

Asciminib Maintains Antibody-Dependent Cellular Cytotoxicity against Leukemic Blasts
 , , and
, , and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Peripheral Blood Mononuclear Cells and Cell Lines
2.2. Tyrosine Kinase Inhibitors (TKI)
2.3. Flow Cytometry
2.4. Cytotoxicity Assays
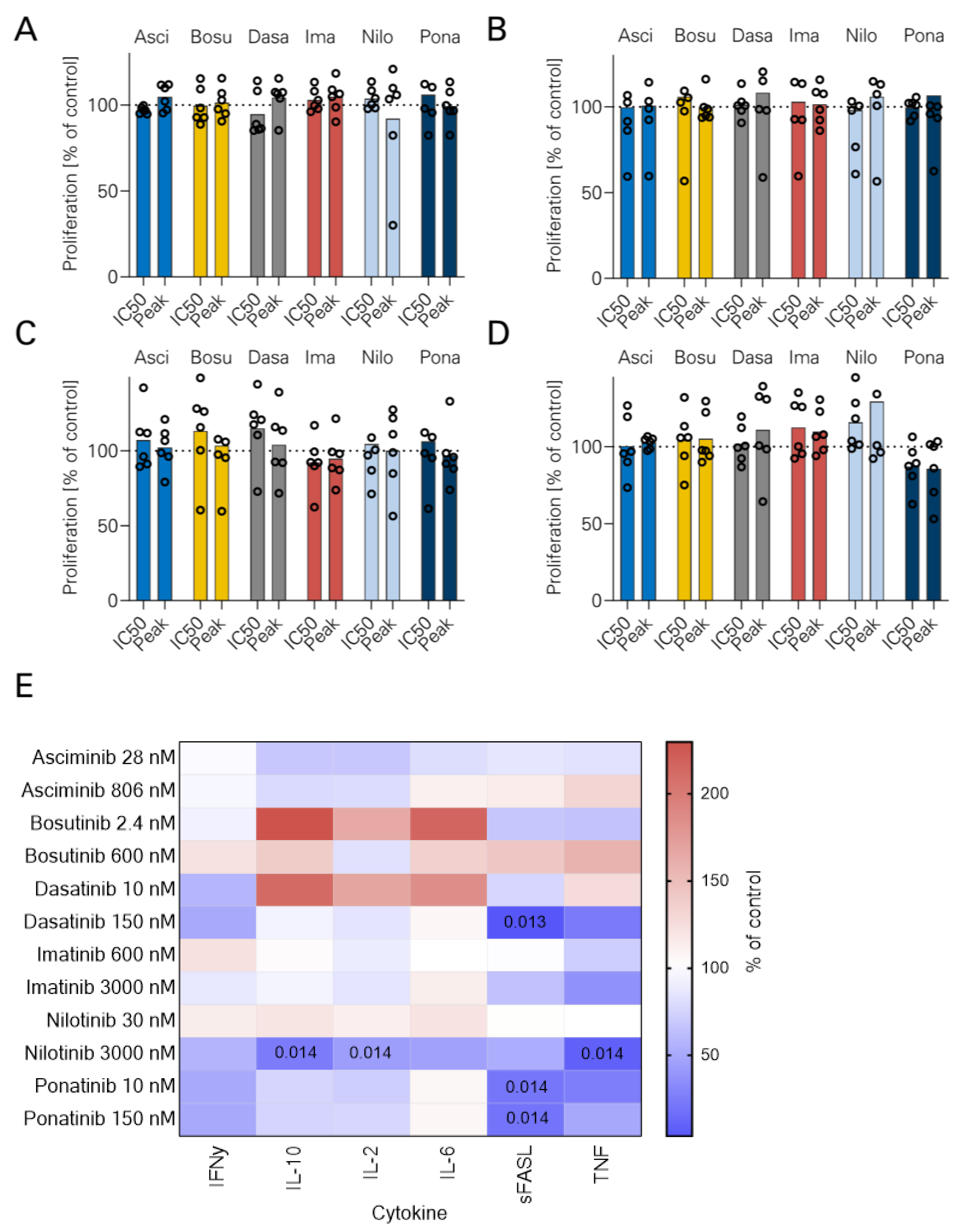
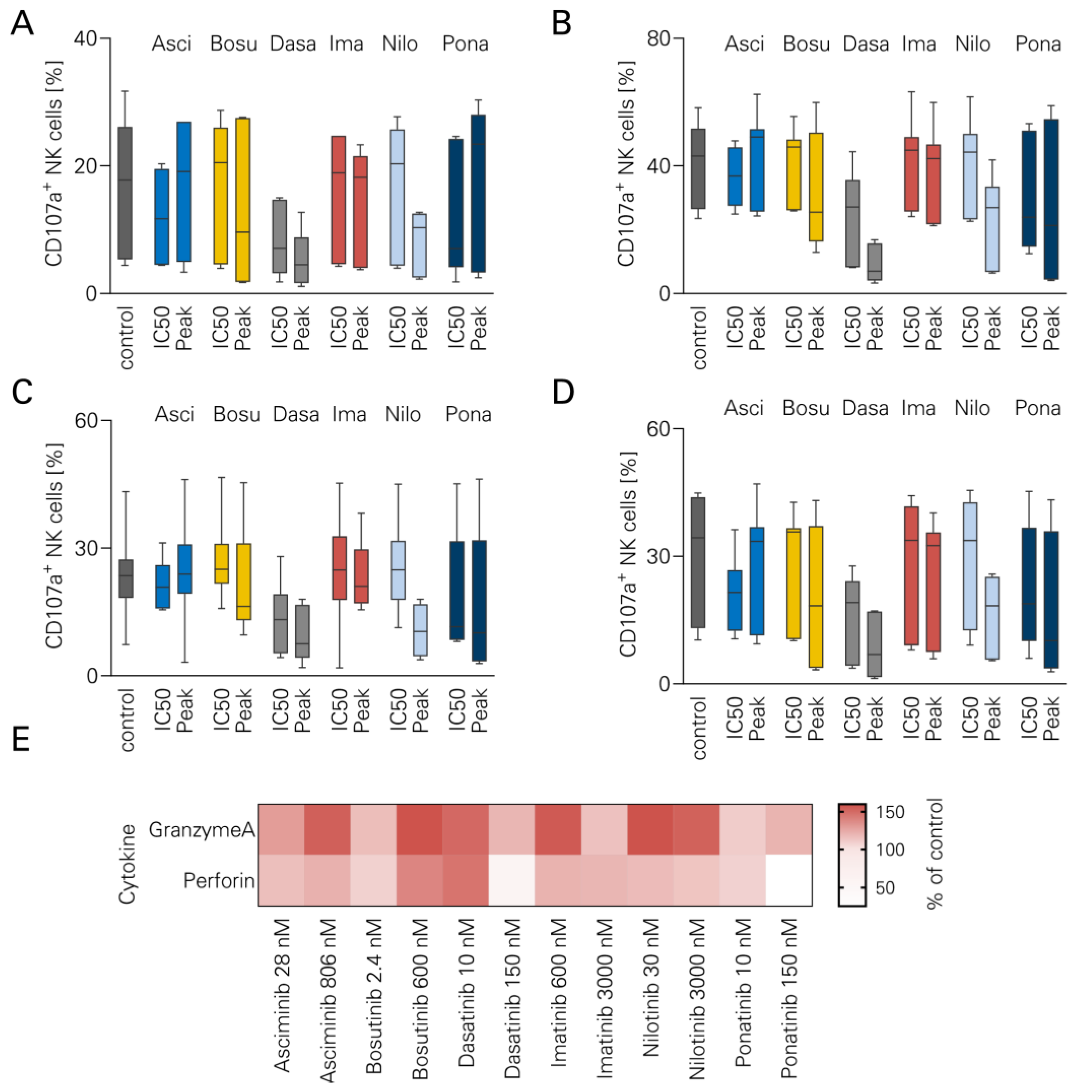
2.5. Legendplex Cytokine Arrays
2.6. Statistical Analysis
3. Results
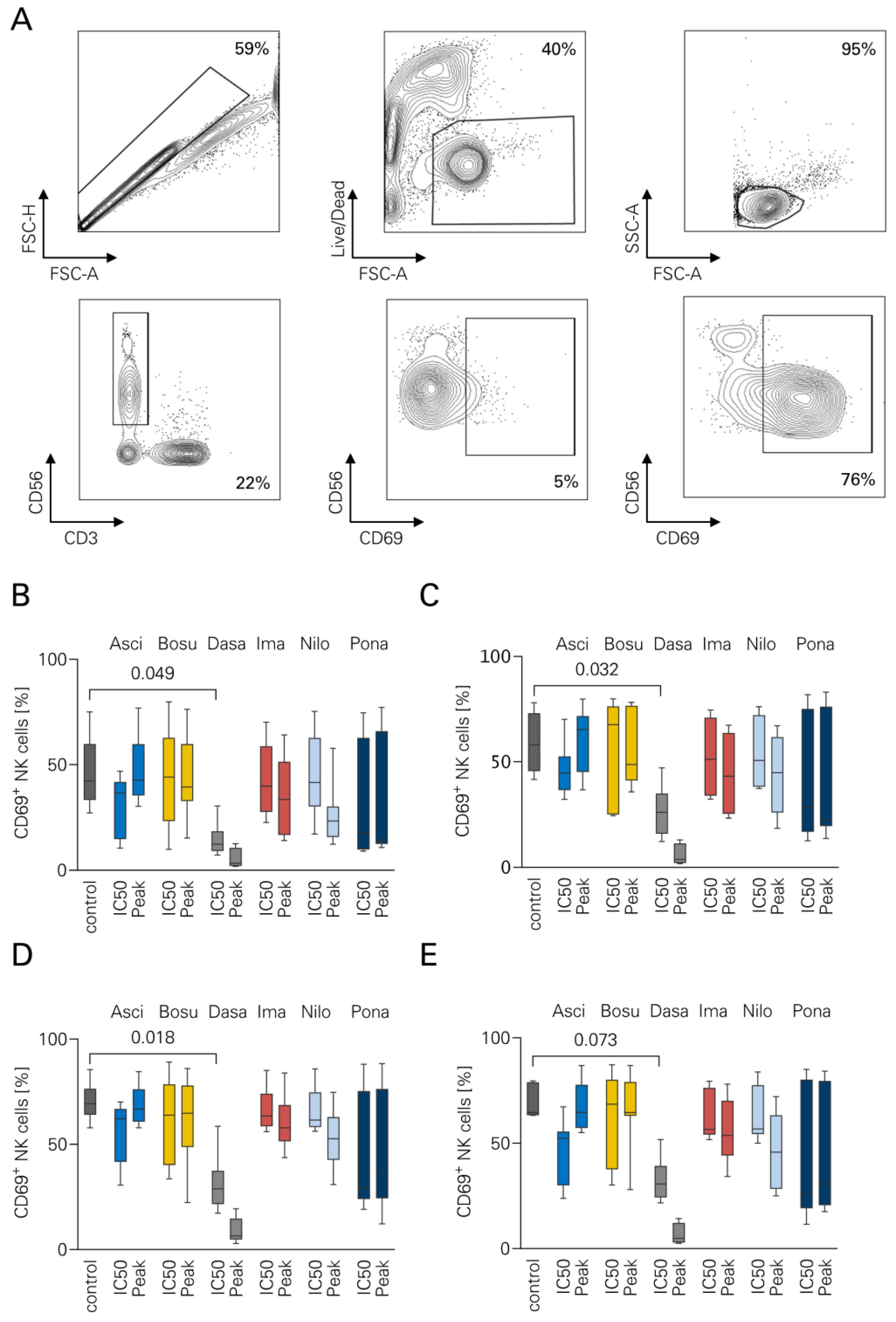
3.1. NK Cell Activation in the Presence of TKI
3.2. NK Cell Proliferation and TKI
3.3. NK Cell Degranulation and TKI
3.4. Short-Term Lysis of Tumor Cells
3.5. Long-Term Lysis in the Presence of TKI
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hunger, S.P.; Mullighan, C.G. Redefining ALL classification: Toward detecting high-risk ALL and implementing precision medicine. Blood J. Am. Soc. Hematol. 2015, 125, 3977–3987. [Google Scholar] [CrossRef] [PubMed]
- Komorowski, L.; Fidyt, K.; Patkowska, E.; Firczuk, M. Philadelphia Chromosome-Positive Leukemia in the Lymphoid Lineage—Similarities and Differences with the Myeloid Lineage and Specific Vulnerabilities. Int. J. Mol. Sci. 2020, 21, 5776. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Guilhot, F.; O’Brien, S.G.; Gathmann, I.; Kantarjian, H.; Gattermann, N.; Deininger, M.W.; Silver, R.T.; Goldman, J.M.; Stone, R.M. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 2006, 355, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Douer, D.; Aldoss, I.; Vahdani, G.; Jeong, A.R.; Ghaznavi, Z.; Zhang, S.; Yaghmour, G.; Lee, K.J.; Weissman, A. Combination chemotherapy plus dasatinib leads to comparable overall survival and relapse-free survival rates as allogeneic hematopoietic stem cell transplantation in Philadelphia positive acute lymphoblastic leukemia. Cancer Med. 2019, 8, 2832–2839. [Google Scholar] [CrossRef] [PubMed]
- Domka, K.; Poprzeczko, M.; Urbanska, Z.; Komorowski, L.; Pastorczak, A.; Fidyt, K.; Dabkowska, A.; Lachota, M.; Winiarska, M.; