Syndrome Enhances Tumor Metastasis into Lungs: Role of BRD4 in the Tumor Microenvironment. Int. Immunopharmacol. 2023, 115, 109701. [Google Scholar] [CrossRef] [PubMed]
- Aramini, B.; Masciale, V.; Samarelli, A.V.; Tonelli, R.; Cerri, S.; Clini, E.; Stella, F.; Dominici, M. Biological Effects of COVID-19 on Lung Cancer: Can We Drive Our Decisions. Front. Oncol. 2022, 12, 1029830. [Google Scholar] [CrossRef] [PubMed]
- McDowell, S.A.C.; Luo, R.B.E.; Arabzadeh, A.; Doré, S.; Bennett, N.C.; Breton, V.; Karimi, E.; Rezanejad, M.; Yang, R.R.; Lach, K.D.; et al. Neutrophil Oxidative Stress Mediates Obesity-Associated Vascular Dysfunction and Metastatic Transmigration. Nat. Cancer 2021, 2, 545–562. [Google Scholar] [CrossRef] [PubMed]
- Olson, O.C.; Quail, D.F.; Joyce, J.A. Obesity and the Tumor Microenvironment. Science 2017, 358, 1130–1131. [Google Scholar] [CrossRef] [PubMed]
- Glaser, R.; Kiecolt-Glaser, J.K. Stress-Induced Immune Dysfunction: Implications for Health. Nat. Rev. Immunol. 2005, 5, 243–251. [Google Scholar] [CrossRef]
- Karagiannis, G.S.; Condeelis, J.S.; Oktay, M.H. Chemotherapy-Induced Metastasis: Mechanisms and Translational Opportunities. Clin. Exp. Metastasis 2018, 35, 269–284. [Google Scholar] [CrossRef]
- Bellomo, G.; Rainer, C.; Quaranta, V.; Astuti, Y.; Raymant, M.; Boyd, E.; Stafferton, R.; Campbell, F.; Ghaneh, P.; Halloran, C.M.; et al. Chemotherapy-Induced Infiltration of Neutrophils Promotes Pancreatic Cancer Metastasis via Gas6/AXL Signalling Axis. Gut 2022, 71, 2284–2299. [Google Scholar] [CrossRef]
- Nolan, E.; Bridgeman, V.L.; Ombrato, L.; Karoutas, A.; Rabas, N.; Sewnath, C.A.N.; Vasquez, M.; Rodrigues, F.S.; Horswell, S.; Faull, P.; et al. Radiation Exposure Elicits a Neutrophil-Driven Response in Healthy Lung Tissue that Enhances Metastatic Colonization. Nat. Cancer 2022, 3, 173–187. [Google Scholar] [CrossRef]
- Jin, C.; Lagoudas, G.K.; Zhao, C.; Bullman, S.; Bhutkar, A.; Hu, B.; Ameh, S.; Sandel, D.; Liang, X.S.; Mazzilli, S.; et al. Commensal Microbiota Promote Lung Cancer Development via Γδ T Cells. Cell 2019, 176, 998–1013.e16. [Google Scholar] [CrossRef]
- Correia, A.L. Locally Sourced: Site-Specific Immune Barriers to Metastasis. Nat. Rev. Immunol. 2023, 23, 522–538. [Google Scholar] [CrossRef] [PubMed]
- Aegerter, H.; Lambrecht, B.N.; Jakubzick, C.V. Biology of Lung Macrophages in Health and Disease. Immunity 2022, 55, 1564–1580. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Chintala, N.K.; Vadrevu, S.K.; Patel, J.; Karbowniczek, M.; Markiewski, M.M. Pulmonary Alveolar Macrophages Contribute to the Premetastatic Niche by Suppressing Antitumor T Cell Responses in the Lungs. J. Immunol. 2015, 194, 5529–5538. [Google Scholar] [CrossRef] [PubMed]
- Darwich, L.; Coma, G.; Peña, R.; Bellido, R.; Blanco, E.J.J.; Este, J.A.; Borras, F.E.; Clotet, B.; Ruiz, L.; Rosell, A.; et al. Secretion of Interferon-γ by Human Macrophages Demonstrated at the Single-cell Level after Costimulation with Interleukin (IL)-12 plus IL-18. Immunology 2009, 126, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.T.; Fong, L.Y.; Abdullah, M.N.H. Interferon-Gamma (IFN-γ): Reviewing Its Mechanisms and Signaling Pathways on the Regulation of Endothelial Barrier Function. Cytokine 2023, 166, 156208. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, T.; Naito, H. Cellular Heterogeneity and Stem Cells of Vascular Endothelial Cells in Blood Vessel Formation and Homeostasis: Insights from Single-Cell RNA Sequencing. Front. Cell Dev. Biol. 2023, 11, 1146399. [Google Scholar] [CrossRef] [PubMed]
- Tomita, T.; Kato, M.; Hiratsuka, S. Regulation of Vascular Permeability in Cancer Metastasis. Cancer Sci. 2021, 112, 2966–2974. [Google Scholar] [CrossRef]
- Weis, S.; Cui, J.; Barnes, L.; Cheresh, D. Endothelial Barrier Disruption by VEGF-Mediated Src Activity Potentiates Tumor Cell Extravasation and Metastasis. J. Cell Biol. 2004, 167, 223–229. [Google Scholar] [CrossRef]
- Egan, K.; Cooke, N.; Kenny, D. Living in Shear: Platelets Protect Cancer Cells from Shear Induced Damage. Clin. Exp. Metastasis 2014, 31, 697–704. [Google Scholar] [CrossRef]
- Plantureux, L.; Mège, D.; Crescence, L.; Carminita, E.; Robert, S.; Cointe, S.; Brouilly, N.; Ezzedine, W.; Dignat-George, F.; Dubois, C.; et al. The Interaction of Platelets with Colorectal Cancer Cells Inhibits Tumor Growth but Promotes Metastasis. Cancer Res. 2020, 80, 291–303. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, C.; Pan, F.; Chen, Y.; Xiong, L.; Li, Y.; Chu, X.; Huang, G. Platelets in the Tumor Microenvironment and Their Biological Effects on Cancer Hallmarks. Front. Oncol. 2023, 13, 1121401. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; He, J.; Zhang, H.; Xia, Y.; Hu, Z.; Loughran, P.; Billiar, T.; Huang, H.; Tsung, A. Platelet TLR4-ERK5 Axis Facilitates NET-Mediated Capturing of Circulating Tumor Cells and Distant Metastasis after Surgical Stress. Cancer Res. 2021, 81, 2373–2385. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Hynes, R.O. The Initial Hours of Metastasis: The Importance of Cooperative Host–Tumor Cell Interactions during Hematogenous Dissemination. Cancer Discov. 2012, 2, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Kaltenmeier, C.; Simmons, R.L.; Tohme, S.; Yazdani, H.O. Neutrophil Extracellular Traps (NETs) in Cancer Metastasis. Cancers 2021, 13, 6131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Y.; Qu, M.; Li, W.; Wu, D.; Cata, J.P.; Miao, C. Neutrophil, Neutrophil Extracellular Traps and Endothelial Cell Dysfunction in Sepsis. Clin. Transl. Med. 2023, 13, e1170. [Google Scholar] [CrossRef]
