
Paracrine Regulation and Immune System Pathways in the Inflammatory Tumor Microenvironment of Lung Cancer: Insights into Oncogenesis and Immunotherapeutic Strategies


Abstract
:Simple Summary
Abstract
1. Introduction
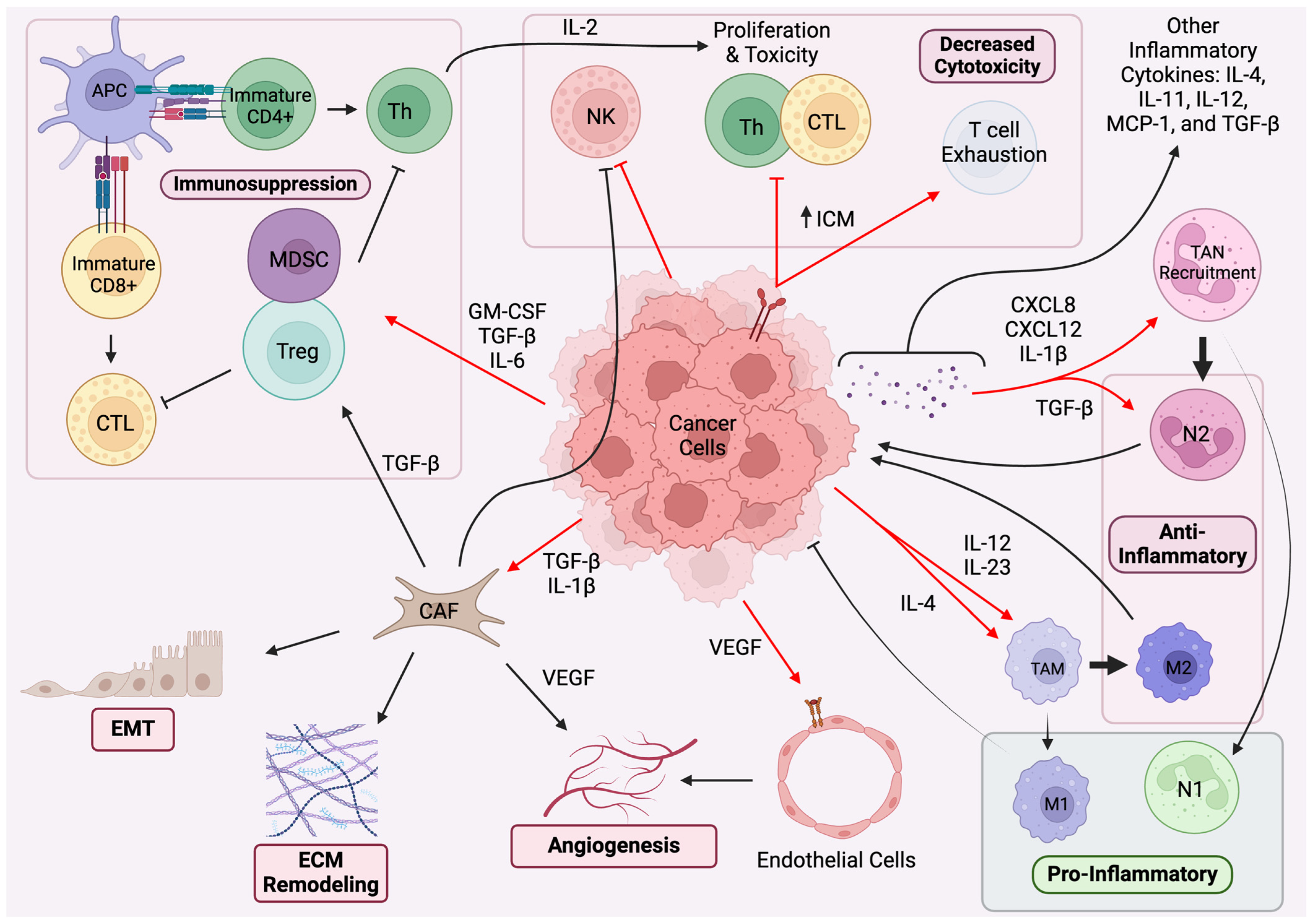
2. Paracrine Regulation and Immune System Pathways
2.1. Paracrine Regulation in TME
2.2. Immune System Pathways
3. Molecular Mechanisms of Oncogenesis
3.1. COX-2/PGE2
3.2. TGF-β
3.3. EGF
3.4. FGF
3.5. Factors of Angiogenesis: VEGFA, HIF-1α, CSF, and PDGF
3.6. Other Cytokines

{kind=link}
| Cell | Cytokine Released | Effect |
|---|---|---|
| CAF | FGF | PD-L1 expression TAM recruitment Treg generation T cell depletion MDSC survival EMT VEGF- and PDGF-mediated angiogenesis Apoptosis regulation in angiogenesis via bcl-2 |
| TGF-β | EMT Angiogenesis via MMP9 M2 and N2 polarization of TAM and TAN Treg recruitment NK cell inhibition | |
| PGE2 | ZEB1-, Snail-, and MMP2-mediated ECM dysfunction immunosuppression of DCs Stabilization of Treg | |
| IL-6 | STAT3-mediated cancer cell proliferation and invasion | |
| VEGF | Angiogenesis | |
| TAM (M2) | IL-1β | EMT Invasion VEGF- and COX-2-mediated angiogenesis MDSC-induced immunosuppression TGF-β stimulation Induction of TP53 mutations |
| TGF-β | EMT Angiogenesis via MMP9 M2 and N2 polarization of other TAM and TAN Treg recruitment | |
| IL-10 | Immunosuppression | |
| IL-6 | Stimulation of pro-cancer fibroblast signaling Angiogenesis Proliferation Immunosuppression Apoptotic evasion EMT Invasion Metastasis | |
| TNF-α | Chronic inflammation NF-κB-mediated tumor cell proliferation Apoptotic evasion Release of angiogenic factors | |
| TAN (N2) | MMP9 VEGF IL-8 | Production of pro-angiogenic factors |
| Treg | IL-10 | Immunosuppression |
| TGF-β | Treg recruitment | |
| MDSC | IL-10 | Immunosuppression |
| Cancer Cells | PGE2 | ZEB1-, Snail-, and MMP2-mediated ECM dysfunction immunosuppression of DCs Stabilization of Treg |
| TGF-β | “reverse Warburg effects” in CAF EMT Aangiogenesis via MMP9 M2 and N2 polarization of TAM and TAN Treg recruitment NK cell inhibition | |
| EGF | Apoptotic evasion via survivin, bcl-2, and BAX Production of angiogenic factors | |
| FGF | TAM recruitment MDSC survival EMT Angiogenesis T cell depletion Treg generation | |
| CSF | WBC proliferation Angiogenesis Increased tumor aggression M2 polarization Increasing MDSC and Treg phenotypes Metastasis via Ly6G+Ly6C+ granulocyte mobilization | |
| VEGF | Aangiogenesis | |
| IL-1β | Protumorigenic signaling in CAF TAN recruitment Angiogenesis Induction of MDSC Leukocyte adhesion on endothelial cells IL-22 production | |
| IL-4 | Regulation of immune response | |
| IL-6 | Treg and MDSC upregulation Immunosuppressive modulation of NK, neutrophil, and T cell activity Suppression of apoptosis Stimulation of CAF growth factor release | |
| IL-8 | Recruitment of MDSC and immunosuppressive neutrophils EMT Angiogenesis | |
| IL-12/23 | TAN recruitment |
3.7. Key Intracellular Signals
4. Immunotherapeutic Strategies
4.1. Immune Checkpoint Inhibitors
4.2. CAR T Cell Therapy
4.3. CAFs, TAMs, and TANs
4.4. Oncolytic Viruses
4.5. Tumor-Infiltrating Lymphocytes (TILs)
4.6. IL-1β
4.7. NF-kB
4.8. IL-6
4.9. STAT3
4.10. TNF-α
4.11. IL-8
4.12. IL-10
5. Challenges and Future Directions
5.1. Case-Specific Variations in Efficacy
5.2. Resistance Mechanisms
5.3. Bringing Scientific Success into the Clinic Going Forward
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fang, J.; Lu, Y.; Zheng, J.; Jiang, X.; Shen, H.; Shang, X.; Lu, Y.; Fu, P. Exploring the Crosstalk between Endothelial Cells, Immune Cells, and Immune Checkpoints in the Tumor Microenvironment: New Insights and Therapeutic Implications. Cell Death Dis. 2023, 14, 586. [Google Scholar] [CrossRef] [PubMed]
- Poncette, L.; Bluhm, J.; Blankenstein, T. The Role of CD4 T Cells in Rejection of Solid Tumors. Curr. Opin. Immunol. 2022, 74, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Veeramachaneni, N. Targeting Interleukin-1β and Inflammation in Lung Cancer. Biomark. Res. 2022, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Nishikawa, H. Mechanisms of Regulatory T Cell Infiltration in Tumors: Implications for Innovative Immune Precision Therapies. J. Immunother. Cancer 2021, 9, e002591. [Google Scholar] [CrossRef] [PubMed]
- Yao, P.-L.; Lin, Y.-C.; Wang, C.-H.; Huang, Y.-C.; Liao, W.-Y.; Wang, S.-S.; Chen, J.J.W.; Yang, P.-C. Autocrine and Paracrine Regulation of Interleukin-8 Expression in Lung Cancer Cells. Am. J. Respir. Cell Mol. Biol. 2005, 32, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.K.; Wong, B.H.S.; Poh, Z.S.; Udayakumar, A.; Verma, R.; Goh, R.K.J.; Duggan, S.P.; Shelat, V.G.; Chandy, K.G.; Grigoropoulos, N.F. Obstacles for T-Lymphocytes in the Tumour Microenvironment: Therapeutic Challenges, Advances and Opportunities beyond Immune Checkpoint. eBioMedicine 2022, 83, 104216. [Google Scholar] [CrossRef] [PubMed]
- Shang, G.-S.; Liu, L.; Qin, Y.-W. IL-6 and TNF-α Promote Metastasis of Lung Cancer by Inducing Epithelial-Mesenchymal Transition. Oncol. Lett. 2017, 13, 4657–4660. [Google Scholar] [CrossRef]
- Chen, W.-J.; Ho, C.-C.; Chang, Y.-L.; Chen, H.-Y.; Lin, C.-A.; Ling, T.-Y.; Yu, S.-L.; Yuan, S.-S.; Louisa Chen, Y.-J.; Lin, C.-Y.; et al. Cancer-Associated Fibroblasts Regulate the Plasticity of Lung Cancer Stemness via Paracrine Signalling. Nat. Commun. 2014, 5, 3472. [Google Scholar] [CrossRef]
- Wong, K.Y.; Cheung, A.H.; Chen, B.; Chan, W.N.; Yu, J.; Lo, K.W.; Kang, W.; To, K.F. Cancer-associated Fibroblasts in Nonsmall Cell Lung Cancer: From Molecular Mechanisms to Clinical Implications. Int. J. Cancer 2022, 151, 1195–1215. [Google Scholar] [CrossRef]
- Mishra, P.; Banerjee, D.; Ben-Baruch, A. Chemokines at the Crossroads of Tumor-Fibroblast Interactions That Promote Malignancy. J. Leukoc. Biol. 2011, 89, 31–39. [Google Scholar] [CrossRef]
- Ren, Y.; Jia, H.-H.; Xu, Y.-Q.; Zhou, X.; Zhao, X.-H.; Wang, Y.-F.; Song, X.; Zhu, Z.-Y.; Sun, T.; Dou, Y.; et al. Paracrine and Epigenetic Control of CAF-Induced Metastasis: The Role of HOTAIR Stimulated by TGF-SS1 Secretion. Mol. Cancer 2018, 17, 5. [Google Scholar] [CrossRef] [PubMed]
- Kogue, Y.; Kobayashi, H.; Nakamura, Y.; Takano, T.; Furuta, C.; Kawano, O.; Yasuma, T.; Nishimura, T.; D’Alessandro-Gabazza, C.N.; Fujimoto, H.; et al. Prognostic Value of CXCL12 in Non-Small Cell Lung Cancer Patients Undergoing Tumor Resection. Pharmaceuticals 2023, 16, 255. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.M.; Webster, S.J.; Flower, D.; Woll, P.J. Interleukin-8/CXCL8 Is a Growth Factor for Human Lung Cancer Cells. Br. J. Cancer 2004, 91, 1970–1976. [Google Scholar] [CrossRef] [PubMed]
- Inoue, C.; Miki, Y.; Saito, R.; Hata, S.; Abe, J.; Sato, I.; Okada, Y.; Sasano, H. PD-L1 Induction by Cancer-Associated Fibroblast-Derived Factors in Lung Adenocarcinoma Cells. Cancers 2019, 11, 1257. [Google Scholar] [CrossRef]
- Erdogan, B.; Webb, D.J. Cancer-Associated Fibroblasts Modulate Growth Factor Signaling and Extracellular Matrix Remodeling to Regulate Tumor Metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef]
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, Z.; Xu, X.; Yu, Z.; Mi, J. The Influence of Microenvironment on Tumor Immunotherapy. FEBS J. 2019, 286, 4160–4175. [Google Scholar] [CrossRef]
- de Rodas, M.L.; Nagineni, V.; Ravi, A.; Datar, I.J.; Mino-Kenudson, M.; Corredor, G.; Barrera, C.; Behlman, L.; Rimm, D.L.; Herbst, R.S.; et al. Role of Tumor Infiltrating Lymphocytes and Spatial Immune Heterogeneity in Sensitivity to PD-1 Axis Blockers in Non-Small Cell Lung Cancer. J. Immunother. Cancer 2022, 10, e004440. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Garcia-Lora, A.; Algarra, I.; Garrido, F. MHC Class I Antigens, Immune Surveillance, and Tumor Immune Escape. J. Cell. Physiol. 2003, 195, 346–355. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt Signal Transduction for Cancer Therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xu, C. Immune Checkpoint Signaling and Cancer Immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Sanz, L.; Muñoz, P. Tumor-Infiltrating Immunosuppressive Cells in Cancer-Cell Plasticity, Tumor Progression and Therapy Response. Cancer Microenviron. 2019, 12, 119–132. [Google Scholar] [CrossRef]
- Anichini, A.; Perotti, V.E.; Sgambelluri, F.; Mortarini, R. Immune Escape Mechanisms in Non Small Cell Lung Cancer. Cancers 2020, 12, 3605. [Google Scholar] [CrossRef] [PubMed]
- Pawelczyk, K.; Piotrowska, A.; Ciesielska, U.; Jablonska, K.; Glatzel-Plucinska, N.; Grzegrzolka, J.; Podhorska-Okolow, M.; Dziegiel, P.; Nowinska, K. Role of PD-L1 Expression in Non-Small Cell Lung Cancer and Their Prognostic Significance According to Clinicopathological Factors and Diagnostic Markers. Int. J. Mol. Sci. 2019, 20, 824. [Google Scholar] [CrossRef]
- Hung, C.-N.; Chen, M.; DeArmond, D.T.; Chiu, C.H.-L.; Limboy, C.A.; Tan, X.; Kusi, M.; Chou, C.-W.; Lin, L.-L.; Zhang, Z.; et al. AXL-Initiated Paracrine Activation of pSTAT3 Enhances Mesenchymal and Vasculogenic Supportive Features of Tumor-Associated Macrophages. Cell Rep. 2023, 42, 113067. [Google Scholar] [CrossRef]
- Tan, Z.; Xue, H.; Sun, Y.; Zhang, C.; Song, Y.; Qi, Y. The Role of Tumor Inflammatory Microenvironment in Lung Cancer. Front. Pharmacol. 2021, 12, 688625. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor Microenvironment Complexity and Therapeutic Implications at a Glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Wang, X.; Qiu, L.; Li, Z.; Wang, X.-Y.; Yi, H. Understanding the Multifaceted Role of Neutrophils in Cancer and Autoimmune Diseases. Front. Immunol. 2018, 9, 2456. [Google Scholar] [CrossRef]
- Fang, Y.; Li, X.; Jiang, Y.; Ge, Z. Blocking TGF-β Expression Attenuates Tumor Growth in Lung Cancers, Potentially Mediated by Skewing Development of Neutrophils. J. Oncol. 2022, 2022, 3447185. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, A.; Salvi, V.; Soriani, A.; Laffranchi, M.; Sozio, F.; Bosisio, D.; Sozzani, S. Dendritic Cell Subsets in Cancer Immunity and Tumor Antigen Sensing. Cell Mol. Immunol. 2023, 20, 432–447. [Google Scholar] [CrossRef] [PubMed]
- Bronte, G.; Petracci, E.; De Matteis, S.; Canale, M.; Zampiva, I.; Priano, I.; Cravero, P.; Andrikou, K.; Burgio, M.A.; Ulivi, P.; et al. High Levels of Circulating Monocytic Myeloid-Derived Suppressive-Like Cells Are Associated With the Primary Resistance to Immune Checkpoint Inhibitors in Advanced Non-Small Cell Lung Cancer: An Exploratory Analysis. Front. Immunol. 2022, 13, 866561. [Google Scholar] [CrossRef]
- Bronte, G.; Calabrò, L.; Olivieri, F.; Procopio, A.D.; Crinò, L. The Prognostic Effects of Circulating Myeloid-Derived Suppressor Cells in Non-Small Cell Lung Cancer: Systematic Review and Meta-Analysis. Clin. Exp. Med. 2023, 23, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Bayik, D.; Lee, J.; Lathia, J.D. The Role of Myeloid-Derived Suppressor Cells in Tumor Growth and Metastasis. In Interaction of Immune and Cancer Cells; Klink, M., Szulc-Kielbik, I., Eds.; Experientia Supplementum; Springer International Publishing: Cham, Switzerland, 2022; pp. 189–217. ISBN 978-3-030-91311-3. [Google Scholar]
- Zhou, Y.; Cheng, L.; Liu, L.; Li, X. NK Cells Are Never Alone: Crosstalk and Communication in Tumour Microenvironments. Mol. Cancer 2023, 22, 34. [Google Scholar] [CrossRef] [PubMed]
