
Sperm-Associated Antigen 5 Knockout Reduces Doxorubicin and Docetaxel Resistance in Triple-Negative Breast Cancer MDA-MB-231 and BT549 Cells



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Transfection and Isolation of SPAG5 Gene Knockout Clones
2.3. DNA Extraction, PCR and Sequencing
2.4. Western Blotting
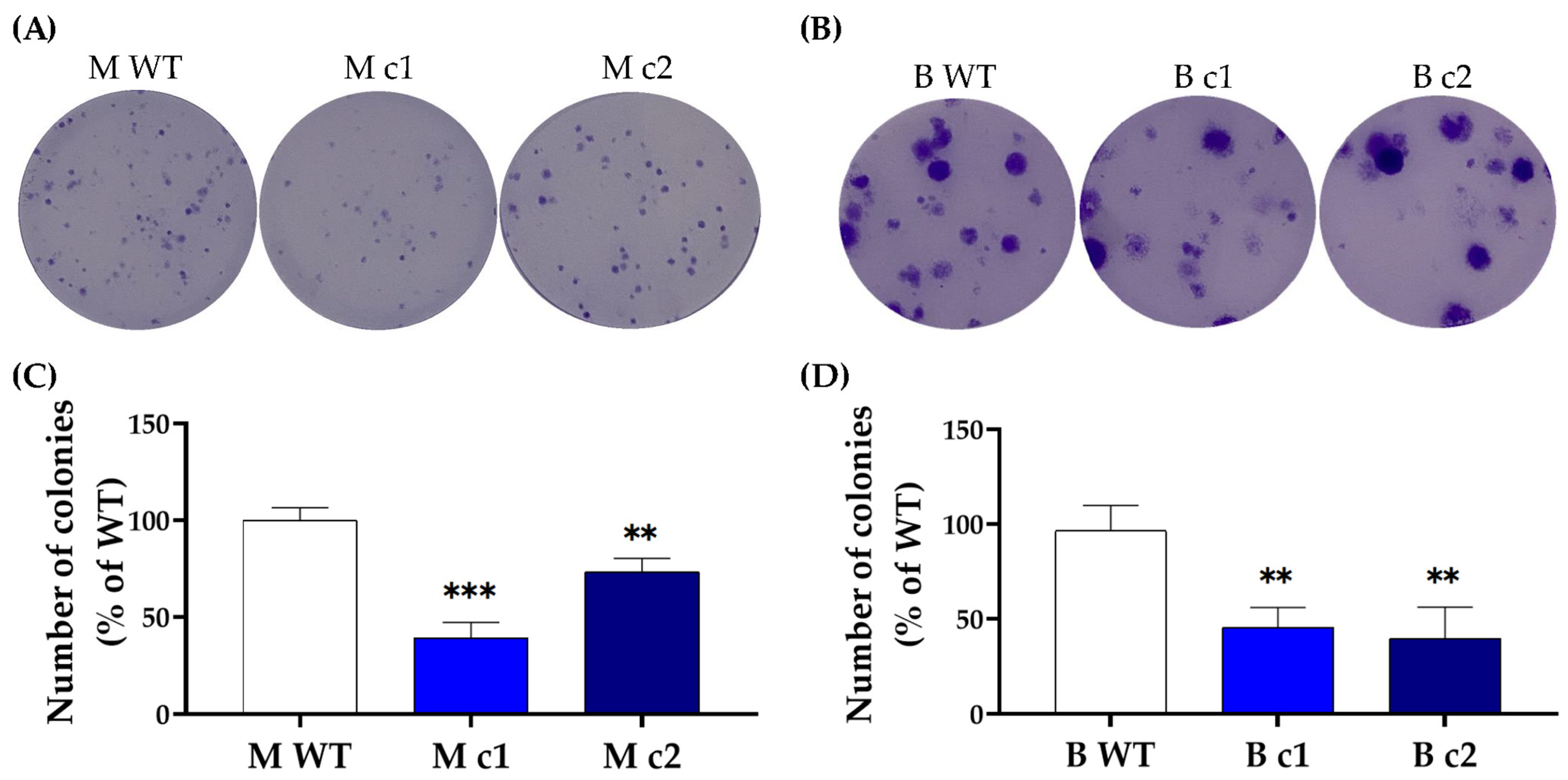
2.5. Colony-Formation Assay
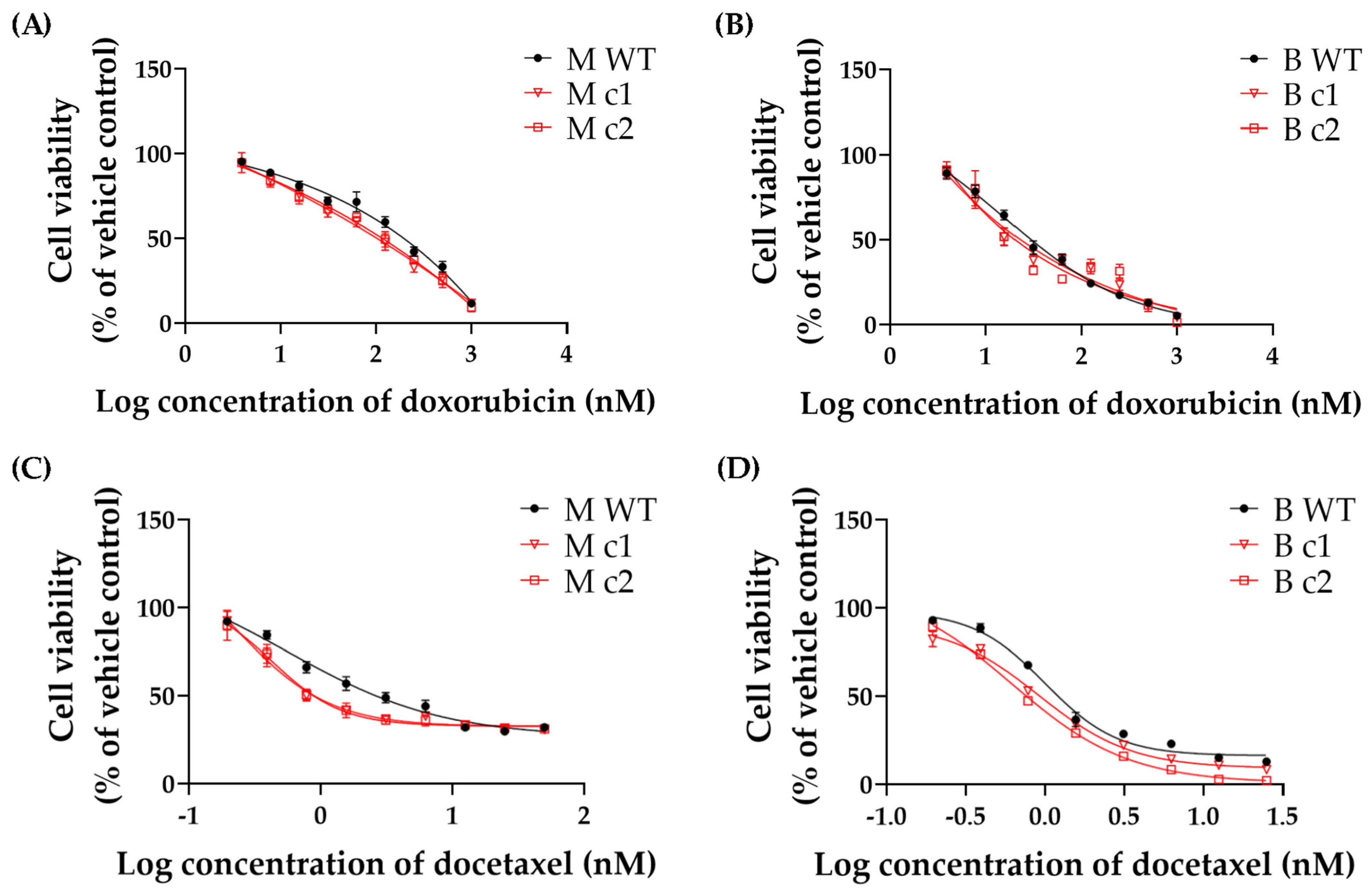
2.6. Cytotoxicity Assay
2.7. Caspase 3/7 Activity Analysis
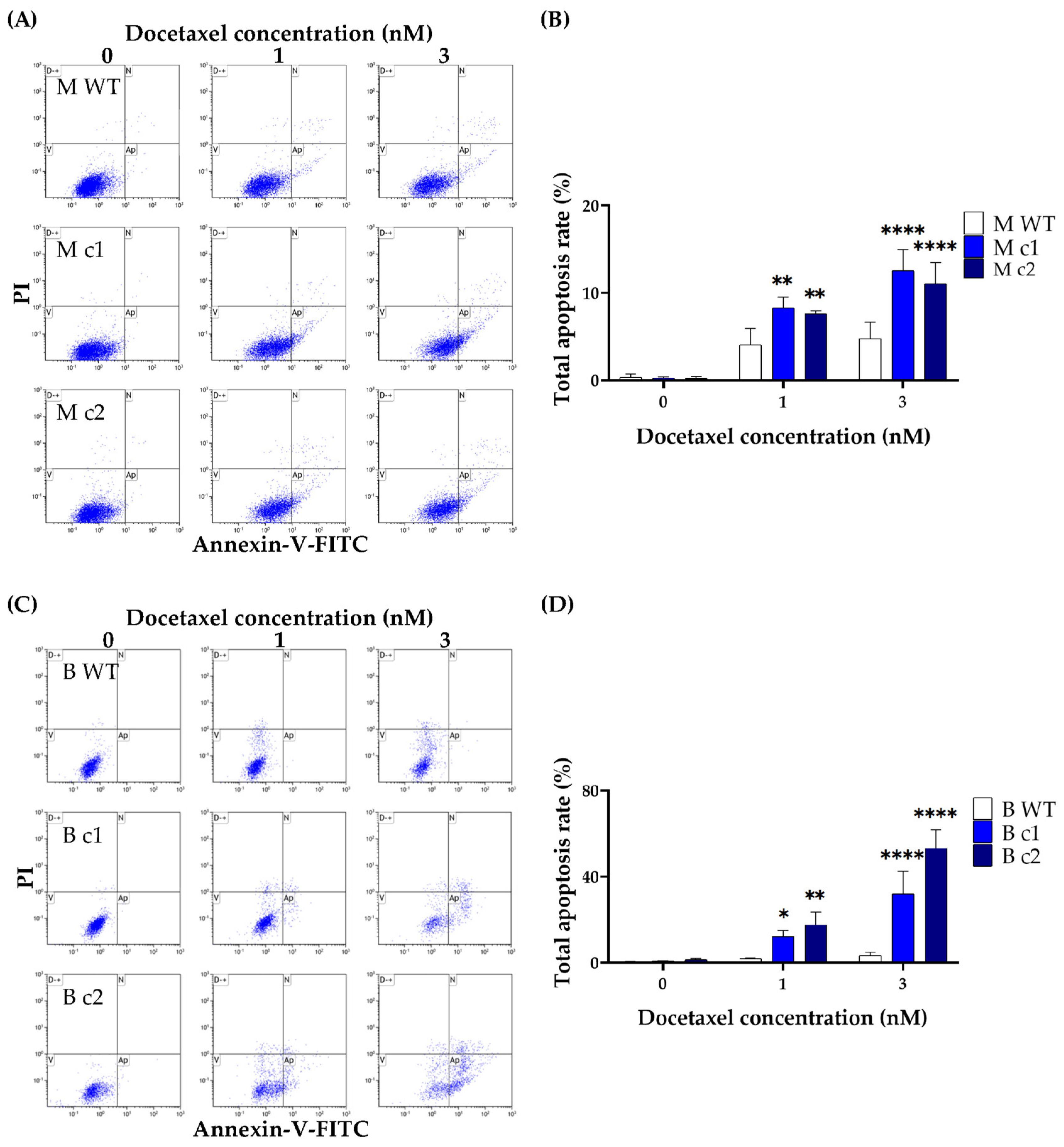
2.8. Apoptosis Detection
2.9. Mitochondrial Membrane Potential Assay
2.10. Statistical Analysis
3. Results
3.1. Deletion of SPAG5 Expression in SPAG5 Knockout Clones
3.2. SPAG5 Knockout Sensitised TNBC Cells to Doxorubicin- and Docetaxel-Induced Growth Inhibition
3.3. SPAG5 Knockout Increased Doxorubicin- and Docetaxel-Induced Apoptosis Rate
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chang, M.S.; Huang, C.J.; Chen, M.L.; Chen, S.T.; Fan, C.C.; Chu, J.M.; Lin, W.C.; Yang, Y.C. Cloning and characterization of hMAP126, a new member of mitotic spindle-associated proteins. Biochem. Biophys. Res. Commun. 2001, 287, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Mack, G.J.; Compton, D.A. Analysis of mitotic microtubule-associated proteins using mass spectrometry identifies astrin, a spindle-associated protein. Proc. Natl. Acad. Sci. USA 2001, 98, 14434–14439. [Google Scholar] [CrossRef] [PubMed]
- Thein, K.H.; Kleylein-Sohn, J.; Nigg, E.A.; Gruneberg, U. Astrin is required for the maintenance of sister chromatid cohesion and centrosome integrity. J. Cell Biol. 2007, 178, 345–354. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Green, A.R.; Li, Y.; Chan, S.Y.T.; Liu, D.X. SPAG5: An Emerging Oncogene. Trends Cancer 2020, 6, 543–547. [Google Scholar] [CrossRef]
- Yuan, L.J.; Li, J.D.; Zhang, L.; Wang, J.H.; Wan, T.; Zhou, Y.; Tu, H.; Yun, J.P.; Luo, R.Z.; Jia, W.H.; et al. SPAG5 upregulation predicts poor prognosis in cervical cancer patients and alters sensitivity to taxol treatment via the mTOR signaling pathway. Cell Death Dis. 2014, 5, e1247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, S.; Yang, X.; Qiao, B.; Zhang, Z.; Xu, Y. miR-539 inhibits prostate cancer progression by directly targeting SPAG5. J. Exp. Clin. Cancer Res. 2016, 35, 60. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Hu, J.; Wei, R.; Zhou, L.; Pan, H.; Zhu, H.; Huang, M.; Luo, J.; Xu, W. SPAG5 promotes hepatocellular carcinoma progression by downregulating SCARA5 through modifying beta-catenin degradation. J. Exp. Clin. Cancer Res. 2018, 37, 229. [Google Scholar] [CrossRef]
- Liu, J.Y.; Zeng, Q.H.; Cao, P.G.; Xie, D.; Yang, F.; He, L.Y.; Dai, Y.B.; Li, J.J.; Liu, X.M.; Zeng, H.L.; et al. SPAG5 promotes proliferation and suppresses apoptosis in bladder urothelial carcinoma by upregulating Wnt3 via activating the AKT/mTOR pathway and predicts poorer survival. Oncogene 2018, 37, 3937–3952. [Google Scholar] [CrossRef]
- Abdel-Fatah, T.M.A.; Agarwal, D.; Liu, D.X.; Russell, R.; Rueda, O.M.; Liu, K.; Xu, B.; Moseley, P.M.; Green, A.R.; Pockley, A.G.; et al. SPAG5 as a prognostic biomarker and chemotherapy sensitivity predictor in breast cancer: A retrospective, integrated genomic, transcriptomic, and protein analysis. Lancet Oncol. 2016, 17, 1004–1018. [Google Scholar] [CrossRef]
- Li, X.A.; Moughan, J.; White, J.R.; Freedman, G.M.; Arthur, D.W.; Galvin, J.; Xiao, Y.; McNulty, S.; Lyons, J.A.; Kavadi, V.S.; et al. Patterns of Failure Observed in the 2-Step Institution Credentialing Process for NRG Oncology/Radiation Therapy Oncology Group 1005 (NCT01349322) and Lessons Learned. Pract. Radiat. Oncol. 2020, 10, 265–273. [Google Scholar] [CrossRef]
- Thedieck, K.; Holzwarth, B.; Prentzell, M.T.; Boehlke, C.; Klasener, K.; Ruf, S.; Sonntag, A.G.; Maerz, L.; Grellscheid, S.N.; Kremmer, E.; et al. Inhibition of mTORC1 by astrin and stress granules prevents apoptosis in cancer cells. Cell 2013, 154, 859–874. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Hsu, Y.T.; Wu, C.C.; Chen, H.T.; Chang, M.S. Silencing of astrin induces the p53-dependent apoptosis by suppression of HPV18 E6 expression and sensitizes cells to paclitaxel treatment in HeLa cells. Biochem. Biophys. Res. Commun. 2006, 343, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ginther, C.; Kim, J.; Mosher, N.; Chung, S.; Slamon, D.; Vadgama, J.V. Expression of Wnt3 activates Wnt/beta-catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells. Mol. Cancer Res. 2012, 10, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.F.; Zhang, M.F.; Tian, Q.H.; Fu, J.; Yang, X.; Zhang, C.Z.; Yang, H. SPAG5 interacts with CEP55 and exerts oncogenic activities via PI3K/AKT pathway in hepatocellular carcinoma. Mol. Cancer 2018, 17, 117. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Severson, E.; Pignon, J.C.; Zhao, H.; Li, T.; Novak, J.; Jiang, P.; Shen, H.; Aster, J.C.; Rodig, S.; et al. Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 2016, 17, 174. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Menyhart, O.; Gyorffy, B.; He, X. The prognostic association of SPAG5 gene expression in breast cancer patients with systematic therapy. BMC Cancer 2019, 19, 1046. [Google Scholar] [CrossRef]
- Li, M.; Li, A.; Zhou, S.; Lv, H.; Yang, W. SPAG5 upregulation contributes to enhanced c-MYC transcriptional activity via interaction with c-MYC binding protein in triple-negative breast cancer. J. Hematol. Oncol. 2019, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wang, J.; He, X.; Ma, W.; Sun, L.; Zhou, Q.; Li, M.; Yu, S. High expression of SPAG5 sustains the malignant growth and invasion of breast cancer cells through the activation of Wnt/beta-catenin signalling. Clin. Exp. Pharmacol. Physiol. 2019, 46, 597–606. [Google Scholar] [CrossRef]
- Canu, V.; Donzelli, S.; Sacconi, A.; Lo Sardo, F.; Pulito, C.; Bossel, N.; Di Benedetto, A.; Muti, P.; Botti, C.; Domany, E.; et al. Aberrant transcriptional and post-transcriptional regulation of SPAG5, a YAP-TAZ-TEAD downstream effector, fuels breast cancer cell proliferation. Cell Death Differ. 2021, 28, 1493–1511. [Google Scholar] [CrossRef]