- Lin, Y.-J.; Wei, K.-C.; Chen, P.-Y.; Lim, M.; Hwang, T.-L. Roles of Neutrophils in Glioma and Brain Metastases. Front. Immunol. 2021, 12, 701383. [Google Scholar] [CrossRef]
- Bhattarai, S.; Subedi, U.; Manikandan, S.; Sharma, S.; Sharma, P.; Miller, C.; Bhuiyan, M.S.; Kidambi, S.; Aidinis, V.; Sun, H.; et al. Endothelial Specific Deletion of Autotaxin Improves Stroke Outcomes. Cells 2023, 12, 511. [Google Scholar] [CrossRef]
- Bhattarai, S.; Sharma, S.; Subedi, U.; Ara, H.; Shum, A.; Milena, M.; Bhuiyan, M.S.; Kidambi, S.; Sun, H.; Miriyala, S.; et al. The ATX–LPA Axis Regulates Vascular Permeability during Cerebral Ischemic-Reperfusion. Int. J. Mol. Sci. 2022, 23, 4138. [Google Scholar] [CrossRef]
- Salminen, A.T.; McCloskey, M.C.; Ahmad, S.D.; Romanick, S.S.; Chen, K.; Houlihan, W.; Klaczko, M.E.; Flax, J.; Waugh, R.E.; McGrath, J.L. Molecular Mechanisms Underlying the Heterogeneous Barrier Responses of Two Primary Endothelial Cell Types to Sphingosine-1-Phosphate. Eur. J. Cell Biol. 2022, 101, 151233. [Google Scholar] [CrossRef]
- Zhang, L.; Zeng, M.; Fan, J.; Tarbell, J.M.; Curry, F.E.; Fu, B.M. Sphingosine-1-phosphate Maintains Normal Vascular Permeability by Preserving Endothelial Surface Glycocalyx in Intact Microvessels. Microcirculation 2016, 23, 301–310. [Google Scholar] [CrossRef]
- Kerage, D.; Brindley, D.N.; Hemmings, D.G. Review: Novel Insights into the Regulation of Vascular Tone by Sphingosine 1-Phosphate. Placenta 2014, 35, S86–S92. [Google Scholar] [CrossRef] [PubMed]
- Hemmings, D.G.; Brindley, D.N. Signalling by Lysophosphatidate and Its Health Implications. Essays Biochem. 2020, 64, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.K.; Tang, X.; Dewald, J.; Dong, W.; Mackey, J.R.; Hemmings, D.G.; McMullen, T.P.W.; Brindley, D.N. Tumor-induced Inflammation in Mammary Adipose Tissue Stimulates a Vicious Cycle of Autotaxin Expression and Breast Cancer Progression. FASEB J. 2015, 29, 3990–4000. [Google Scholar] [CrossRef]
- Engel, N.; Adamus, A.; Frank, M.; Kraft, K.; Kühn, J.; Müller, P.; Nebe, B.; Kasten, A.; Seitz, G. First Evidence of SGPL1 Expression in the Cell Membrane Silencing the Extracellular S1P Siren in Mammary Epithelial Cells. PLoS ONE 2018, 13, e0196854. [Google Scholar] [CrossRef] [PubMed]
- Goddard, L.; Iruela-Arispe, L. Cellular and Molecular Regulation of Vascular Permeability. Thromb. Haemost. 2013, 109, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Bussard, K.M.; Venzon, D.J.; Mastro, A.M. Osteoblasts Are a Major Source of Inflammatory Cytokines in the Tumor Microenvironment of Bone Metastatic Breast Cancer. J. Cell. Biochem. 2010, 111, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Harding, I.C.; Mitra, R.; Mensah, S.A.; Nersesyan, A.; Bal, N.N.; Ebong, E.E. Endothelial Barrier Reinforcement Relies on Flow-Regulated Glycocalyx, a Potential Therapeutic Target. Biorheology 2019, 56, 131–149. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Schmidt, E.P. The Endothelial Glycocalyx. Tissue Barriers 2013, 1, e23494. [Google Scholar] [CrossRef]
- Vink, H.; Duling, B.R. Capillary Endothelial Surface Layer Selectively Reduces Plasma Solute Distribution Volume. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H285–H289. [Google Scholar] [CrossRef]
- Curry, F.E.; Adamson, R.H. Endothelial Glycocalyx: Permeability Barrier and Mechanosensor. Ann. Biomed. Eng. 2012, 40, 828–839. [Google Scholar] [CrossRef]
- Rangarajan, S.; Richter, J.R.; Richter, R.P.; Bandari, S.K.; Tripathi, K.; Vlodavsky, I.; Sanderson, R.D. Heparanase-Enhanced Shedding of Syndecan-1 and Its Role in Driving Disease Pathogenesis and Progression. J. Histochem. Cytochem. 2020, 68, 823–840. [Google Scholar] [CrossRef] [PubMed]
- Mensah, S.A.; Harding, I.C.; Zhang, M.; Jaeggli, M.P.; Torchilin, V.P.; Niedre, M.J.; Ebong, E.E. Metastatic Cancer Cell Attachment to Endothelium Is Promoted by Endothelial Glycocalyx Sialic Acid Degradation. AIChE J. 2019, 65, e16634. [Google Scholar] [CrossRef] [PubMed]
- Weinbaum, S.; Cancel, L.M.; Fu, B.M.; Tarbell, J.M. The Glycocalyx and Its Role in Vascular Physiology and Vascular Related Diseases. Cardiovasc. Eng. Technol. 2021, 12, 37–71. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.; Finkelstein, D.M.; Barrios, C.; Martin, M.; Iwata, H.; Hegg, R.; Glaspy, J.; Periañez, A.M.; Tonkin, K.; Deleu, I.; et al. Adjuvant Denosumab in Early Breast Cancer (D-CARE): An International, Multicentre, Randomised, Controlled, Phase 3 Trial. Lancet Oncol. 2020, 21, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Lipton, A.; Uzzo, R.; Amato, R.J.; Ellis, G.K.; Hakimian, B.; Roodman, G.D.; Smith, M.R. The Science and Practice of Bone Health in Oncology: Managing Bone Loss and Metastasis in Patients with Solid Tumors. J. Natl. Compr. Cancer Netw. 2009, 7, S-1–S-29. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.-H.; Zhang, S.; Yang, H.; Yi, Z.-B.; Ouyang, Q.-C.; Yan, M.; Wang, X.-J.; Hu, X.-C.; Jiang, Z.-F.; Huang, T.; et al. Molecular Subtypes Predict the Preferential Site of Distant Metastasis in Advanced Breast Cancer: A Nationwide Retrospective Study. Front. Oncol. 2023, 13, 978985. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Zheng, S.; Yang, A.; Zhang, X.; Zou, Y.; Tang, H.; Xie, X. Breast Cancer Subtypes and the Risk of Distant Metastasis at Initial Diagnosis: A Population-Based Study. Cancer Manag. Res. 2018, 10, 5329–5338. [Google Scholar] [CrossRef] [PubMed]
- Shemanko, C.S.; Cong, Y.; Forsyth, A. What Is Breast in the Bone? Int. J. Mol. Sci. 2016, 17, 1764. [Google Scholar] [CrossRef]
- Miller, S.L.; Antico, G.; Raghunath, P.N.; Tomaszewski, J.E.; Clevenger, C.V. Nek3 Kinase Regulates Prolactin-Mediated Cytoskeletal Reorganization and Motility of Breast Cancer Cells. Oncogene 2007, 26, 4668–4678. [Google Scholar] [CrossRef]
- Bertucci, F.; Finetti, P.; Cervera, N.; Maraninchi, D.; Viens, P.; Birnbaum, D. Gene Expression Profiling and Clinical Outcome in Breast Cancer. OMICS J. Integr. Biol. 2006, 10, 429–443. [Google Scholar] [CrossRef]
- Perotti, C.; Liu, R.; Parusel, C.T.; Böcher, N.; Schultz, J.; Bork, P.; Pfitzner, E.; Groner, B.; Shemanko, C.S. Heat Shock Protein-90-Alpha, a Prolactin-STAT5 Target Gene Identified in Breast Cancer Cells, Is Involved in Apoptosis Regulation. Breast Cancer Res. 2008, 10, R94. [Google Scholar] [CrossRef] [PubMed]
- Atici, Ö.K.; Urbanska, A.; Gopinathan, S.G.; Boutillon, F.; Goffin, V.; Shemanko, C.S. ATM Is Required for the Prolactin-Induced HSP90-Mediated Increase in Cellular Viability and Clonogenic Growth after DNA Damage. Endocrinology 2017, 159, 907–930. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, A.; Forsyth, A.; Cong, Y.; Grant, L.; Juan, T.-H.; Lee, J.K.; Klimowicz, A.; Petrillo, S.K.; Hu, J.; Chan, A.; et al. The Role of Prolactin in Bone Metastasis and Breast Cancer Cell–Mediated Osteoclast Differentiation. JNCI J. Natl. Cancer Inst. 2016, 108, djv338. [Google Scholar] [CrossRef] [PubMed]
- Clément-Lacroix, P.; Ormandy, C.; Lepescheux, L.; Ammann, P.; Damotte, D.; Goffin, V.; Bouchard, B.; Amling, M.; Gaillard-Kelly, M.; Binart, N.; et al. Osteoblasts Are a New Target for Prolactin: Analysis of Bone Formation in Prolactin Receptor Knockout Mice. Endocrinology 1999, 140, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Amarasekara, D.S.; Kim, S.; Rho, J. Regulation of Osteoblast Differentiation by Cytokine Networks. Int. J. Mol. Sci. 2021, 22, 2851. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, N.K.; Lee, S.Y. Current Understanding of RANK Signaling in Osteoclast Differentiation and Maturation. Mol. Cells 2017, 40, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Zhao, E.; Xu, H.; Wang, L.; Kryczek, I.; Wu, K.; Hu, Y.; Wang, G.; Zou, W. Bone Marrow and the Control of Immunity. Cell. Mol. Immunol. 2012, 9, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Place, D.E.; Malireddi, R.K.S.; Kim, J.; Vogel, P.; Yamamoto, M.; Kanneganti, T.-D. Osteoclast Fusion and Bone Loss Are Restricted by Interferon Inducible Guanylate Binding Proteins. Nat. Commun. 2021, 12, 496. [Google Scholar] [CrossRef]
- Baccala, R.; Welch, M.J.; Gonzalez-Quintial, R.; Walsh, K.B.; Teijaro, J.R.; Nguyen, A.; Ng, C.T.; Sullivan, B.M.; Zarpellon, A.; Ruggeri, Z.M.; et al. Type I Interferon Is a Therapeutic Target for Virus-Induced Lethal Vascular Damage. Proc. Natl. Acad. Sci. USA 2014, 111, 8925–8930. [Google Scholar] [CrossRef]
- Owen, K.L.; Gearing, L.J.; Zanker, D.J.; Brockwell, N.K.; Khoo, W.H.; Roden, D.L.; Cmero, M.; Mangiola, S.; Hong, M.K.; Spurling, A.J.; et al. Prostate Cancer Cell-intrinsic Interferon Signaling Regulates Dormancy and Metastatic Outgrowth in Bone. EMBO Rep. 2020, 21, e50162. [Google Scholar] [CrossRef]
- Arellano, D.L.; Juárez, P.; Verdugo-Meza, A.; Almeida-Luna, P.S.; Corral-Avila, J.A.; Drescher, F.; Olvera, F.; Jiménez, S.; Elzey, B.D.; Guise, T.A.; et al. Bone Microenvironment-Suppressed T Cells Increase Osteoclast Formation and Osteolytic Bone Metastases in Mice. J. Bone Miner. Res. 2022, 37, 1446–1463. [Google Scholar] [CrossRef] [PubMed]
- Chao, X.; Zhang, Y.; Zheng, C.; Huang, Q.; Lu, J.; Pulver, E.M.; Houthuijzen, J.; Hutten, S.; Luo, R.; He, J.; et al. Metastasis of Breast Cancer to Bones Alters the Tumor Immune Microenvironment. Eur. J. Méd. Res. 2023, 28, 119. [Google Scholar] [CrossRef] [PubMed]
- Kadry, H.; Noorani, B.; Cucullo, L. A Blood–Brain Barrier Overview on Structure, Function, Impairment, and Biomarkers of Integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, S.; Iadecola, C. Revisiting the Neurovascular Unit. Nat. Neurosci. 2021, 24, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Muoio, V.; Persson, P.B.; Sendeski, M.M. The Neurovascular Unit—Concept Review. Acta Physiol. 2014, 210, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Kienast, Y.; von Baumgarten, L.; Fuhrmann, M.; Klinkert, W.E.F.; Goldbrunner, R.; Herms, J.; Winkler, F. Real-Time Imaging Reveals the Single Steps of Brain Metastasis Formation. Nat. Med. 2010, 16, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, X.; Tang, K.; Xin, Y.; Hu, G.; Zheng, Y.; Li, K.; Zhang, C.; Tan, Y. Adhesion to the Brain Endothelium Selects Breast Cancer Cells with Brain Metastasis Potential. Int. J. Mol. Sci. 2023, 24, 7087. [Google Scholar] [CrossRef]
- Soto, M.S.; Serres, S.; Anthony, D.C.; Sibson, N.R. Functional Role of Endothelial Adhesion Molecules in the Early Stages of Brain Metastasis. Neuro-Oncol. 2014, 16, 540–551. [Google Scholar] [CrossRef]
- Bos, P.D.; Zhang, X.H.-F.; Nadal, C.; Shu, W.; Gomis, R.R.; Nguyen, D.X.; Minn, A.J.; van de Vijver, M.J.; Gerald, W.L.; Foekens, J.A.; et al. Genes that Mediate Breast Cancer Metastasis to the Brain. Nature 2009, 459, 1005–1009. [Google Scholar] [CrossRef]
- Conrad, C.; Götte, M.; Schlomann, U.; Roessler, M.; Pagenstecher, A.; Anderson, P.; Preston, J.; Pruessmeyer, J.; Ludwig, A.; Li, R.; et al. ADAM8 Expression in Breast Cancer Derived Brain Metastases: Functional Implications on MMP-9 Expression and Transendothelial Migration in Breast Cancer Cells. Int. J. Cancer 2018, 142, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Dong, S.; Higazy, D.; Jin, L.; Zou, Q.; Chen, H.; Inayat, A.; Hu, S.; Cui, M. Inflammatory Environment Promotes the Adhesion of Tumor Cells to Brain Microvascular Endothelial Cells. Front. Oncol. 2021, 11, 691771. [Google Scholar] [CrossRef] [PubMed]
- Saranchova, I.; Han, J.; Zaman, R.; Arora, H.; Huang, H.; Fenninger, F.; Choi, K.B.; Munro, L.; Pfeifer, C.G.; Welch, I.; et al. Type 2 Innate Lymphocytes Actuate Immunity against Tumours and Limit Cancer Metastasis. Sci. Rep. 2018, 8, 2924. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Döring, A.; Zemp, F.J.; Silva, C.; Lun, X.; Wang, X.; Kelly, J.; Hader, W.; Hamilton, M.; Mercier, P.; et al. Therapeutic Activation of Macrophages and Microglia to Suppress Brain Tumor-Initiating Cells. Nat. Neurosci. 2014, 17, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Pukrop, T.; Dehghani, F.; Chuang, H.; Lohaus, R.; Bayanga, K.; Heermann, S.; Regen, T.; Rossum, D.V.; Klemm, F.; Schulz, M.; et al. Microglia Promote Colonization of Brain Tissue by Breast Cancer Cells in a Wnt-dependent Way. GLIA 2010, 58, 1477–1489. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of Pro-Inflammatory Cytokines Released from Microglia in Neurodegenerative Diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Thanasupawat, T.; Natarajan, S.; Rommel, A.; Glogowska, A.; Bergen, H.; Krcek, J.; Pitz, M.; Beiko, J.; Krawitz, S.; Verma, I.M.; et al. Dovitinib Enhances Temozolomide Efficacy in Glioblastoma Cells. Mol. Oncol. 2017, 11, 1078–1098. [Google Scholar] [CrossRef] [PubMed]
- Lorger, M.; Felding-Habermann, B. Capturing Changes in the Brain Microenvironment during Initial Steps of Breast Cancer Brain Metastasis. Am. J. Pathol. 2010, 176, 2958–2971. [Google Scholar] [CrossRef]
- Burn, L.; Gutowski, N.; Whatmore, J.; Giamas, G.; Pranjol, M.Z.I. The Role of Astrocytes in Brain Metastasis at the Interface of Circulating Tumour Cells and the Blood Brain Barrier. Front. Biosci. Landmark 2021, 26, 590. [Google Scholar] [CrossRef]
- Gong, X.; Hou, Z.; Endsley, M.P.; Gronseth, E.I.; Rarick, K.R.; Jorns, J.M.; Yang, Q.; Du, Z.; Yan, K.; Bordas, M.L.; et al. Interaction of Tumor Cells and Astrocytes Promotes Breast Cancer Brain Metastases through TGF-Β2/ANGPTL4 Axes. npj Precis. Oncol. 2019, 3, 24. [Google Scholar] [CrossRef]
- Wrobel, J.K.; Toborek, M. Blood-Brain Barrier Remodeling during Brain Metastasis Formation. Mol. Med. 2016, 22, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.S.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma–Astrocyte Gap Junctions Promote Brain Metastasis by CGAMP Transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Kucharz, K.; Kristensen, K.; Johnsen, K.B.; Lund, M.A.; Lønstrup, M.; Moos, T.; Andresen, T.L.; Lauritzen, M.J. Post-Capillary Venules Are the Key Locus for Transcytosis-Mediated Brain Delivery of Therapeutic Nanoparticles. Nat. Commun. 2021, 12, 4121. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The Movers and Shapers in Immune Privilege of the CNS. Nat. Immunol. 2017, 18, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, Z.; Sinha, D.; Kaur, P. Pericyte Biology in Disease. Adv. Exp. Med. Biol. 2019, 1147, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Babak, M.V.; Zalutsky, M.R.; Balyasnikova, I.V. Heterogeneity and Vascular Permeability of Breast Cancer Brain Metastases. Cancer Lett. 2020, 489, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Lockman, P.R.; Mittapalli, R.K.; Taskar, K.S.; Rudraraju, V.; Gril, B.; Bohn, K.A.; Adkins, C.E.; Roberts, A.; Thorsheim, H.R.; Gaasch, J.A.; et al. Heterogeneous Blood–Tumor Barrier Permeability Determines Drug Efficacy in Experimental Brain Metastases of Breast Cancer. Clin. Cancer Res. 2010, 16, 5664–5678. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, A.; Peereboom, D.M.; Thorsheim, H.R.; Samala, R.; Balyan, R.; Murphy, C.G.; Lockman, P.R.; Simmons, A.; Weil, R.J.; Tabar, V.; et al. Capecitabine and Lapatinib Uptake in Surgically Resected Brain Metastases from Metastatic Breast Cancer Patients: A Prospective Study. Neuro-Oncol. 2015, 17, 289–295. [Google Scholar] [CrossRef]
- Yonemori, K.; Tsuta, K.; Ono, M.; Shimizu, C.; Hirakawa, A.; Hasegawa, T.; Hatanaka, Y.; Narita, Y.; Shibui, S.; Fujiwara, Y. Disruption of the Blood Brain Barrier by Brain Metastases of Triple-negative and Basal-type Breast Cancer but Not HER2/Neu-positive Breast Cancer. Cancer 2010, 116, 302–308. [Google Scholar] [CrossRef]
- Lau, L.W.; Cua, R.; Keough, M.B.; Haylock-Jacobs, S.; Yong, V.W. Pathophysiology of the Brain Extracellular Matrix: A New Target for Remyelination. Nat. Rev. Neurosci. 2013, 14, 722–729. [Google Scholar] [CrossRef]
- Mohiuddin, E.; Wakimoto, H. Extracellular Matrix in Glioblastoma: Opportunities for Emerging Therapeutic Approaches. Am. J. Cancer Res. 2021, 11, 3742–3754. [Google Scholar] [PubMed]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of Extracellular Matrix Remodelling in Tumour Progression and Metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef] [PubMed]
- Kai, F.; Drain, A.P.; Weaver, V.M. The Extracellular Matrix Modulates the Metastatic Journey. Dev. Cell 2019, 49, 332–346. [Google Scholar] [CrossRef] [PubMed]
- Chhichholiya, Y.; Ruthuparna, M.; Velagaleti, H.; Munshi, A. Brain Metastasis in Breast Cancer: Focus on Genes and Signaling Pathways Involved, Blood–Brain Barrier and Treatment Strategies. Clin. Transl. Oncol. 2023, 25, 1218–1241. [Google Scholar] [CrossRef] [PubMed]
- Klemm, F.; Bleckmann, A.; Siam, L.; Chuang, H.N.; Rietkötter, E.; Behme, D.; Schulz, M.; Schaffrinski, M.; Schindler, S.; Trümper, L.; et al. β-Catenin-Independent WNT Signaling in Basal-like Breast Cancer and Brain Metastasis. Carcinogenesis 2011, 32, 434–442. [Google Scholar] [CrossRef] [PubMed]
- McGowan, P.M.; Simedrea, C.; Ribot, E.J.; Foster, P.J.; Palmieri, D.; Steeg, P.S.; Allan, A.L.; Chambers, A.F. Notch1 Inhibition Alters the CD44hi/CD24lo Population and Reduces the Formation of Brain Metastases from Breast Cancer. Mol. Cancer Res. 2011, 9, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Smid, M.; Wang, Y.; Zhang, Y.; Sieuwerts, A.M.; Yu, J.; Klijn, J.G.M.; Foekens, J.A.; Martens, J.W.M. Subtypes of Breast Cancer Show Preferential Site of Relapse. Cancer Res. 2008, 68, 3108–3114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Huang, W.-C.; Zhang, L.; Zhang, C.; Lowery, F.J.; Ding, Z.; Guo, H.; Wang, H.; Huang, S.; Sahin, A.A.; et al. Src Family Kinases as Novel Therapeutic Targets to Treat Breast Cancer Brain Metastases. Cancer Res. 2013, 73, 5764–5774. [Google Scholar] [CrossRef]
- Ippen, F.M.; Alvarez-Breckenridge, C.A.; Kuter, B.M.; Fink, A.L.; Bihun, I.V.; Lastrapes, M.; Penson, T.; Schmidt, S.P.; Wojtkiewicz, G.R.; Ning, J.; et al. The Dual PI3K/MTOR-Pathway Inhibitor GDC-0084 Achieves Antitumor Activity in PIK3CA-Mutant Breast Cancer Brain Metastases. Clin. Cancer Res. 2019, 25, 3374–3383. [Google Scholar] [CrossRef]
- Gallardo, A.; Lerma, E.; Escuin, D.; Tibau, A.; Muñoz, J.; Ojeda, B.; Barnadas, A.; Adrover, E.; Sánchez-Tejada, L.; Giner, D.; et al. Increased Signalling of EGFR and IGF1R, and Deregulation of PTEN/PI3K/Akt Pathway Are Related with Trastuzumab Resistance in HER2 Breast Carcinomas. Br. J. Cancer 2012, 106, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.D.; Simpson, P.T.; Smart, C.E.; Cocciardi, S.; Waddell, N.; Lane, A.; Morrison, B.J.; Vargas, A.C.; Healey, S.; Beesley, J.; et al. HER3 and Downstream Pathways Are Involved in Colonization of Brain Metastases from Breast Cancer. Breast Cancer Res. 2010, 12, R46. [Google Scholar] [CrossRef] [PubMed]
- Momeny, M.; Saunus, J.M.; Marturana, F.; Reed, A.E.M.; Black, D.; Sala, G.; Iacobelli, S.; Holland, J.D.; Yu, D.; Silva, L.D.; et al. Heregulin-HER3-HER2 Signaling Promotes Matrix Metalloproteinase-Dependent Blood-Brain-Barrier Transendothelial Migration of Human Breast Cancer Cell Lines. Oncotarget 2015, 6, 3932–3946. [Google Scholar] [CrossRef]
- Breast Cancer: Statistics; American Society of Clinical Oncology: Alexandria, VA, USA, 2012.
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast Cancer. Lancet 2021, 397, 1750–1769. [Google Scholar] [CrossRef]
- Riemsma, R.; Forbes, C.A.; Kessels, A.; Lykopoulos, K.; Amonkar, M.M.; Rea, D.W.; Kleijnen, J. Systematic Review of Aromatase Inhibitors in the First-Line Treatment for Hormone Sensitive Advanced or Metastatic Breast Cancer. Breast Cancer Res. Treat. 2010, 123, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Shimoi, T.; Sagara, Y.; Hara, F.; Toyama, T.; Iwata, H. First-Line Endocrine Therapy for Postmenopausal Patients with Hormone Receptor-Positive, HER2−Negative Metastatic Breast Cancer: A Systematic Review and Meta-Analysis. Breast Cancer 2020, 27, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Sáez, O.; Chic, N.; Pascual, T.; Adamo, B.; Vidal, M.; González-Farré, B.; Sanfeliu, E.; Schettini, F.; Conte, B.; Brasó-Maristany, F.; et al. Frequency and Spectrum of PIK3CA Somatic Mutations in Breast Cancer. Breast Cancer Res. 2020, 22, 45. [Google Scholar] [CrossRef]
- Shimelis, H.; LaDuca, H.; Hu, C.; Hart, S.N.; Na, J.; Thomas, A.; Akinhanmi, M.; Moore, R.M.; Brauch, H.; Cox, A.; et al. Triple-Negative Breast Cancer Risk Genes Identified by Multigene Hereditary Cancer Panel Testing. JNCI J. Natl. Cancer Inst. 2018, 110, 855–862. [Google Scholar] [CrossRef]
- Samadi, N.; Gaetano, C.; Goping, I.S.; Brindley, D.N. Autotaxin Protects MCF-7 Breast Cancer and MDA-MB-435 Melanoma Cells against Taxol-Induced Apoptosis. Oncogene 2009, 28, 1028–1039. [Google Scholar] [CrossRef]
- Venkatraman, G.; Benesch, M.G.K.; Tang, X.; Dewald, J.; McMullen, T.P.W.; Brindley, D.N. Lysophosphatidate Signaling Stabilizes Nrf2 and Increases the Expression of Genes Involved in Drug Resistance and Oxidative Stress Responses: Implications for Cancer Treatment. FASEB J. 2015, 29, 772–785. [Google Scholar] [CrossRef]
- Tang, X.; Wang, X.; Zhao, Y.Y.; Curtis, J.M.; Brindley, D.N. Doxycycline Attenuates Breast Cancer Related Inflammation by Decreasing Plasma Lysophosphatidate Concentrations and Inhibiting NF-ΚB Activation. Mol. Cancer 2017, 16, 36. [Google Scholar] [CrossRef] [PubMed]
- Fingleton, B. MMP Inhibitor Clinical Trials—The Past, Present, and Future. In The Cancer Degradome: Proteases and Cancer Biology; Edwards, D., Høyer-Hansen, G., Blasi, F., Sloane, B.F., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 759–785. [Google Scholar]
- Hadjimichael, A.C.; Foukas, A.F.; Savvidou, O.D.; Mavrogenis, A.F.; Psyrri, A.K.; Papagelopoulos, P.J. The Anti-Neoplastic Effect of Doxycycline in Osteosarcoma as a Metalloproteinase (MMP) Inhibitor: A Systematic Review. Clin. Sarcoma Res. 2020, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Winer, A.; Adams, S.; Mignatti, P. Matrix Metalloproteinase Inhibitors in Cancer Therapy: Turning Past Failures into Future Successes. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xu, L.; Zhang, F.; Vlashi, E. Doxycycline Inhibits the Cancer Stem Cell Phenotype and Epithelial-to-Mesenchymal Transition in Breast Cancer. Cell Cycle 2017, 16, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Habli, Z.; AlChamaa, W.; Saab, R.; Kadara, H.; Khraiche, M.L. Circulating Tumor Cell Detection Technologies and Clinical Utility: Challenges and Opportunities. Cancers 2020, 12, 1930. [Google Scholar] [CrossRef] [PubMed]
- Millner, L.M.; Linder, M.W.; Valdes, R. Circulating Tumor Cells: A Review of Present Methods and the Need to Identify Heterogeneous Phenotypes. Ann. Clin. Lab. Sci. 2013, 43, 295–304. [Google Scholar] [PubMed]
- Castro-Giner, F.; Aceto, N. Tracking Cancer Progression: From Circulating Tumor Cells to Metastasis. Genome Med. 2020, 12, 31. [Google Scholar] [CrossRef] [PubMed]
- Hristozova, T.; Konschak, R.; Stromberger, C.; Fusi, A.; Liu, Z.; Weichert, W.; Stenzinger, A.; Budach, V.; Keilholz, U.; Tinhofer, I. The Presence of Circulating Tumor Cells (CTCs) Correlates with Lymph Node Metastasis in Nonresectable Squamous Cell Carcinoma of the Head and Neck Region (SCCHN). Ann. Oncol. 2011, 22, 1878–1885. [Google Scholar] [CrossRef]
- Schuster, E.; Taftaf, R.; Reduzzi, C.; Albert, M.K.; Romero-Calvo, I.; Liu, H. Better Together: Circulating Tumor Cell Clustering in Metastatic Cancer. Trends Cancer 2021, 7, 1020–1032. [Google Scholar] [CrossRef]
- Kucerova, P.; Cervinkova, M. Spontaneous Regression of Tumour and the Role of Microbial Infection—Possibilities for Cancer Treatment. Anti-Cancer Drugs 2016, 27, 269–277. [Google Scholar] [CrossRef]
- Jiang, T.; Zhou, C. The Past, Present and Future of Immunotherapy against Tumor. Transl. Lung Cancer Res. 2014, 4, 253–264. [Google Scholar] [CrossRef]
- Guha, P.; Heatherton, K.R.; O’Connell, K.P.; Alexander, I.S.; Katz, S.C. Assessing the Future of Solid Tumor Immunotherapy. Biomedicines 2022, 10, 655. [Google Scholar] [CrossRef]
- Cui, J.; Sim, T.H.-F.; Gong, Z.; Shen, H.-M. Generation of Transgenic Zebrafish with Liver-Specific Expression of EGFP-Lc3: A New in Vivo Model for Investigation of Liver Autophagy. Biochem. Biophys. Res. Commun. 2012, 422, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Wherry, E.J.; Ahmed, R.; Freeman, G.J. The Function of Programmed Cell Death 1 and Its Ligands in Regulating Autoimmunity and Infection. Nat. Immunol. 2007, 8, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and Cellular Insights into T Cell Exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Zajac, A.J.; Blattman, J.N.; Murali-Krishna, K.; Sourdive, D.J.D.; Suresh, M.; Altman, J.D.; Ahmed, R. Viral Immune Evasion due to Persistence of Activated T Cells without Effector Function. J. Exp. Med. 1998, 188, 2205–2213. [Google Scholar] [CrossRef] [PubMed]
- Ando, M.; Ito, M.; Srirat, T.; Kondo, T.; Yoshimura, A. Memory T Cell, Exhaustion, and Tumor Immunity. Immunol. Med. 2020, 43, 1–9. [Google Scholar] [CrossRef]
- Curiel, T.J.; Wei, S.; Dong, H.; Alvarez, X.; Cheng, P.; Mottram, P.; Krzysiek, R.; Knutson, K.L.; Daniel, B.; Zimmermann, M.C.; et al. Blockade of B7-H1 Improves Myeloid Dendritic Cell–Mediated Antitumor Immunity. Nat. Med. 2003, 9, 562–567. [Google Scholar] [CrossRef]
- Hirano, F.; Kaneko, K.; Tamura, H.; Dong, H.; Wang, S.; Ichikawa, M.; Rietz, C.; Flies, D.B.; Lau, J.S.; Zhu, G.; et al. Blockade of B7-H1 and PD-1 by Monoclonal Antibodies Potentiates Cancer Therapeutic Immunity. Cancer Res. 2005, 65, 1089–1096. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2015, 37, 457–495. [Google Scholar] [CrossRef]
- Savas, P.; Salgado, R.; Denkert, C.; Sotiriou, C.; Darcy, P.K.; Smyth, M.J.; Loi, S. Clinical Relevance of Host Immunity in Breast Cancer: From TILs to the Clinic. Nat. Rev. Clin. Oncol. 2016, 13, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Miremadi, A.; Pinder, S.E.; Ellis, I.O.; Caldas, C. An Immune Response Gene Expression Module Identifies a Good Prognosis Subtype in Estrogen Receptor Negative Breast Cancer. Genome Biol. 2007, 8, R157. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Rosenberg, S.A. Adoptive-Cell-Transfer Therapy for the Treatment of Patients with Cancer. Nat. Rev. Cancer 2003, 3, 666–675. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Lotze, M.T.; Muul, L.M.; Leitman, S.; Chang, A.E.; Ettinghausen, S.E.; Matory, Y.L.; Skibber, J.M.; Shiloni, E.; Vetto, J.T.; et al. Observations on the Systemic Administration of Autologous Lymphokine-Activated Killer Cells and Recombinant Interleukin-2 to Patients with Metastatic Cancer. N. Engl. J. Med. 1985, 313, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Deng, J.; Rao, S.; Guo, S.; Shen, J.; Du, F.; Wu, X.; Chen, Y.; Li, M.; Chen, M.; et al. Tumor Infiltrating Lymphocyte (TIL) Therapy for Solid Tumor Treatment: Progressions and Challenges. Cancers 2022, 14, 4160. [Google Scholar] [CrossRef] [PubMed]
- Zacharakis, N.; Chinnasamy, H.; Black, M.; Xu, H.; Lu, Y.-C.; Zheng, Z.; Pasetto, A.; Langhan, M.; Shelton, T.; Prickett, T.; et al. Immune Recognition of Somatic Mutations Leading to Complete Durable Regression in Metastatic Breast Cancer. Nat. Med. 2018, 24, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.; Schüßler-Lenz, M.; Bondanza, A.; Buchholz, C.J. Clinical Development of CAR T Cells—Challenges and Opportunities in Translating Innovative Treatment Concepts. EMBO Mol. Med. 2017, 9, 1183–1197. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Yang, Y.-H.; Liu, J.-W.; Lu, C.; Wei, J.-F. CAR-T Cell Therapy for Breast Cancer: From Basic Research to Clinical Application. Int. J. Biol. Sci. 2022, 18, 2609–2626. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A Phase I Study on Adoptive Immunotherapy Using Gene-Modified T Cells for Ovarian Cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Rivière, I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. TRUCKs: The Fourth Generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric Antigen Receptor (CAR) T Cells Engineered with an Inducible Cytokine to Modulate the Tumor Stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Priceman, S.J.; Tilakawardane, D.; Jeang, B.; Aguilar, B.; Murad, J.P.; Park, A.K.; Chang, W.-C.; Ostberg, J.R.; Neman, J.; Jandial, R.; et al. Regional Delivery of Chimeric Antigen Receptor–Engineered T Cells Effectively Targets HER2+ Breast Cancer Metastasis to the Brain. Clin. Cancer Res. 2018, 24, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Shi, H.; Liu, C.; Liu, J.; Liu, X.; Sun, Y. Construction and Evaluation of a Novel Humanized HER2−Specific Chimeric Receptor. Breast Cancer Res. 2014, 16, R61. [Google Scholar] [CrossRef] [PubMed]
- Debien, V.; Caluwé, A.D.; Wang, X.; Piccart-Gebhart, M.; Tuohy, V.K.; Romano, E.; Buisseret, L. Immunotherapy in Breast Cancer: An Overview of Current Strategies and Perspectives. NPJ Breast Cancer 2023, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Söderberg-Nauclér, C. New Mechanistic Insights of the Pathogenicity of High-Risk Cytomegalovirus (CMV) Strains Derived from Breast Cancer: Hope for New Cancer Therapy Options. eBioMedicine 2022, 81, 104103. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Cecil, D.L. Breast Cancer Vaccines for Treatment and Prevention. Breast Cancer Res. Treat. 2022, 191, 481–489. [Google Scholar] [CrossRef]
- Zhu, S.-Y.; Yu, K.-D. Breast Cancer Vaccines: Disappointing or Promising? Front. Immunol. 2022, 13, 828386. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Type of Metastatic Breast Cancer | Intervention/Treatment | Mechanisms of Action | Phase | Identifier |
|---|---|---|---|---|
| HER2− | Utidelone vs. docetaxel | Microtubule stabilizers | 3 | NCT05430399 |
| HER2− | Alpelisib in combination with chemotherapy (nab-paclitaxel) and L-NMMA Combination regimen | PI3K inhibitor (alpelisib); microtubule stabilizer (nab-paclitaxel); iNOS inhibitor (L-NMMA); | 2 | NCT05660083 |
| ER+ HER2− | Enobosarm in combination with abemaciclib | Selective androgen receptor modulator (enobosarm); CDK4/6 inhibitor (abemaciclib) | 3 | NCT05065411 |
| ER+ HER2− | Combination therapy with anastrozole, fulvestrant, and abemaciclib | Aromatase inhibitor (anastrazole); selective estrogen receptor down-regulator (fulvestrant); CDK4/6 inhibitor (abemaciclib) | 2 | NCT05524584 |
| ER+ HER2− | ARV-471 in combination with everolimus | Selective estrogen receptor down-regulator (ARV-471); mTOR inhibitor (everolimus) | 1 | NCT05501769 |
