
A Phase I Study of the Oral Dual-Acting Pan-PI3K/mTOR Inhibitor Bimiralisib in Patients with Advanced Solid Tumors
 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Study Design
2.3. Assessments
2.4. Statistics
3. Results
3.1. Patient Demographics and Characteristics
3.2. Dose Escalation, DLTs, and MTD Determination
3.3. Pharmacokinetics of Bimiralisib
3.4. Safety of Bimiralisib
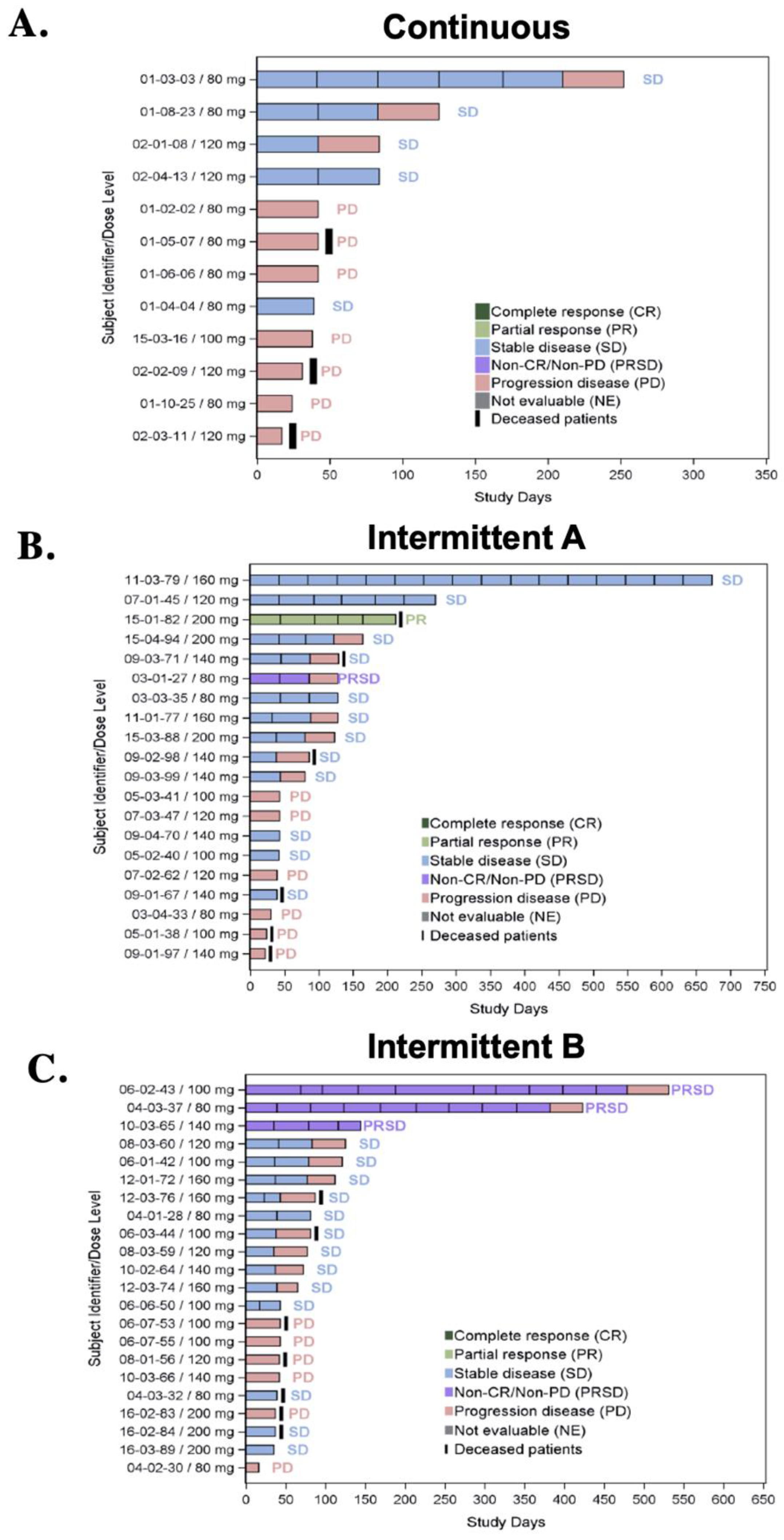
3.5. Efficacy
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Wang, Y.; Zhou, C.; Mei, W.; Zeng, C. PI3K/Akt/mTOR Pathway and Its Role in Cancer Therapeutics: Are We Making Headway? Front. Oncol. 2022, 12, 819128. [Google Scholar] [CrossRef] [PubMed]
- Castel, P.; Toska, E.; Engelman, J.A.; Scaltriti, M. The present and future of PI3K inhibitors for cancer therapy. Nat. Cancer 2021, 2, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Castel, P.; Toska, E.; Engelman, J.A.; Scaltriti, M. PI3K inhibitors are finally coming of age. Nat. Rev. Drug Discov. 2021, 20, 741–769. [Google Scholar]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef]
- Beaufils, F.; Cmiljanovic, N.; Cmiljanovic, V.; Bohnacker, T.; Melone, A.; Marone, R.; Jackson, E.; Zhang, X.; Sele, A.; Borsari, C.; et al. 5-(4,6-Dimorpholino-1,3,5-triazin-2-yl)-4-(trifluoromethyl)pyridin-2-amine (PQR309), a Potent, Brain-Penetrant, Orally Bioavailable, Pan-Class I PI3K/mTOR Inhibitor as Clinical Candidate in Oncology. J. Med. Chem. 2017, 60, 7524–7538. [Google Scholar] [CrossRef]
- Tarantelli, C.; Gaudio, E.; Arribas, A.J.; Kwee, I.; Hillmann, P.; Rinaldi, A.; Cascione, L.; Spriano, F.; Bernasconi, E.; Guidetti, F.; et al. PQR309 Is a Novel Dual PI3K/mTOR Inhibitor with Preclinical Antitumor Activity in Lymphomas as a Single Agent and in Combination Therapy. Clin. Cancer Res. 2018, 24, 120–129. [Google Scholar] [CrossRef]
- Yang, K.; Tang, X.J.; Xu, F.F.; Liu, J.H.; Tan, Y.Q.; Gao, L.; Sun, Q.; Ding, X.; Liu, B.H.; Chen, Q.X. PI3K/mTORC1/2 inhibitor PQR309 inhibits proliferation and induces apoptosis in human glioblastoma cells. Oncol. Rep. 2020, 43, 773–782. [Google Scholar] [CrossRef]
- Rodon, J.; Dienstmann, R.; Serra, V.; Tabernero, J. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 2013, 10, 143–153. [Google Scholar] [CrossRef]
- Tasian, S.K.; Teachey, D.T.; Rheingold, S.R. Targeting the PI3K/mTOR Pathway in Pediatric Hematologic Malignancies. Front. Oncol. 2014, 4, 108. [Google Scholar] [CrossRef]
- Bellmunt, J.; Szczylik, C.; Feingold, J.; Strahs, A.; Berkenblit, A. Temsirolimus safety profile and management of toxic effects in patients with advanced renal cell carcinoma and poor prognostic features. Ann. Oncol. 2008, 19, 1387–1392. [Google Scholar] [CrossRef]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A., 3rd; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in Postmenopausal Hormone-Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2011, 366, 520–529. [Google Scholar] [CrossRef]
- Furman, R.R.; Sharman, J.P.; Coutre, S.E.; Cheson, B.D.; Pagel, J.M.; Hillmen, P.; Barrientos, J.C.; Zelenetz, A.D.; Kipps, T.J.; Flinn, I.; et al. Idelalisib and Rituximab in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2014, 370, 997–1007. [Google Scholar] [CrossRef]
- Gopal, A.K.; Kahl, B.S.; de Vos, S.; Wagner-Johnston, N.D.; Schuster, S.J.; Jurczak, W.J.; Flinn, I.W.; Flowers, C.R.; Martin, P.; Viardot, A.; et al. PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N. Engl. J. Med. 2014, 370, 1008–1018. [Google Scholar] [CrossRef]
- Flinn, I.W.; Miller, C.B.; Ardeshna, K.M.; Tetreault, S.; Assouline, S.E.; Mayer, J.; Merli, M.; Lunin, S.D.; Pettitt, A.R.; Nagy, Z.; et al. DYNAMO: A Phase II Study of Duvelisib (IPI-145) in Patients With Refractory Indolent Non-Hodgkin Lymphoma. J. Clin. Oncol. 2019, 37, 912–922. [Google Scholar] [CrossRef]
- Rao, V.K.; Webster, S.; Šedivá, A.; Plebani, A.; Schuetz, C.; Shcherbina, A.; Conlon, N.; Coulter, T.; Dalm, V.A.; Trizzino, A.; et al. A randomized, placebo-controlled phase 3 trial of the PI3Kδ inhibitor leniolisib for activated PI3Kδ syndrome. Blood 2023, 141, 971–983. [Google Scholar] [CrossRef]
- Dreyling, M.; Santoro, A.; Mollica, L.; Leppä, S.; Follows, G.A.; Lenz, G.; Kim, W.S.; Nagler, A.; Panayiotidis, P.; Demeter, J.; et al. Phosphatidylinositol 3-Kinase Inhibition by Copanlisib in Relapsed or Refractory Indolent Lymphoma. J. Clin. Oncol. 2017, 35, 3898–3905. [Google Scholar] [CrossRef] [PubMed]