- Castelao, J.E.; Bart, R.D.; DiPerna, C.A.; Sievers, E.M.; Bremner, R.M. Lung Cancer and Cyclooxygenase-2. Ann. Thorac. Surg. 2003, 76, 1327–1335. [Google Scholar] [CrossRef]
- Ochiai, M.; Oguri, T.; Isobe, T.; Ishioka, S.; Yamakido, M. Cyclooxygenase-2 (COX-2) mRNA Expression Levels in Normal Lung Tissues and Non-Small Cell Lung Cancers. Jpn. J. Cancer Res. 1999, 90, 1338–1343. [Google Scholar] [CrossRef]
- Dohadwala, M.; Batra, R.K.; Luo, J.; Lin, Y.; Krysan, K.; Põld, M.; Sharma, S.; Dubinett, S.M. Autocrine/Paracrine Prostaglandin E2 Production by Non-Small Cell Lung Cancer Cells Regulates Matrix Metalloproteinase-2 and CD44 in Cyclooxygenase-2-Dependent Invasion. J. Biol. Chem. 2002, 277, 50828–50833. [Google Scholar] [CrossRef]
- Gomperts, B.N.; Spira, A.; Massion, P.P.; Walser, T.C.; Wistuba, I.I.; Minna, J.D.; Dubinett, S.M. Evolving Concepts in Lung Carcinogenesis. Semin. Respir. Crit. Care Med. 2011, 32, 32–43. [Google Scholar] [CrossRef]
- Dohadwala, M.; Yang, S.-C.; Luo, J.; Sharma, S.; Batra, R.K.; Huang, M.; Lin, Y.; Goodglick, L.; Krysan, K.; Fishbein, M.C.; et al. Cyclooxygenase-2–Dependent Regulation of E-Cadherin: Prostaglandin E2 Induces Transcriptional Repressors ZEB1 and Snail in Non–Small Cell Lung Cancer. Cancer Res. 2006, 66, 5338–5345. [Google Scholar] [CrossRef]
- Baratelli, F.; Lin, Y.; Zhu, L.; Yang, S.-C.; Heuzé-Vourc’h, N.; Zeng, G.; Reckamp, K.; Dohadwala, M.; Sharma, S.; Dubinett, S.M. Prostaglandin E2 Induces FOXP3 Gene Expression and T Regulatory Cell Function in Human CD4+ T Cells1. J. Immunol. 2005, 175, 1483–1490. [Google Scholar] [CrossRef] [PubMed]
- Põld, M.; Zhu, L.X.; Sharma, S.; Burdick, M.D.; Lin, Y.; Lee, P.P.N.; Põld, A.; Luo, J.; Krysan, K.; Dohadwala, M.; et al. Cyclooxygenase-2-Dependent Expression of Angiogenic CXC Chemokines ENA-78/CXC Ligand (CXCL) 5 and Interleukin-8/CXCL8 in Human Non-Small Cell Lung Cancer. Cancer Res. 2004, 64, 1853–1860. [Google Scholar] [CrossRef] [PubMed]
- Gavin, M.A.; Rasmussen, J.P.; Fontenot, J.D.; Vasta, V.; Manganiello, V.C.; Beavo, J.A.; Rudensky, A.Y. Foxp3-Dependent Programme of Regulatory T-Cell Differentiation. Nature 2007, 445, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Stolina, M.; Yang, S.-C.; Baratelli, F.; Lin, J.F.; Atianzar, K.; Luo, J.; Zhu, L.; Lin, Y.; Huang, M.; et al. Tumor Cyclooxygenase 2-Dependent Suppression of Dendritic Cell Function. Clin. Cancer Res. 2003, 9, 961–968. [Google Scholar] [PubMed]
- Krysan, K.; Merchant, F.H.; Zhu, L.; Dohadwala, M.; Luo, J.; Lin, Y.; Heuze-Vourc’h, N.; Põld, M.; Seligson, D.; Chia, D.; et al. COX-2-Dependent Stabilization of Survivin in Non-Small Cell Lung Cancer. FASEB J. 2004, 18, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.-S.; Jen, J. TGF-β Signaling and the Role of Inhibitory Smads in Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2010, 5, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Horie, M.; Nagase, T. TGF-β Signaling in Lung Health and Disease. Int. J. Mol. Sci. 2018, 19, 2460. [Google Scholar] [CrossRef]
- Seoane, J.; Gomis, R.R. TGF-β Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb. Perspect. Biol. 2017, 9, a022277. [Google Scholar] [CrossRef]
- Ahmadi, A.; Najafi, M.; Farhood, B.; Mortezaee, K. Transforming Growth Factor-β Signaling: Tumorigenesis and Targeting for Cancer Therapy. J. Cell. Physiol. 2019, 234, 12173–12187. [Google Scholar] [CrossRef]
- Shi, X.; Yang, J.; Deng, S.; Xu, H.; Wu, D.; Zeng, Q.; Wang, S.; Hu, T.; Wu, F.; Zhou, H. TGF-β Signaling in the Tumor Metabolic Microenvironment and Targeted Therapies. J. Hematol. Oncol. 2022, 15, 135. [Google Scholar] [CrossRef]
- Saito, A.; Suzuki, H.I.; Horie, M.; Ohshima, M.; Morishita, Y.; Abiko, Y.; Nagase, T. An Integrated Expression Profiling Reveals Target Genes of TGF-β and TNF-α Possibly Mediated by microRNAs in Lung Cancer Cells. PLoS ONE 2013, 8, e56587. [Google Scholar] [CrossRef]
- Peng, D.; Fu, M.; Wang, M.; Wei, Y.; Wei, X. Targeting TGF-β Signal Transduction for Fibrosis and Cancer Therapy. Mol. Cancer 2022, 21, 104. [Google Scholar] [CrossRef] [PubMed]
- Shintani, Y.; Kimura, T.; Funaki, S.; Ose, N.; Kanou, T.; Fukui, E. Therapeutic Targeting of Cancer-Associated Fibroblasts in the Non-Small Cell Lung Cancer Tumor Microenvironment. Cancers 2023, 15, 335. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The Reverse Warburg Effect: Aerobic Glycolysis in Cancer Associated Fibroblasts and the Tumor Stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Ngaha, T.Y.S.; Zhilenkova, A.V.; Essogmo, F.E.; Uchendu, I.K.; Abah, M.O.; Fossa, L.T.; Sangadzhieva, Z.D.; Sanikovich, D.V.; Rusanov, S.A.; Pirogova, Y.N.; et al. Angiogenesis in Lung Cancer: Understanding the Roles of Growth Factors. Cancers 2023, 15, 4648. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.-C.; Jin, X.; Wang, Y.; Wang, K. Role of Epidermal Growth Factor Receptor in Lung Cancer and Targeted Therapies. Am. J. Cancer Res. 2017, 7, 187–202. [Google Scholar]
- Siegelin, M.D.; Borczuk, A.C. Epidermal Growth Factor Receptor Mutations in Lung Adenocarcinoma. Lab. Investig. 2014, 94, 129–137. [Google Scholar] [CrossRef]
- Takata, S.; Takigawa, N.; Segawa, Y.; Kubo, T.; Ohashi, K.; Kozuki, T.; Teramoto, N.; Yamashita, M.; Toyooka, S.; Tanimoto, M.; et al. STAT3 Expression in Activating EGFR-Driven Adenocarcinoma of the Lung. Lung Cancer 2012, 75, 24–29. [Google Scholar] [CrossRef]
- Busser, B.; Sancey, L.; Josserand, V.; Niang, C.; Khochbin, S.; Favrot, M.C.; Coll, J.-L.; Hurbin, A. Amphiregulin Promotes Resistance to Gefitinib in Nonsmall Cell Lung Cancer Cells by Regulating Ku70 Acetylation. Mol. Ther. 2010, 18, 536–543. [Google Scholar] [CrossRef]