| ER+ HER2− | Gedatolisib plus fulvestrant with or without palbociclib | A dual inhibitor, targets both PI3K and mTOR (gedatolisib), selective estrogen receptor down-regulator (fulvestrant), CDK4/6 inhibitor (palbociclib) | 3 | NCT05501886 |
| HER2+ | YH32367 | HER2/4-1BB bispecific antibody (BsAb) | 1/2 | NCT05523947 |
| HER2+ | Tucatinib in combination with pegylated liposomal doxorubicin (Doxil) | HER2 tyrosine kinase inhibitor (tucatinib); DNA intercalation and inhibition of topoisomerase II-driven DNA repair (doxil) | 2 | NCT05748834 |
| PIK3CA-Mutant HER2+ | Combination of alpelisb with tucatinib | PI3K inhibitor (alpelisb); HER2 tyrosine kinase inhibitor (tucatinib); | 1/2 | NCT05230810 |
| TNBC or HER2+ with brain metastasis | Dendritic cell vaccines against Her2/Her3 and pembrolizumab | Booster of immune response against tumor cells (dendritic cell vaccine); PD-1 receptor monoclonal antibody (pembrolizumab) | 2 | NCT04348747 |
| TNBC | CDX-301 and CDX-1140 in combination with the standard chemotherapy (pegylated liposomal doxorubicin (Doxil)) | Recombinant FMS-like tyrosine kinase 3 ligand (CDX-301); monoclonal antibody as the agonist of CD40 (CDX-1140); DNA intercalation and inhibition of topoisomerase II-driven DNA repair (doxil) | 1 | NCT05029999 |
| TNBC | ASTX727 (cedazuridine, decitabine) to chemotherapy (paclitaxel) and immunotherapy (pembrolizumab) | ASTX727 composed of decitabine as a hypomethylating agent protected against deamination by the cytidine deaminase inhibitor component, cedazuridine; microtubule stabilizer (paclitaxel); PD-1 inhibitor (pembrolizumab) | 1 | NCT05673200 |
| TNBC refractory to anthracycline with PI3KCA or PTEN alterations | Alpelisib in combination with nab-paclitaxel | PI3K inhibitor (alpelisib); microtubule stabilizer (nab-paclitaxel); | 2 | NCT04216472 |
| TNBC with either PI3KCA mutation or PTEN loss | Alpelisib in combination with nab-paclitaxel | PI3K inhibitor (alpelisib); microtubule stabilizer (nab-paclitaxel); | 3 | NCT04251533 |
| MUC1* positive breast cancer | Autologous huMNC2-CAR44 T cells | Chimeric antigen receptor (CAR)-modified T cells that target specifically the cancerous form of cleaved MUC1 (called MUC1*), which is known as a growth factor receptor of many solid tumors. | 1 | NCT04020575 |
| Breast Cancer Stage | Immunotherapeutic | Therapy Type | Reference | ||||
|---|---|---|---|---|---|---|---|
| PD-L1 Inhibitor | PD-1 Inhibitor | CTLA-4 Inhibitor | Mono-Therapy | Multi-Therapy | |||
| Phase 1 | Early | ||||||
| Atezolizumab | • | NCT03802604 | |||||
| Locally Advanced | |||||||
| Atezolizumab | • | NCT03800836 | |||||
| Durvalumab | • | NCT03356860 | |||||
| M7824 | • | NCT02699515 | |||||
| Pembrolizumab | • | NCT03310957 | |||||
| Metastatic | |||||||
| Atezolizumab | • | NCT03853707 | |||||
| Avelumab | • | NCT04360941 | |||||
| Nivolumab | • | NCT02393794 | |||||
| Pembrolizumab | • | NCT03362060 NCT03272334 | |||||
| Not Specified | |||||||
| Pembrolizumab | • | NCT06246968 | |||||
| Phase 2 | Early | ||||||
| Avelumab | • | NCT04841148 | |||||
| Pembrolizumab | • | NCT05675579 | |||||
| Locally Advanced | |||||||
| Atezolizumab | • | NCT02924883 NCT03424005 | |||||
| Pembrolizumab | • | 3 | |||||
| Metastatic | |||||||
| Atezolizumab | • | NCTT0294883 | |||||
| Avelumab | • | NCT04215146 NT03147287 | |||||
| Ipilimumab | • | NCT03789110 | |||||
| Nivolumab | • | NCT03316586 | |||||
| Pembrolizumab | • | • | NCT03139851 NCT02447003 | ||||
| Not Specified | |||||||
| Atezolizumab | • | NCT03170960 | |||||
| Ipilimumab | • | NCT03815890 | |||||
| Nivolumab | • | NCT03815890 NCT03742968 | |||||
| Pembrolizumab | • | NCT03025035 | |||||
| Phase 3 | Early | ||||||
| Atezolizumab | • | NCT03726879 NCT03595592 | |||||
| Nivolumab | • | NCT04109066 | |||||
| Pembrolizumab | • | NCT03725059 | |||||
| Locally Advanced | |||||||
| Atezolizumab | • | NCT04148911 NCT03125902 | |||||
| Pembrolizumab | • | NCT05382286 NCT03036488 | |||||
| Metastatic | |||||||
| Atezolizumab | • | NCT04177108 NCT04740918 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonni, S.; Brindley, D.N.; Chamberlain, M.D.; Daneshvar-Baghbadorani, N.; Freywald, A.; Hemmings, D.G.; Hombach-Klonisch, S.; Klonisch, T.; Raouf, A.; Shemanko, C.S.; et al. Breast Tumor Metastasis and Its Microenvironment: It Takes Both Seed and Soil to Grow a Tumor and Target It for Treatment. Cancers 2024, 16, 911. https://doi.org/10.3390/cancers16050911
Bonni S, Brindley DN, Chamberlain MD, Daneshvar-Baghbadorani N, Freywald A, Hemmings DG, Hombach-Klonisch S, Klonisch T, Raouf A, Shemanko CS, et al. Breast Tumor Metastasis and Its Microenvironment: It Takes Both Seed and Soil to Grow a Tumor and Target It for Treatment. Cancers. 2024; 16(5):911. https://doi.org/10.3390/cancers16050911
Chicago/Turabian StyleBonni, Shirin, David N. Brindley, M. Dean Chamberlain, Nima Daneshvar-Baghbadorani, Andrew Freywald, Denise G. Hemmings, Sabine Hombach-Klonisch, Thomas Klonisch, Afshin Raouf, Carrie Simone Shemanko, and et al. 2024. "Breast Tumor Metastasis and Its Microenvironment: It Takes Both Seed and Soil to Grow a Tumor and Target It for Treatment" Cancers 16, no. 5: 911. https://doi.org/10.3390/cancers16050911
APA StyleBonni, S., Brindley, D. N., Chamberlain, M. D., Daneshvar-Baghbadorani, N., Freywald, A., Hemmings, D. G., Hombach-Klonisch, S., Klonisch, T., Raouf, A., Shemanko, C. S., Topolnitska, D., Visser, K., Vizeacoumar, F. J., Wang, E., & Gibson, S. B. (2024). Breast Tumor Metastasis and Its Microenvironment: It Takes Both Seed and Soil to Grow a Tumor and Target It for Treatment. Cancers, 16(5), 911. https://doi.org/10.3390/cancers16050911