- Narayan, P.; Prowell, T.M.; Gao, J.J.; Fernandes, L.L.; Li, E.; Jiang, X.; Qiu, J.; Fan, J.; Song, P.; Yu, J.; et al. FDA Approval Summary: Alpelisib Plus Fulvestrant for Patients with HR-positive, HER2-negative, PIK3CA-mutated, Advanced or Metastatic Breast Cancer. Clin. Cancer Res. 2021, 27, 1842–1849. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.L.; Ditzel, H.J. Drugging the PI3K/AKT/mTOR Pathway in ER+ Breast Cancer. Int. J. Mol. Sci. 2023, 24, 4522. [Google Scholar] [CrossRef] [PubMed]
- Canaud, G.; Gutierrez, J.C.L.; Irvine, A.D.; Vabres, P.; Hansford, J.R.; Ankrah, N.; Branle, F.; Papadimitriou, A.; Ridolfi, A.; O’connell, P.; et al. Alpelisib for treatment of patients with PIK3CA-related overgrowth spectrum (PROS). Genet. Med. 2023, 25, 100969. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Oliveira, M.; Howell, S.J.; Dalenc, F.; Cortes, J.; Moreno, H.L.G.; Hu, X.; Jhaveri, K.; Krivorotko, P.; Loibl, S.; et al. Capivasertib in Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2023, 388, 2058–2070. [Google Scholar] [CrossRef]
- Mishra, R.; Patel, H.; Alanazi, S.; Kilroy, M.K.; Garrett, J.T. PI3K Inhibitors in Cancer: Clinical Implications and Adverse Effects. Int. J. Mol. Sci. 2021, 22, 3464. [Google Scholar] [CrossRef]
- Shaffer, A. PI3K Inhibitors Face Challenges in Hematologic Malignancies. Oncol. Live® 2022, 23, 35–39. [Google Scholar]
- Bohnacker, T.; Prota, A.E.; Beaufils, F.; Burke, J.E.; Melone, A.; Inglis, A.J.; Rageot, D.; Sele, A.M.; Cmiljanovic, V.; Cmiljanovic, N.; et al. Deconvolution of Buparlisib’s mechanism of action defines specific PI3K and tubulin inhibitors for therapeutic intervention. Nat. Commun. 2017, 8, 14683. [Google Scholar] [CrossRef]
- Wicki, A.; Brown, N.; Xyrafas, A.; Bize, V.; Hawle, H.; Berardi, S.; Cmiljanović, N.; Cmiljanović, V.; Stumm, M.; Dimitrijević, S.; et al. First-in human, phase 1, dose-escalation pharmacokinetic and pharmacodynamic study of the oral dual PI3K and mTORC1/2 inhibitor PQR309 in patients with advanced solid tumors (SAKK 67/13). Eur. J. Cancer 2018, 96, 6–16. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE) Version 4.0; U.S. Department of Health and Human Services: Washington, DC, USA, 2009.