- Tsao, M.S.; Zhu, H.; Viallet, J. Autocrine Growth Loop of the Epidermal Growth Factor Receptor in Normal and Immortalized Human Bronchial Epithelial Cells. Exp. Cell Res. 1996, 223, 268–273. [Google Scholar] [CrossRef]
- Dong, Z.; Zhang, L.; Xu, W.; Zhang, G. EGFR May Participate in Immune Evasion through Regulation of B7-H5 Expression in Non-small Cell Lung Carcinoma. Mol. Med. Rep. 2018, 18, 3769–3779. [Google Scholar] [CrossRef] [PubMed]
- Bruns, C.J.; Solorzano, C.C.; Harbison, M.T.; Ozawa, S.; Tsan, R.; Fan, D.; Abbruzzese, J.; Traxler, P.; Buchdunger, E.; Radinsky, R.; et al. Blockade of the Epidermal Growth Factor Receptor Signaling by a Novel Tyrosine Kinase Inhibitor Leads to Apoptosis of Endothelial Cells and Therapy of Human Pancreatic Carcinoma. Cancer Res. 2000, 60, 2926–2935. [Google Scholar] [PubMed]
- Schelch, K.; Vogel, L.; Schneller, A.; Brankovic, J.; Mohr, T.; Mayer, R.L.; Slany, A.; Gerner, C.; Grusch, M. EGF Induces Migration Independent of EMT or Invasion in A549 Lung Adenocarcinoma Cells. Front. Cell Dev. Biol. 2021, 9, 634371. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zeng, Y.; Du, W.; Zhu, J.; Shen, D.; Liu, Z.; Huang, J.-A. The EGFR Pathway Is Involved in the Regulation of PD-L1 Expression via the IL-6/JAK/STAT3 Signaling Pathway in EGFR-Mutated Non-Small Cell Lung Cancer. Int. J. Oncol. 2016, 49, 1360–1368. [Google Scholar] [CrossRef]
- Behrens, C.; Lin, H.Y.; Lee, J.J.; Raso, M.G.; Hong, W.K.; Wistuba, I.I.; Lotan, R. Immunohistochemical Expression of Basic Fibroblast Growth Factor and Fibroblast Growth Factor Receptors 1 and 2 in the Pathogenesis of Lung Cancer. Clin. Cancer Res. 2008, 14, 6014–6022. [Google Scholar] [CrossRef]
- Ruan, R.; Li, L.; Li, X.; Huang, C.; Zhang, Z.; Zhong, H.; Zeng, S.; Shi, Q.; Xia, Y.; Zeng, Q.; et al. Unleashing the Potential of Combining FGFR Inhibitor and Immune Checkpoint Blockade for FGF/FGFR Signaling in Tumor Microenvironment. Mol. Cancer 2023, 22, 60. [Google Scholar] [CrossRef]
- Hegab, A.E.; Ozaki, M.; Kameyama, N.; Gao, J.; Kagawa, S.; Yasuda, H.; Soejima, K.; Yin, Y.; Guzy, R.D.; Nakamura, Y.; et al. Effect of FGF/FGFR Pathway Blocking on Lung Adenocarcinoma and Its Cancer-Associated Fibroblasts. J. Pathol. 2019, 249, 193–205. [Google Scholar] [CrossRef]
- Murakami, M.; Simons, M. Fibroblast Growth Factor Regulation of Neovascularization. Curr. Opin. Hematol. 2008, 15, 215–220. [Google Scholar] [CrossRef]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental Regulation of Tumour Angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis. Genes. Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef]
- Fontanini, G.; Boldrini, L.; Vignati, S.; Chinè, S.; Basolo, F.; Silvestri, V.; Lucchi, M.; Mussi, A.; Angeletti, C.A.; Bevilacqua, G. Bcl2 and P53 Regulate Vascular Endothelial Growth Factor (VEGF)-Mediated Angiogenesis in Non-Small Cell Lung Carcinoma. Eur. J. Cancer 1998, 34, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Markovic, M.; Mitrovic, S.; Dagovic, A.; Jovanovic, D.; Nikolic, T.; Ivosevic, A.; Milosavljevic, M.Z.; Vojinovic, R.; Petrovic, M. Does the Expression of Vascular Endothelial Growth Factor (VEGF) and Bcl-2 Have a Prognostic Significance in Advanced Non-Small Cell Lung Cancer? Healthcare 2023, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Pezzuto, A.; Carico, E. Role of HIF-1 in Cancer Progression: Novel Insights. A Review. Curr. Mol. Med. 2018, 18, 343–351. [Google Scholar] [CrossRef]
- Li, T.; Qiao, T. Unraveling Tumor Microenvironment of Small-Cell Lung Cancer: Implications for Immunotherapy. Semin. Cancer Biol. 2022, 86, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, L.A.; Semenza, G.L. HIF and the Lung. Am. J. Respir. Crit. Care Med. 2011, 183, 152–156. [Google Scholar] [CrossRef]
- Karagiannidis, I.; Salataj, E.; Said Abu Egal, E.; Beswick, E.J. G-CSF in Tumors: Aggressiveness, Tumor Microenvironment and Immune Cell Regulation. Cytokine 2021, 142, 155479. [Google Scholar] [CrossRef]
- Kowanetz, M.; Wu, X.; Lee, J.; Tan, M.; Hagenbeek, T.; Qu, X.; Yu, L.; Ross, J.; Korsisaari, N.; Cao, T.; et al. Granulocyte-Colony Stimulating Factor Promotes Lung Metastasis through Mobilization of Ly6G+Ly6C+ Granulocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 21248–21255. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- KATOH, M. FGFR Inhibitors: Effects on Cancer Cells, Tumor Microenvironment and Whole-Body Homeostasis (Review). Int. J. Mol. Med. 2016, 38, 3–15. [Google Scholar] [CrossRef]
- Zeltz, C.; Primac, I.; Erusappan, P.; Alam, J.; Noel, A.; Gullberg, D. Cancer-Associated Fibroblasts in Desmoplastic Tumors: Emerging Role of Integrins. Semin. Cancer Biol. 2020, 62, 166–181. [Google Scholar] [CrossRef]
- Garon, E.B.; Chih-Hsin Yang, J.; Dubinett, S.M. The Role of Interleukin 1β in the Pathogenesis of Lung Cancer. JTO Clin. Res. Rep. 2020, 1, 100001. [Google Scholar] [CrossRef]
- Zhang, W.; Borcherding, N.; Kolb, R. IL-1 Signaling in Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1240, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking IL-1β Reverses the Immunosuppression in Mouse Breast Cancer and Synergizes with Anti-PD-1 for Tumor Abrogation. Proc. Natl. Acad. Sci. USA 2019, 116, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Landvik, N.E.; Hart, K.; Skaug, V.; Stangeland, L.B.; Haugen, A.; Zienolddiny, S. A Specific Interleukin-1B Haplotype Correlates with High Levels of IL1B mRNA in the Lung and Increased Risk of Non-Small Cell Lung Cancer. Carcinogenesis 2009, 30, 1186–1192. [Google Scholar] [CrossRef] [PubMed]
- Essogmo, F.E.; Zhilenkova, A.V.; Tchawe, Y.S.N.; Owoicho, A.M.; Rusanov, A.S.; Boroda, A.; Pirogova, Y.N.; Sangadzhieva, Z.D.; Sanikovich, V.D.; Bagmet, N.N.; et al. Cytokine Profile in Lung Cancer Patients: Anti-Tumor and Oncogenic Cytokines. Cancers 2023, 15, 5383. [Google Scholar] [CrossRef] [PubMed]