- Yu, J.S.L.; Yusa, K. Genome-wide CRISPR-Cas9 screening in mammalian cells. Methods 2019, 164–165, 29–35. [Google Scholar] [CrossRef]
- He, J.; Biswas, R.; Bugde, P.; Li, J.; Liu, D.X.; Li, Y. Application of CRISPR-Cas9 System to Study Biological Barriers to Drug Delivery. Pharmaceutics 2022, 14, 894. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.; Pereira, A.M.; Lopes, A.L.; Coimbra, S. The best CRISPR/Cas9 versus RNA interference approaches for Arabinogalactan proteins’ study. Mol. Biol. Rep. 2020, 47, 2315–2325. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, M.; McManus, M.T. Choosing the Right Tool for the Job: RNAi, TALEN, or CRISPR. Mol. Cell 2015, 58, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Janes, K.A. An analysis of critical factors for quantitative immunoblotting. Sci. Signal 2015, 8, rs2. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Ma, Q.; Sun, S.; Li, N.; Wang, H.; Ying, Z.; Ke, S. Exon-intron boundary inhibits m(6)A deposition, enabling m(6)A distribution hallmark, longer mRNA half-life and flexible protein coding. Nat. Commun. 2023, 14, 4172. [Google Scholar] [CrossRef] [PubMed]
- Carey, J.P.W.; Karakas, C.; Bui, T.; Chen, X.; Vijayaraghavan, S.; Zhao, Y.; Wang, J.; Mikule, K.; Litton, J.K.; Hunt, K.K.; et al. Synthetic Lethality of PARP Inhibitors in Combination with MYC Blockade Is Independent of BRCA Status in Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 742–757. [Google Scholar] [CrossRef] [PubMed]
- Mosca, L.; Ilari, A.; Fazi, F.; Assaraf, Y.G.; Colotti, G. Taxanes in cancer treatment: Activity, chemoresistance and its overcoming. Drug Resist. Updates 2021, 54, 100742. [Google Scholar] [CrossRef]
- Zhang, X.; Li, L.; Li, Y.; Li, Z.; Zhai, W.; Sun, Q.; Yang, X.; Roth, M.; Lu, S. mTOR regulates PRMT1 expression and mitochondrial mass through STAT1 phosphorylation in hepatic cell. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119017. [Google Scholar] [CrossRef]
- He, J.; Fortunati, E.; Liu, D.X.; Li, Y. Pleiotropic Roles of ABC Transporters in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 3199. [Google Scholar] [CrossRef]
- Barman, P.; Joshi, S.; Sharma, S.; Preet, S.; Sharma, S.; Saini, A. Strategic Approaches to Improvise Peptide Drugs as Next Generation Therapeutics. Int. J. Pept. Res. Ther. 2023, 29, 61. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Ding, H.; Tan, P.; Fu, S.; Tian, X.; Zhang, H.; Ma, X.; Gu, Z.; Luo, K. Preparation and application of pH-responsive drug delivery systems. J. Control. Release 2022, 348, 206–238. [Google Scholar] [CrossRef]
- Song, L.; Dai, Z.; Zhang, S.; Zhang, H.; Liu, C.; Ma, X.; Liu, D.; Zan, Y.; Yin, X. MicroRNA-1179 suppresses cell growth and invasion by targeting sperm-associated antigen 5-mediated Akt signaling in human non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2018, 504, 164–170. [Google Scholar] [CrossRef]
- Conti, A.; Di Micco, R. p53 activation: A checkpoint for precision genome editing? Genome Med. 2018, 10, 66. [Google Scholar] [CrossRef]
- Sinha, S.; Barbosa, K.; Cheng, K.; Leiserson, M.D.M.; Jain, P.; Deshpande, A.; Wilson, D.M., 3rd; Ryan, B.M.; Luo, J.; Ronai, Z.A.; et al. A systematic genome-wide mapping of oncogenic mutation selection during CRISPR-Cas9 genome editing. Nat. Commun. 2021, 12, 6512. [Google Scholar] [CrossRef]
- Mirgayazova, R.; Khadiullina, R.; Chasov, V.; Mingaleeva, R.; Miftakhova, R.; Rizvanov, A.; Bulatov, E. Therapeutic Editing of the TP53 Gene: Is CRISPR/Cas9 an Option? Genes 2020, 11, 704. [Google Scholar] [CrossRef]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef]
- Papathanasiou, S.; Markoulaki, S.; Blaine, L.J.; Leibowitz, M.L.; Zhang, C.Z.; Jaenisch, R.; Pellman, D. Whole chromosome loss and genomic instability in mouse embryos after CRISPR-Cas9 genome editing. Nat. Commun. 2021, 12, 5855. [Google Scholar] [CrossRef]