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Gibaldi, M.; Perrier, D. Non-compartmental analysis based on statistical moment therapy. In Pharmacokinetics, 2nd ed.; Marcel Dekker: New York, NY, USA, 1982. [Google Scholar]
- Collins, G.P.; Eyre, T.A.; Schmitz-Rohmer, D.; Townsend, W.; Popat, R.; Giulino-Roth, L.; Fields, P.A.; Krasniqi, F.; Soussain, C.; Stathis, A.; et al. A Phase II Study to Assess the Safety and Efficacy of the Dual mTORC1/2 and PI3K Inhibitor Bimiralisib (PQR309) in Relapsed, Refractory Lymphoma. HemaSphere 2021, 5, e656. [Google Scholar] [CrossRef]
- Juric, D.; de Bono, J.S.; LoRusso, P.M.; Nemunaitis, J.; Heath, E.I.; Kwak, E.L.; Macarulla Mercadé, T.; Geuna, E.; Jose de Miguel-Luken, M.; Patel, C.; et al. A First-in-Human, Phase I, Dose-Escalation Study of TAK-117, a Selective PI3Kalpha Isoform Inhibitor, in Patients with Advanced Solid Malignancies. Clin. Cancer Res. 2017, 23, 5015–5023. [Google Scholar] [CrossRef]
- Johnson, F.M.; Janku, F.; Gouda, M.A.; Tran, H.T.; Kawedia, J.D.; Schmitz, D.; Streefkerk, H.; Lee, J.J.; Andersen, C.R.; Deng, D.; et al. Inhibition of the Phosphatidylinositol-3 Kinase Pathway Using Bimiralisib in Loss-of-Function NOTCH1-Mutant Head and Neck Cancer. Oncologist 2022, 27, 1004-e926. [Google Scholar] [CrossRef] [PubMed]
- Sambandam, V.; Frederick, M.J.; Shen, L.; Tong, P.; Rao, X.; Peng, S.; Singh, R.; Mazumdar, T.; Huang, C.; Li, Q.; et al. PDK1 Mediates NOTCH1-Mutated Head and Neck Squamous Carcinoma Vulnerability to Therapeutic PI3K/mTOR Inhibition. Clin. Cancer Res. 2019, 25, 3329–3340. [Google Scholar] [CrossRef]
 : Target lesion.
: Target lesion.
: Target lesion.
: Target lesion.

{kind=link}
{kind=link}
| Dose Level | Bimiralisib * |
|---|---|
| −1 ** | 60 mg daily |
| 1 (starting) | 80 mg daily |
| 2 | 120 mg daily |
| 3 | 140 mg daily |
| 4 | 160 mg daily |
| 5 | 180 mg daily |
| Intermittent Schedules A and B Dose Level | Bimiralisib * |
|---|---|
| 1 (starting) | MTD qd of continuous schedule |
| 2 | MTD qd + 20 mg |
| 3 | MTD qd + 40 mg |
| 4 | MTD qd + 60 mg |
| 5 | MTD qd + 80 mg |
| Characteristic | Continuous (n = 21) | Intermittent A (n = 25) | Intermittent B (n = 24) | Total (n = 70) |
|---|---|---|---|---|
| Median age, years (range) | 58 (41–78) | 56 (22–81) | 56 (19–83) | 57 (19–83) |
| Female, n (%) | 14 (66.7) | 17 (68) | 18 (75) | 49 (70) |
| Male, n (%) | 7 (33.3) | 8 (32) | 6 (25) | 21 (30) |
| Race, n (%) | ||||
| White | 20 (95.2) | 22 (88) | 23 (95.8) | 65 (92.9) |
| Black or African American | 1 (4.8) | 2 (8) | 0 (0) | 3 (4.3) |
| Asian | 0 (0) | 1 (4) | 1 (4.2) | 2 (2.9) |
| ECOG performance status, n (%) | ||||
| 0 | 9 (42.9) | 9 (36) | 9 (37.5) | 27 (38.6) |
| 1 | 12 (57.1) | 16 (64) | 15 (62.5) | 43 (61.4) |
| Smoking history, n (%) | ||||
| Yes | 11 (52.4) | 11 (44) | 12 (50) | 34 (48.6) |
| No | 10 (47.6) | 15 (56) | 12 (50) | 36 (51.4) |
| Disease primary diagnosis, n (%) | ||||
| Lung | 3 | 4 | 0 | 7 |
| Breast | 1 | 6 | 4 | 11 |
| Prostate/GU | 2 | 0 | 1 | 3 |
| Ovarian/Gynecologic | 5 | 5 | 4 | 14 |
| Gastroesophageal | 2 | 0 | 0 | 2 |
| Colorectal | 2 | 3 | 2 | 7 |
| Pancreatic | 1 | 0 | 0 | 1 |
| Anal | 1 | 1 | 1 | 3 |
| Sarcoma/other | 4 | 2 | 2 | 8 |
| Melanoma | 0 | 1 | 0 | 1 |
| Lymphoma | 0 | 2 | 0 | 2 |
| GBM | 0 | 0 | 1 | 1 |
| Disease stage at diagnosis, n (%) | ||||
| Stage III | 4 (19) | 4 (16) | 2 (8.4) | 10 (14.3) |
| Stage IV | 4 (19) | 7 (28) | 6 (25) | 17 (24.3) |
| Disease stage at study entry, n (%) | ||||
| Stage III/IV | 3 (14.3) | 7 (28) | 1 (4.2) | 11 (15.7) |
| Recurrent | 0 (0) | 4 (16) | 5 (20.8) | 9 (12.9) |
| Metastatic | 18 (85.7) | 14 (56) | 18 (75) | 50 (71.4) |
| Prior therapy, n (%) | ||||
| Prior surgery or non-radiation procedure | 19 (90.5) | 18 (72) | 15 (62.5) | 42 (60) |
| Prior radiation | 10 (47.6) | 16 (64) | 17 (70.8) | 43 (61.4) |
| Prior antineoplastic therapy | ||||
| 1 | 1 (4.8) | 4 (16) | 3 (12.5) | 8 (11.4) |
| 2 | 2 (8.5) | 2 (8) | 4 (16.7) | 8 (11.4) |
| ≥3 | 18 (85.7) | 19 (76) | 17 (70.8) | 54 (77.2) |
| Best response to last prior antineoplastic therapy, n (%) | ||||
| Partial response | 0 (0) | 6 (24) | 3 (12.5) | 9 (12.9) |
| Stable disease | 9 (42.9) | 6 (24) | 3 (12.5) | 18 (25.7) |
| Progressive disease | 12 (57.1) | 11 (44) | 12 (50) | 35 (50.0) |
| Unknown | 0 | 2 (8) | 6 (25) | 8 (11.4) |
| AE, n (%) | Continuous (n = 21) | Intermittent A (n = 25) | Intermittent B (n = 24) | Total n = 70 |
|---|---|---|---|---|
| Any TEAE | 21 (100) | 24 (96) | 24 (100) | 69 (98.9) |
| Any drug-related AE | 21 (100) | 22 (88) | 21 (88) | 63 (91) |
| Common TEAE (≥10% of patients) | ||||
| Nausea | 12 (57.1) | 14 (56) | 15 (62.5) | 41 (58.6) |
| Fatigue | 13 (61.9) | 12 (48) | 10 (41.7) | 35 (50.0) |
| Hyperglycemia | 16 (76.2) | 6 (24) | 9 (37.5) | 31 (44.3) |
| Decreased appetite | 11 (52.4) | 9 (36) | 8 (33.3) | 28 (40.0) |
| Diarrhea | 10 (47.6) | 11 (44) | 7 (29.2) | 28 (40.0) |
| Vomiting | 10 (47.6) | 5 (20) | 7 (29.2) | 22 (31.4) |
| Weight decrease | 11 (52.4) | 4 (16) | 3 (12.5) | 18 (25.7) |
| Dry Mouth | 10 (47.6) | 3 (12) | 4 (16.7) | 17 (24.3) |
| Anxiety | 5 (23.8) | 5 (20) | 6 (25) | 16 (22.9) |
| Constipation | 3 (14.3) | 3 (12) | 8 (33.3) | 14 (20.0) |
| Rash | 4 (19.1) | 5 (20) | 5 (20.8) | 14 (20.0) |
| Back pain | 4 (19.1) | 4 (16) | 5 (20.8) | 13 (18.6) |
| Disturbance attention | 11 (52.4) | 1 (4) | 1 (4.2) | 13 (18.6) |
| Pruritus | 6 (28.6) | 2 (8) | 5 (20.8) | 13 (18.6) |
| AST elevation | 3 (14.3) | 4 (16) | 5 (20.8) | 12 (17.1) |
| Asthenia | 6 (28.6) | 2 (8) | 4 (16.7) | 12 (17.1) |
| Disease progression | 3 (14.3) | 5 (20) | 3 (12.5) | 11 (15.7) |
| Dry skin | 7 (33.3) | 3 (12) | 1 (4.2) | 11 (15.7) |
| Headache | 0 (0) | 3 (12) | 7 (29.2) | 10 (14.3) |
| Hyperinsulinemia | 7 (33.3) | 1 (4) | 1 (4.2) | 9 (12.9) |
| Hypokalemia | 6 (28.6) | 1 (4) | 2 (8.3) | 9 (12.9) |
| Abdominal pain | 1 (4.76) | 4 (16) | 3 (12.5) | 8 (11.4) |
| Anemia | 0 (0) | 3 (12) | 5 (20) | 8 (11.4) |
| Dyspepsia | 1 (4.76) | 3 (12) | 4 (16.7) | 8 (11.4) |
| ALT elevation | 1 (4.76) | 3 (12) | 3 (12.5) | 7 (10) |
| Cough | 2 (9.5) | 3 (12) | 2 (8.3) | 7 (10) |
| Dehydration | 6 (28.6) | 1 (4) | 0 (0) | 7 (10) |
| Depression | 5 (23.8) | 0 (0) | 2 (8.3) | 7 (10) |
| Dizziness | 3 (14.3) | 1 (4) | 3 (12.5) | 7 (10) |
| Dyspnea | 4 (19.1) | 2 (8) | 1 (4.2) | 7 (10) |
| Muscular weakness | 3 (14.3) | 0 (0) | 4 (16.7) | 7 (10) |
| Peripheral edema | 3 (14.3) | 2 (8) | 2 (8.3) | 7 (10) |
| Tachycardia | 4 (19.1) | 2 (8) | 1 (4.2) | 7 (10) |
| Dysgeusia | 4 (19.1) | 0 (0) | 2 (8.3) | 6 (8.6) |
| Irritability | 0 (0) | 3 (12) | 2 (8.3) | 5 (7.1) |
| Upper respiratory tract infection | 1 (4.76) | 3 (12) | 1 (4.2) | 5 (7.1) |
| Cachexia | 4 (19.1) | 0 (0) | 0 (0) | 4 (5.7) |
| Hyperkalemia | 0 (0) | 3 (12) | 1 (4.2) | 4 (5.7) |
| Hyperphosphatasemia | 1 (4.76) | 3 (12) | 0 (0) | 4 (5.7) |
| Malaise | 4 (19.1) | 0 (0) | 0 (0) | 4 (5.7) |
| Ascites | 0 (0) | 3 (12) | 0 (0) | 3 (4.3) |
| Confusional state | 3 (14.3) | 0 (0) | 0 (0) | 3 (4.3) |
| Early satiety | 3 (14.3) | 0 (0) | 0 (0) | 3 (4.3) |
| Hallucination | 3 (14.3) | 0 (0) | 0 (0) | 3 (4.3) |
| Mucosal Inflammation | 0 (0) | 3 (12) | 0 (0) | 3 (4.3) |
| Neck pain | 0 (0) | 0 (0) | 3 (12.5) | 3 (4.3) |
| Any grade ≥ 3 TEAE | 17 (81) | 14 (56) | 16 (66.7) | 47 (67.1) |
| Grade ≥ 3 TEAEs (≥5% of patients) | ||||
| Hyperglycemia | 6 (28.6) | 3 (12) | 3 (12.5) | 12 (17.1) |
| Fatigue | 7 (33.3) | 0 (0) | 2 (8.3) | 9 (12.9) |
| AST elevation | 3 (14.3) | 1 (4) | 1 (4.2) | 4 (5.7) |
| ALT elevation | 1 (4.8) | 1 (4) | 2 (8.3) | 4 (5.7) |
| Dehydration | 4 (19) | 0 (0) | 0 (0) | 4 (5.7) |
| Any serious AE | 9 (43) | 8 (32) | 11 (46) | 28 (40) |
| Any drug-related serious AE | 2 (10) | 0 (0) | 2 (8) | 4 (5.7) |
| Treatment-related death | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Any drug-related grade ≥ 3 AE | 11 (52.4) | 5 (20) | 10 (41.7) | 26 (37.1) |
| Dosing Schedule | Dose Level | Patients Enrolled | No. of Patients with ≥1 DLT Events | Grade of DLT | TEAE Resulting in Dose Interruption | TEAE Resulting in Dose Reduction |
|---|---|---|---|---|---|---|
| Continuous Dosing | 80 mg | 11 * | 0 (0%) | NA | 4 (36%) | 0 (0%) |
| 100 mg | 5 | 1 (20%) | Grade 3 fatigue | 3 (60%) | 1 (20%) | |
| 120 mg | 5 | 3 (60%) | Grade 3 fatigue | 4 (80%) | 1 (20%) | |
| Total per schedule | 21 | 4 (19%) | NA | 11 (52%) | 2 (10%) | |
| Intermittent Schedule A | 80 mg | 4 | 0 (0%) | NA | 1 (25%) | 0 (0%) |
| 100 mg | 3 | 0 (0%) | NA | 1 (33%) | 0 (0%) | |
| 120 mg | 4 | 0 (0%) | NA | 1 (25%) | 0 (0%) | |
| 140 mg | 6 | 0 (0%) | NA | 2 (33%) | 0 (0%) | |
| 160 mg | 3 | 0 (0%) | NA | 1 (33%) | 0 (0%) | |
| 200 mg | 5 | 0 (0%) | NA | 3 (60%) | 0 (0%) | |
| Total per schedule | 21 | 0 (0%) | NA | 9 (36%) | 0 (0%) | |
| Intermittent Schedule B | 80 mg | 4 | 0 (0%) | NA | 2 (50%) | 0 (0%) |
| 100 mg | 7 | 1 (14.3%) | Grade 4 Lipase elevation | 3 (43%) | 0 (0%) | |
| 120 mg | 3 | 0 (0%) | NA | 0 (0%) | 0 (0%) | |
| 140 mg | 3 | 0 (0%) | NA | 2 (67%) | 1 (33%) | |
| 160 mg | 3 | 0 (0%) | NA | 1 (33%) | 0 (0%) | |
| 200 mg | 4 | 0 (0%) | NA | 1 (25%) | 0 (0%) | |
| Total per schedule: | 24 | 1 (4.2%) | NA | 9 (38%) | 1 (4%) |
| AE, n (%) | Continuous (n = 21) | Intermittent A (n = 25) | Intermittent B (n = 24) | Total n = 70 |
|---|---|---|---|---|
| Patients discontinuing study due to AE | 5 (24.0%) | 1 (4.0%) | 3 (12.5%) | 9 (12.9%) |
| Fatigue | 3 (14.3%) | 1 (4.2%) | 4 (5.7%) | |
| ALT Increased | 2 (8.3%) | 2 (2.8%) | ||
| Dehydration | 2 (9.5%) | 2 (2.8%) | ||
| AST Increased | 1 (4.2%) | 1 (1.4%) | ||
| Ejection Fraction decreased | 1 (4.8%) | 1 (1.4%) | ||
| Musculoskeletal chest pain | 1 (4.2%) | 1 (1.4%) | ||
| Myalgia | 1 (4.2%) | 1 (1.4%) | ||
| Polyuria | 1 (4.2%) | 1 (1.4%) | ||
| Pollakiuria | 1 (4.2%) | 1 (1.4%) | ||
| Renal Failure | 1 (4.0%) | 1 (1.4%) | ||
| Tumor pain | 1 (4.8%) | 1 (1.4%) | ||
| Urinary Tract Infection | 1 (4.0%) | 1 (1.4%) |
| Dose Level (mg) | Period | Patients (n) | Tmax (h) Median, (Range) | Cmax, (ng/mL) Mean (%CV) | AUC0–8, (h·ng/mL) Geometric Mean (%CV) | AUC0–24, (h·ng/mL) Geometric Mean (%CV) |
|---|---|---|---|---|---|---|
| Continuous Dosing | ||||||
| 80 | Day -3 | 11 | 2 (0.5–24) | 407 (47) | 1530 (48) | 4290 (50) |
| Day 15 | 7 | 1 (0.5–8) | 766 (60) | 4090 (64) | NA | |
| 100 | Day 1 | 5 | 1.7 (1–8) | 441 (49) | 1690 (29) | 4660 (24) |
| Day 15 | 2 | 2 (2–2) | 1110 (ND) | 5750 (ND) | NA | |
| 120 | Day 1 | 5 | 2 (1–48) | 876 (79) | 2990 (65) | 7600 (52) |
| Day 15 | 1 | 8 | 982 (ND) | 6430 (ND) | NA | |
| Intermittent Dose Schedule A | ||||||
| 80 | Day -3 | 4 | 1.5 (1–2) | 410 (50) | 1580 (53) | 3540 (59) |
| Day 8 | 3 | 1 (1–1) | 614 (47) | 2350 (10) | 6740 (n = 1) | |
| 100 | Day -3 | 4 | 1 (1–1) | 543 (70) | 1970 (59) | 4170 (52) |
| Day 8 | 3 | 1 (1–2) | 966 (10) | 3910 (22) | 12,400 (n = 2) | |
| 120 | Day -3 | 4 | 3 (1–4) | 549 (44) | 2590 (54) | 5590 (62) |
| Day 8 | 3 | 1 (1–24) | 872 (79) | 3710 (78) | 8940 (69) | |
| 140 | Day -3 | 6 | 1 (1–4) | 709 (60) | 2580 (48) | 5810 (42) |
| Day 8 | 6 | 1 (0.5–2) | 923 (48) | 3220 (28) | 7590 (30; n = 3) | |
| 160 | Day -3 | 3 | 1 (1–2) | 539 (25) | 2480 (16) | 5830 (20) |
| Day 8 | 3 | 2 (1–2) | 857 (28) | 4080 (41) | 11,100 (64) | |
| 200 | Day -3 | 4 | 2 (1–2) | 1030 (34) | 3480 (37) | 7050 (37) |
| Day 8 | 4 | 2 (1–4) | 963 (61) | 4460 (56) | 15,900 (n = 2) | |
| Intermittent Dose Schedule B | ||||||
| 80 | Day 1 | 4 | 3 (1–6) | 341 (17) | 1680 (40) | 4000 (55) |
| Day 8 | 4 | 1.5 (1–8) | 681 (30) | 2600 (34) | 6350 (44; n = 3) | |
| 100 | Day 1 | 7 | 2 (0.5–8) | 562 (98) | 1920 (67) | 4520 (54) |
| Day 8 | 6 | 1.5 (0.5–6) | 714 (69) | 2880 (54) | 6200 (67; n = 4) | |
| 120 | Day 1 | 3 | 1 (1–2) | 471 (35) | 1530 (5) | 3340 (4) |
| Day 8 | 3 | 1 (1–8) | 594 (34) | 2210 (8) | 5850 (24) | |
| 140 | Day 1 | 3 | 1 (0.5–1) | 1010 (37) | 4150 (63) | 9740 (68) |
| Day 8 | 3 | 1 (1–2) | 1560 (30) | 5160 (44) | 12,600 (n = 2) | |
| 160 | Day 1 | 3 | 12 (1–12) | 317 (63) | 1050 (54) | 3700 (29) |
| Day 8 | 3 | 8 (2–22.3) | 325 (83) | 1770 (97) | ND | |
| 200 | Day 1 | 4 | 1.5 (1–8) | 734 (66) | 3190 (72) | 8010 (68) |
| Day 8 | 4 | 1.5 (1–6) | 1200 (46) | 5800 (38) | 14,700 (58; n = 3) | |
| Best Overall Response (BOR) | Treatment Group/Number of Patients | |||
|---|---|---|---|---|
| Continuous | Intermittent A | Intermittent B | All Schedules | |
| n = 21 | n = 25 | n = 24 | n = 70 | |
| Complete response (CR) | 0 | 0 | 0 | 0 |
| Partial response | 0 | 1 (4%) | 0 | 1 (1.4%) |
| Stable disease | 5 (23.8%) | 12 (48%) | 13 (54.2%) | 30 (43%) |
| Non-CR/Non-PD | 0 | 1 (4%) | 3 (12.5%) | 4 (5.7%) |
| Progressive disease (PD) | 7 (33.3%) | 6 (24%) | 6 (25%) | 19 (27.1%) |
| Missing | 9 (42.9%) | 5 (20%) | 2 (8.3%) | 16 (22.9%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janku, F.; Choong, G.M.; Opyrchal, M.; Dowlati, A.; Hierro, C.; Rodon, J.; Wicki, A.; Forster, M.D.; Blagden, S.P.; Yin, J.; et al. A Phase I Study of the Oral Dual-Acting Pan-PI3K/mTOR Inhibitor Bimiralisib in Patients with Advanced Solid Tumors. Cancers 2024, 16, 1137. https://doi.org/10.3390/cancers16061137
Janku F, Choong GM, Opyrchal M, Dowlati A, Hierro C, Rodon J, Wicki A, Forster MD, Blagden SP, Yin J, et al. A Phase I Study of the Oral Dual-Acting Pan-PI3K/mTOR Inhibitor Bimiralisib in Patients with Advanced Solid Tumors. Cancers. 2024; 16(6):1137. https://doi.org/10.3390/cancers16061137
Chicago/Turabian StyleJanku, Filip, Grace M. Choong, Mateusz Opyrchal, Afshin Dowlati, Cinta Hierro, Jordi Rodon, Andreas Wicki, Martin D. Forster, Sarah P. Blagden, Jun Yin, and et al. 2024. "A Phase I Study of the Oral Dual-Acting Pan-PI3K/mTOR Inhibitor Bimiralisib in Patients with Advanced Solid Tumors" Cancers 16, no. 6: 1137. https://doi.org/10.3390/cancers16061137