- Medrano, R.F.V.; Hunger, A.; Mendonça, S.A.; Barbuto, J.A.M.; Strauss, B.E. Immunomodulatory and Antitumor Effects of Type I Interferons and Their Application in Cancer Therapy. Oncotarget 2017, 8, 71249–71284. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.S.; Femel, J.; Breazeale, A.P.; Loo, C.P.; Thibault, G.; Kaempf, A.; Mori, M.; Tsujikawa, T.; Chang, Y.H.; Lund, A.W. IFNγ-Activated Dermal Lymphatic Vessels Inhibit Cytotoxic T Cells in Melanoma and Inflamed Skin. J. Exp. Med. 2018, 215, 3057–3074. [Google Scholar] [CrossRef] [PubMed]
- Werner, E.R.; Bitterlich, G.; Fuchs, D.; Hausen, A.; Reibnegger, G.; Szabo, G.; Dierich, M.P.; Wachter, H. Human Macrophages Degrade Tryptophan upon Induction by Interferon-Gamma. Life Sci. 1987, 41, 273–280. [Google Scholar] [CrossRef]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in Tumor Progression and Regression: A Review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef]
- Vahl, J.M.; Friedrich, J.; Mittler, S.; Trump, S.; Heim, L.; Kachler, K.; Balabko, L.; Fuhrich, N.; Geppert, C.-I.; Trufa, D.I.; et al. Interleukin-10-Regulated Tumour Tolerance in Non-Small Cell Lung Cancer. Br. J. Cancer 2017, 117, 1644–1655. [Google Scholar] [CrossRef]
- Tazzyman, S.; Lewis, C.E.; Murdoch, C. Neutrophils: Key Mediators of Tumour Angiogenesis. Int. J. Exp. Pathol. 2009, 90, 222–231. [Google Scholar] [CrossRef]
- Ramachandran, S.; Verma, A.K.; Dev, K.; Goyal, Y.; Bhatt, D.; Alsahli, M.A.; Rahmani, A.H.; Almatroudi, A.; Almatroodi, S.A.; Alrumaihi, F.; et al. Role of Cytokines and Chemokines in NSCLC Immune Navigation and Proliferation. Oxid. Med. Cell. Longev. 2021, 2021, 5563746. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.; Groth, C.; Lasser, S.; Arkhypov, I.; Petrova, V.; Altevogt, P.; Utikal, J.; Umansky, V. IL-6 as a Major Regulator of MDSC Activity and Possible Target for Cancer Immunotherapy. Cell. Immunol. 2021, 359, 104254. [Google Scholar] [CrossRef]
- Rašková, M.; Lacina, L.; Kejík, Z.; Venhauerová, A.; Skaličková, M.; Kolář, M.; Jakubek, M.; Rosel, D.; Smetana, K.; Brábek, J. The Role of IL-6 in Cancer Cell Invasiveness and Metastasis—Overview and Therapeutic Opportunities. Cells 2022, 11, 3698. [Google Scholar] [CrossRef]
- Benoot, T.; Piccioni, E.; De Ridder, K.; Goyvaerts, C. TNFα and Immune Checkpoint Inhibition: Friend or Foe for Lung Cancer? Int. J. Mol. Sci. 2021, 22, 8691. [Google Scholar] [CrossRef] [PubMed]
- Guttridge, D.C.; Albanese, C.; Reuther, J.Y.; Pestell, R.G.; Baldwin, A.S. NF-kappaB Controls Cell Growth and Differentiation through Transcriptional Regulation of Cyclin D1. Mol. Cell. Biol. 1999, 19, 5785–5799. [Google Scholar] [CrossRef]
- Chen, C.; Edelstein, L.C.; Gélinas, C. The Rel/NF-κB Family Directly Activates Expression of the Apoptosis Inhibitor Bcl-xL. Mol. Cell. Biol. 2000, 20, 2687–2695. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear Factor-kappaB in Cancer Development and Progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, X.; Mi, Z.; Jiang, X.; Sun, L.; Zheng, B.; Wang, J.; Meng, M.; Zhang, L.; Wang, Z.; et al. STAT3/miR-135b/NF-κB Axis Confers Aggressiveness and Unfavorable Prognosis in Non-Small-Cell Lung Cancer. Cell Death Dis. 2021, 12, 493. [Google Scholar] [CrossRef]
- Rébé, C.; Ghiringhelli, F. STAT3, a Master Regulator of Anti-Tumor Immune Response. Cancers 2019, 11, 1280. [Google Scholar] [CrossRef]
- Rasmi, R.R.; Sakthivel, K.M.; Guruvayoorappan, C. NF-κB Inhibitors in Treatment and Prevention of Lung Cancer. Biomed. Pharmacother. 2020, 130, 110569. [Google Scholar] [CrossRef] [PubMed]
- Bonizzi, G.; Karin, M. The Two NF-kappaB Activation Pathways and Their Role in Innate and Adaptive Immunity. Trends Immunol. 2004, 25, 280–288. [Google Scholar] [CrossRef]
- Tabruyn, S.P.; Griffioen, A.W. NF-κB: A New Player in Angiostatic Therapy. Angiogenesis 2008, 11, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.S. Regulation of Cell Death and Autophagy by IKK and NF-κB: Critical Mechanisms in Immune Function and Cancer. Immunol. Rev. 2012, 246, 327–345. [Google Scholar] [CrossRef] [PubMed]
- Helin, K. Regulation of Cell Proliferation by the E2F Transcription Factors. Curr. Opin. Genet. Dev. 1998, 8, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T.; Pines, J. Cyclins and Cancer. II: Cyclin D and CDK Inhibitors Come of Age. Cell 1994, 79, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Deveraux, Q.L.; Reed, J.C. IAP Family Proteins—Suppressors of Apoptosis. Genes Dev. 1999, 13, 239–252. [Google Scholar] [CrossRef]
- Gyrd-Hansen, M.; Meier, P. IAPs: From Caspase Inhibitors to Modulators of NF-kappaB, Inflammation and Cancer. Nat. Rev. Cancer 2010, 10, 561–574. [Google Scholar] [CrossRef]
- Fan, Y.; Mao, R.; Yang, J. NF-κB and STAT3 Signaling Pathways Collaboratively Link Inflammation to Cancer. Protein Cell 2013, 4, 176–185. [Google Scholar] [CrossRef]
- Huynh, J.; Chand, A.; Gough, D.; Ernst, M. Therapeutically Exploiting STAT3 Activity in Cancer—Using Tissue Repair as a Road Map. Nat. Rev. Cancer 2019, 19, 82–96. [Google Scholar] [CrossRef]
- Waldner, M.J.; Foersch, S.; Neurath, M.F. Interleukin-6—A Key Regulator of Colorectal Cancer Development. Int. J. Biol. Sci. 2012, 8, 1248–1253. [Google Scholar] [CrossRef]
- Grivennikov, S.; Karin, M. Dangerous Liaisons: STAT3 and NF-κB Collaboration and Crosstalk in Cancer. Cytokine Growth Factor. Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef]
- Mitochondrial STAT3 Supports Ras-Dependent Oncogenic Transformation. Available online: https://pubmed.ncbi.nlm.nih.gov/19556508/ (accessed on 25 October 2023).
- Lahiri, A.; Maji, A.; Potdar, P.D.; Singh, N.; Parikh, P.; Bisht, B.; Mukherjee, A.; Paul, M.K. Lung Cancer Immunotherapy: Progress, Pitfalls, and Promises. Mol. Cancer 2023, 22, 40. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.-W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef]
- Paz-Ares, L.G.; Ciuleanu, T.-E.; Cobo, M.; Bennouna, J.; Schenker, M.; Cheng, Y.; Juan-Vidal, O.; Mizutani, H.; Lingua, A.; Reyes-Cosmelli, F.; et al. First-Line Nivolumab Plus Ipilimumab With Chemotherapy Versus Chemotherapy Alone for Metastatic NSCLC in CheckMate 9LA: 3-Year Clinical Update and Outcomes in Patients With Brain Metastases or Select Somatic Mutations. J. Thorac. Oncol. 2023, 18, 204–222. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay Şenler, F.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Sezer, A.; Kilickap, S.; Gümüş, M.; Bondarenko, I.; Özgüroğlu, M.; Gogishvili, M.; Turk, H.M.; Cicin, I.; Bentsion, D.; Gladkov, O.; et al. Cemiplimab Monotherapy for First-Line Treatment of Advanced Non-Small-Cell Lung Cancer with PD-L1 of at Least 50%: A Multicentre, Open-Label, Global, Phase 3, Randomised, Controlled Trial. Lancet 2021, 397, 592–604. [Google Scholar] [CrossRef]
- Gogishvili, M.; Melkadze, T.; Makharadze, T.; Giorgadze, D.; Dvorkin, M.; Penkov, K.; Laktionov, K.; Nemsadze, G.; Nechaeva, M.; Rozhkova, I.; et al. Cemiplimab plus Chemotherapy versus Chemotherapy Alone in Non-Small Cell Lung Cancer: A Randomized, Controlled, Double-Blind Phase 3 Trial. Nat. Med. 2022, 28, 2374–2380. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- Johnson, M.L.; Cho, B.C.; Luft, A.; Alatorre-Alexander, J.; Geater, S.L.; Laktionov, K.; Kim, S.-W.; Ursol, G.; Hussein, M.; Lim, F.L.; et al. Durvalumab With or Without Tremelimumab in Combination With Chemotherapy as First-Line Therapy for Metastatic Non–Small-Cell Lung Cancer: The Phase III POSEIDON Study. JCO 2023, 41, 1213–1227. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Mei, Q.; Chen, L.; Zhou, J. Chimeric Antigen Receptor (CAR)-T-Cell Therapy in Non-Small-Cell Lung Cancer (NSCLC): Current Status and Future Perspectives. Cancer Immunol. Immunother. 2021, 70, 619–631. [Google Scholar] [CrossRef]
- Chen, X.W.; Yu, T.J.; Zhang, J.; Li, Y.; Chen, H.L.; Yang, G.F.; Yu, W.; Liu, Y.Z.; Liu, X.X.; Duan, C.F.; et al. CYP4A in Tumor-Associated Macrophages Promotes Pre-Metastatic Niche Formation and Metastasis. Oncogene 2017, 36, 5045–5057. [Google Scholar] [CrossRef]
- Ford, K.; Hanley, C.J.; Mellone, M.; Szyndralewiez, C.; Heitz, F.; Wiesel, P.; Wood, O.; Machado, M.; Lopez, M.-A.; Ganesan, A.-P.; et al. NOX4 Inhibition Potentiates Immunotherapy by Overcoming Cancer-Associated Fibroblast-Mediated CD8 T-Cell Exclusion from Tumors. Cancer Res. 2020, 80, 1846–1860. [Google Scholar] [CrossRef] [PubMed]
- Knaapen, A.M.; Güngör, N.; Schins, R.P.F.; Borm, P.J.A.; Van Schooten, F.J. Neutrophils and Respiratory Tract DNA Damage and Mutagenesis: A Review. Mutagenesis 2006, 21, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Andzinski, L.; Kasnitz, N.; Stahnke, S.; Wu, C.-F.; Gereke, M.; von Köckritz-Blickwede, M.; Schilling, B.; Brandau, S.; Weiss, S.; Jablonska, J. Type I IFNs Induce Anti-Tumor Polarization of Tumor Associated Neutrophils in Mice and Human. Int. J. Cancer 2016, 138, 1982–1993. [Google Scholar] [CrossRef] [PubMed]
- Schott, A.F.; Goldstein, L.J.; Cristofanilli, M.; Ruffini, P.A.; McCanna, S.; Reuben, J.M.; Perez, R.P.; Kato, G.; Wicha, M. Phase Ib Pilot Study to Evaluate Reparixin in Combination with Weekly Paclitaxel in Patients with HER-2-Negative Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 5358–5365. [Google Scholar] [CrossRef]
- Taucher, E.; Taucher, V.; Fink-Neuboeck, N.; Lindenmann, J.; Smolle-Juettner, F.-M. Role of Tumor-Associated Neutrophils in the Molecular Carcinogenesis of the Lung. Cancers 2021, 13, 5972. [Google Scholar] [CrossRef]
- Zhang, H.; Yue, X.; Chen, Z.; Liu, C.; Wu, W.; Zhang, N.; Liu, Z.; Yang, L.; Jiang, Q.; Cheng, Q.; et al. Define Cancer-Associated Fibroblasts (CAFs) in the Tumor Microenvironment: New Opportunities in Cancer Immunotherapy and Advances in Clinical Trials. Mol. Cancer 2023, 22, 159. [Google Scholar] [CrossRef]
- Eli Lilly and Company. A Phase 1b/2 Dose Escalation and Cohort Expansion Study of the Safety, Tolerability and Efficacy of a Novel Transforming Growth Factor-Beta Receptor I Kinase Inhibitor (Galunisertib) Administered in Combination With Anti-PD-1 (Nivolumab) in Advanced Refractory Solid Tumors (Phase 1b) and in Recurrent or Refractory Non-Small Cell Lung Cancer or Hepatocellular Carcinoma (Phase 2). 2021. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT02423343 (accessed on 28 January 2024).
- Peters, S.; Paz-Ares, L.; Herbst, R.S.; Reck, M. Addressing CPI Resistance in NSCLC: Targeting TAM Receptors to Modulate the Tumor Microenvironment and Future Prospects. J. ImmunoTherapy Cancer 2022, 10, e004863. Available online: https://jitc.bmj.com/content/10/7/e004863 (accessed on 28 October 2023). [CrossRef]
- Oliva, M.; Chepeha, D.; Araujo, D.V.; Diaz-Mejia, J.J.; Olson, P.; Prawira, A.; Spreafico, A.; Bratman, S.V.; Shek, T.; de Almeida, J.; et al. Antitumor Immune Effects of Preoperative Sitravatinib and Nivolumab in Oral Cavity Cancer: SNOW Window-of-Opportunity Study. J. Immunother. Cancer 2021, 9, e003476. [Google Scholar] [CrossRef] [PubMed]
- Leal, T.A.; Berz, D.; Rybkin, I.; Iams, W.T.; Bruno, D.; Blakely, C.; Spira, A.; Patel, M.R.; Waterhouse, D.M.; Richards, D.; et al. 1191O MRTX-500: Phase II Trial of Sitravatinib (Sitra) + Nivolumab (Nivo) in Patients (Pts) with Non-Squamous (NSQ) Non-Small Cell Lung Cancer (NSCLC) Progressing on or after Prior Checkpoint Inhibitor (CPI) Therapy. Ann. Oncol. 2021, 32, S949. [Google Scholar] [CrossRef]
- Borghaei, H.; de Marinis, F.; Dumoulin, D.; Reynolds, C.; Theelen, W.S.M.E.; Percent, I.; Gutierrez Calderon, V.; Johnson, M.L.; Madroszyk-Flandin, A.; Garon, E.B.; et al. SAPPHIRE: Phase III Study of Sitravatinib plus Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. Ann. Oncol. 2024, 35, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Neal, J.W.; Kundu, P.; Tanaka, T.; Enquist, I.; Patel, S.; Balestrini, A.; Wang, J.; Newsom-Davis, T.; Goto, Y.; Pavlakis, N.; et al. CONTACT-01: A Phase III, Randomized Study of Atezolizumab plus Cabozantinib versus Docetaxel in Patients with Metastatic Non-Small Cell Lung Cancer (mNSCLC) Previously Treated with PD-L1/PD-1 Inhibitors and Platinum-Containing Chemotherapy. JCO 2021, 39, TPS9134. [Google Scholar] [CrossRef]
- Neal, J.; Pavlakis, N.; Kim, S.-W.; Goto, Y.; Lim, S.M.; Mountzios, G.; Fountzilas, E.; Mochalova, A.; Christoph, D.C.C.; Bearz, A.; et al. 60 CONTACT-01: Efficacy and Safety from a Phase III Study of Atezolizumab (Atezo) + Cabozantinib (Cabo) vs Docetaxel (Doc) Monotherapy in Patients (Pts) with Metastatic NSCLC (mNSCLC) Previously Treated with Checkpoint Inhibitors and Chemotherapy. J. Thorac. Oncol. 2023, 18, S39–S40. [Google Scholar] [CrossRef]
- Santos Apolonio, J.; Lima de Souza Gonçalves, V.; Cordeiro Santos, M.L.; Silva Luz, M.; Silva Souza, J.V.; Rocha Pinheiro, S.L.; de Souza, W.R.; Sande Loureiro, M.; de Melo, F.F. Oncolytic Virus Therapy in Cancer: A Current Review. World J. Virol. 2021, 10, 229–255. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.-W.; Bell, J.C. Oncolytic Virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef]
- Guan, Y.; Liu, Y.; Zou, Q.; He, Q.; La, Z.; Yang, L.; Hu, Y. Adenovirus-Mediated Wild-Type P53 Gene Transfer in Combination with Bronchial Arterial Infusion for Treatment of Advanced Non-Small-Cell Lung Cancer, One Year Follow-Up. J. Zhejiang Univ. Sci. B 2009, 10, 331–340. [Google Scholar] [CrossRef]
- Wang, S.; Sun, J.; Chen, K.; Ma, P.; Lei, Q.; Xing, S.; Cao, Z.; Sun, S.; Yu, Z.; Liu, Y.; et al. Perspectives of Tumor-Infiltrating Lymphocyte Treatment in Solid Tumors. BMC Med. 2021, 19, 140. [Google Scholar] [CrossRef]
- Ratto, G.B.; Zino, P.; Mirabelli, S.; Minuti, P.; Aquilina, R.; Fantino, G.; Spessa, E.; Ponte, M.; Bruzzi, P.; Melioli, G. A Randomized Trial of Adoptive Immunotherapy with Tumor-Infiltrating Lymphocytes and Interleukin-2 versus Standard Therapy in the Postoperative Treatment of Resected Nonsmall Cell Lung Carcinoma. Cancer 1996, 78, 244–251. [Google Scholar] [CrossRef]
- Creelan, B.C.; Wang, C.; Teer, J.K.; Toloza, E.M.; Yao, J.; Kim, S.; Landin, A.M.; Mullinax, J.E.; Saller, J.J.; Saltos, A.N.; et al. Tumor-Infiltrating Lymphocyte Treatment for Anti-PD-1 Resistant Metastatic Lung Cancer: A Phase I Trial. Nat. Med. 2021, 27, 1410–1418. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Nokihara, H.; Yamamoto, A.; Goto, H.; Ogawa, H.; Kanematsu, T.; Miki, T.; Uehara, H.; Saijo, Y.; Nukiwa, T.; et al. Multifunctional Interleukin-1beta Promotes Metastasis of Human Lung Cancer Cells in SCID Mice via Enhanced Expression of Adhesion-, Invasion- and Angiogenesis-Related Molecules. Cancer Sci. 2003, 94, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Saijo, Y.; Tanaka, M.; Miki, M.; Usui, K.; Suzuki, T.; Maemondo, M.; Hong, X.; Tazawa, R.; Kikuchi, T.; Matsushima, K.; et al. Proinflammatory Cytokine IL-1 Beta Promotes Tumor Growth of Lewis Lung Carcinoma by Induction of Angiogenic Factors: In Vivo Analysis of Tumor-Stromal Interaction. J. Immunol. 2002, 169, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Ong, S.L.; Tran, L.M.; Jing, Z.; Liu, B.; Park, S.J.; Huang, Z.L.; Walser, T.C.; Heinrich, E.L.; Lee, G.; et al. Chronic IL-1β-Induced Inflammation Regulates Epithelial-to-Mesenchymal Transition Memory Phenotypes via Epigenetic Modifications in Non-Small Cell Lung Cancer. Sci. Rep. 2020, 10, 377. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, D.; Wauters, E.; Boeckx, B.; Aibar, S.; Nittner, D.; Burton, O.; Bassez, A.; Decaluwé, H.; Pircher, A.; Van den Eynde, K.; et al. Phenotype Molding of Stromal Cells in the Lung Tumor Microenvironment. Nat. Med. 2018, 24, 1277–1289. [Google Scholar] [CrossRef] [PubMed]
- Watari, K.; Shibata, T.; Kawahara, A.; Sata, K.; Nabeshima, H.; Shinoda, A.; Abe, H.; Azuma, K.; Murakami, Y.; Izumi, H.; et al. Tumor-Derived Interleukin-1 Promotes Lymphangiogenesis and Lymph Node Metastasis through M2-Type Macrophages. PLoS ONE 2014, 9, e99568. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Shapiro, B.; Vucic, E.A.; Vogt, S.; Bar-Sagi, D. Tumor Cell-Derived IL1β Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Res. 2020, 80, 1088–1101. [Google Scholar] [CrossRef]
- Sandler, A.; Gray, R.; Perry, M.C.; Brahmer, J.; Schiller, J.H.; Dowlati, A.; Lilenbaum, R.; Johnson, D.H. Paclitaxel-Carboplatin Alone or with Bevacizumab for Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2006, 355, 2542–2550. [Google Scholar] [CrossRef]
- Reck, M.; von Pawel, J.; Zatloukal, P.; Ramlau, R.; Gorbounova, V.; Hirsh, V.; Leighl, N.; Mezger, J.; Archer, V.; Moore, N.; et al. Phase III Trial of Cisplatin plus Gemcitabine with Either Placebo or Bevacizumab as First-Line Therapy for Nonsquamous Non-Small-Cell Lung Cancer: AVAil. J. Clin. Oncol. 2009, 27, 1227–1234. [Google Scholar] [CrossRef]
- Davies, A.M.; Chansky, K.; Lara, P.N.; Gumerlock, P.H.; Crowley, J.; Albain, K.S.; Vogel, S.J.; Gandara, D.R. Bortezomib plus Gemcitabine/Carboplatin as First-Line Treatment of Advanced Non-Small Cell Lung Cancer: A Phase II Southwest Oncology Group Study (S0339). J. Thorac. Oncol. 2009, 4, 87–92. [Google Scholar] [CrossRef]
- Ando, K.; Takahashi, F.; Kato, M.; Kaneko, N.; Doi, T.; Ohe, Y.; Koizumi, F.; Nishio, K.; Takahashi, K. Tocilizumab, a Proposed Therapy for the Cachexia of Interleukin6-Expressing Lung Cancer. PLoS ONE 2014, 9, e102436. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.L.; Soo, R.A.; Tan, D.S.; Lee, S.C.; Lim, J.S.; Marban, P.C.; Kong, L.R.; Lee, Y.J.; Wang, L.Z.; Thuya, W.L.; et al. Phase I and Biomarker Study of OPB-51602, a Novel Signal Transducer and Activator of Transcription (STAT) 3 Inhibitor, in Patients with Refractory Solid Malignancies. Ann. Oncol. 2015, 26, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Luo, J.; Ai, N.; Kim, R.; Ahn, L.; Biswas, A.; Coker, C.; Ma, W.; Wong, P.; Buonocore, D.J.; et al. Phase I Trial of the TNF-α Inhibitor Certolizumab plus Chemotherapy in Stage IV Lung Adenocarcinomas. Nat. Commun. 2022, 13, 6095. [Google Scholar] [CrossRef]
- Spigel, D.; Jotte, R.; Nemunaitis, J.; Shum, M.; Schneider, J.; Goldschmidt, J.; Eisenstein, J.; Berz, D.; Seneviratne, L.; Socoteanu, M.; et al. Randomized Phase 2 Studies of Checkpoint Inhibitors Alone or in Combination With Pegilodecakin in Patients With Metastatic NSCLC (CYPRESS 1 and CYPRESS 2). J Thorac Oncol 2021, 16, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Shukla, S. Limitations of Immunotherapy in Cancer. Cureus 2022, 14, e30856. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. Cancer Immunotherapy, Part 3: Challenges and Future Trends. Pharm. Ther. 2017, 42, 514–521. [Google Scholar]
- Chu, X.; Niu, L.; Xiao, G.; Peng, H.; Deng, F.; Liu, Z.; Wu, H.; Yang, L.; Tan, Z.; Li, Z.; et al. The Long-Term and Short-Term Efficacy of Immunotherapy in Non-Small Cell Lung Cancer Patients With Brain Metastases: A Systematic Review and Meta-Analysis. Front. Immunol. 2022, 13, 875488. [Google Scholar] [CrossRef]
- Wang, F.; Wang, S.; Zhou, Q. The Resistance Mechanisms of Lung Cancer Immunotherapy. Front. Oncol. 2020, 10, 568059. [Google Scholar] [CrossRef]
- Mamdani, H.; Matosevic, S.; Khalid, A.B.; Durm, G.; Jalal, S.I. Immunotherapy in Lung Cancer: Current Landscape and Future Directions. Front. Immunol. 2022, 13, 823618. [Google Scholar] [CrossRef]
- Hamilton, G.; Rath, B. Immunotherapy for Small Cell Lung Cancer: Mechanisms of Resistance. Expert. Opin. Biol. Ther. 2019, 19, 423–432. [Google Scholar] [CrossRef]
- Tostes, K.; Siqueira, A.P.; Reis, R.M.; Leal, L.F.; Arantes, L.M.R.B. Biomarkers for Immune Checkpoint Inhibitor Response in NSCLC: Current Developments and Applicability. Int. J. Mol. Sci. 2023, 24, 11887. [Google Scholar] [CrossRef] [PubMed]
- Puri, S.; Shafique, M. Combination Checkpoint Inhibitors for Treatment of Non-Small-Cell Lung Cancer: An Update on Dual Anti-CTLA-4 and Anti-PD-1/PD-L1 Therapies. Drugs Context 2020, 9, 2019-9-2. [Google Scholar] [CrossRef] [PubMed]
| Target | Treatment | Mechanism | Phase | Clinical Trial ID |
|---|---|---|---|---|
| CTLA-4 | Iplimumab | Immune Checkpoint Inhibitor | 3 | NCT00527735 |
| PD-1 | Nivolumab | Immune Checkpoint Inhibitor | 2 | NCT02998528 |
| EGFR | N/A | CAR T Cell Therapy | 2 | NCT01869166 |
| N/A | CAR T Cell Therapy | 1 | NCT0415379 | |
| TGF-β | Galunisertib | CAF Inhibition | 1b/2 | NCT02423343 |
| Receptor Tyrosine Kinase | Siravatinib, Nivolumab | TAM Inhibition | 3 | NCT03906071 |
| Tyrosine Kinase | Cabozantinib, Atezolizumab | TAM Inhibition | 3 | NCT04471428 |
| IL-1β | Canakinumbad, Pembrolizumab, Chemotherapy | IL-1β Inhibition | 3 | NCT03631199 |
| Canakinumbad, Chemotherapy | 3 | NCT03626545 | ||
| Canakinumbad, Pebrolizumab | 2 | NCT03968419 | ||
| Canakinumbad | 3 | NCT03447769 | ||
| Canakinumbad, PDR001 | 1b | NCT02900664 | ||
| Canakinumbad, PDR001+ | 1b | NCT03064854 | ||
| P53 Gene | N/A | Oncolytic Virus | 2 | NCT01574729 |
| N/A | N/A | TIL | 3 | N/A |
| N/A | N/A | TIL | 1 | NCT03215810 |
| NF-kB | Bevacimuzab | VEGF Inhibition | 3 | NCT00021060 |
| NF-kB | Bevacimuzab | VEGF Inhibition | 3 | NCT00806923 |
| NF-kB | Bortezomib | NF-kB Inhibition | 2 | NCT00075751 |
| IL-6 | Tocilizumab | IL-6 Antibody | 2 | NCT04940299 |
| IL-6 | Tocilizumab | IL-6 Antibody | 1/2 | NCT04691817 |
| STAT3 | OPB-51602 | STAT3 Inhibitor | 1 | NCT01184807) |
| STAT3 | Danvatirsen, Durvulamab | STAT3 Inhibitor | 2 | NCT02983578 |
| TNF-α | Certizolumab | TNF-α Inhibitor | 1 | NCT02120807 |
| IL-8 | BMS-986253 | IL-8 inhibitor | 2 | NCT04123379 |
| N/A | Pegilodecakin | Recombinant IL-10 | 2 | NCT03382899 |
| N/A | Pegilodecakin | Recombinant IL-10 | 2 | NCT03382912 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batrash, F.; Shaik, A.; Rauf, R.; Kutmah, M.; Zhang, J. Paracrine Regulation and Immune System Pathways in the Inflammatory Tumor Microenvironment of Lung Cancer: Insights into Oncogenesis and Immunotherapeutic Strategies. Cancers 2024, 16, 1113. https://doi.org/10.3390/cancers16061113
Batrash F, Shaik A, Rauf R, Kutmah M, Zhang J. Paracrine Regulation and Immune System Pathways in the Inflammatory Tumor Microenvironment of Lung Cancer: Insights into Oncogenesis and Immunotherapeutic Strategies. Cancers. 2024; 16(6):1113. https://doi.org/10.3390/cancers16061113
Chicago/Turabian StyleBatrash, Firas, Adnan Shaik, Rayaan Rauf, Mahmoud Kutmah, and Jun Zhang. 2024. "Paracrine Regulation and Immune System Pathways in the Inflammatory Tumor Microenvironment of Lung Cancer: Insights into Oncogenesis and Immunotherapeutic Strategies" Cancers 16, no. 6: 1113. https://doi.org/10.3390/cancers16061113
APA StyleBatrash, F., Shaik, A., Rauf, R., Kutmah, M., & Zhang, J. (2024). Paracrine Regulation and Immune System Pathways in the Inflammatory Tumor Microenvironment of Lung Cancer: Insights into Oncogenesis and Immunotherapeutic Strategies. Cancers, 16(6), 1113. https://doi.org/10.3390/cancers16061113