- Borst, P. Looking back at multidrug resistance (MDR) research and ten mistakes to be avoided when writing about ABC transporters in MDR. FEBS Lett. 2020, 594, 4001–4011. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Constructs |
|---|---|
| Puro 5 | 5′-TTGAATGGAAGGATGGAGCTAC-3′ |
| SPAG5R3 | 5′-GACTGACCTTTCCGTAAGTGAC-3′ |
| SPAG5F6 | 5′-GTTAGCCCAGTAAACTGGTAGC-3′ |
| LucR | 5′-GTCTTCGAGTGGGTAGAATGGC-3′ |
| A, Doxorubicin | ||
| Cell type | IC50, nM | p-value |
| M WT | 199.667 ± 47.919 | |
| M c1 | 81.79 ± 15.342 | 0.0046 |
| M c2 | 103.967 ± 2.909 | 0.0123 |
| B, Docetaxel | ||
| Cell type | IC50, nM | p-value |
| M WT | 1.009 ± 0.062 | |
| M c1 | 0.488 ± 0.045 | 0.0004 |
| M c2 | 0.557 ± 0.117 | 0.0008 |
| A, Doxorubicin | ||
| Cell type | IC50, nM | p-value |
| B WT | 30.657 ± 5.512 | |
| B c1 | 19.113 ± 2.048 | 0.0446 |
| B c2 | 8.465 ± 5.847 | 0.0023 |
| B, Docetaxel | ||
| Cell type | IC50, nM | p-value |
| B WT | 0.905 ± 0.107 | |
| B c1 | 0.676 ± 0.116 | 0.0142 |
| B c2 | 0.658 ± 0.112 | 0.0106 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, J.; Li, J.; Liu, Y.; Li, Y. Sperm-Associated Antigen 5 Knockout Reduces Doxorubicin and Docetaxel Resistance in Triple-Negative Breast Cancer MDA-MB-231 and BT549 Cells. Cancers 2024, 16, 1269. https://doi.org/10.3390/cancers16071269
He J, Li J, Liu Y, Li Y. Sperm-Associated Antigen 5 Knockout Reduces Doxorubicin and Docetaxel Resistance in Triple-Negative Breast Cancer MDA-MB-231 and BT549 Cells. Cancers. 2024; 16(7):1269. https://doi.org/10.3390/cancers16071269
Chicago/Turabian StyleHe, Ji, Jiawei Li, Yanbiao Liu, and Yan Li. 2024. "Sperm-Associated Antigen 5 Knockout Reduces Doxorubicin and Docetaxel Resistance in Triple-Negative Breast Cancer MDA-MB-231 and BT549 Cells" Cancers 16, no. 7: 1269. https://doi.org/10.3390/cancers16071269
APA StyleHe, J., Li, J., Liu, Y., & Li, Y. (2024). Sperm-Associated Antigen 5 Knockout Reduces Doxorubicin and Docetaxel Resistance in Triple-Negative Breast Cancer MDA-MB-231 and BT549 Cells. Cancers, 16(7), 1269. https://doi.org/10.3390/cancers16071269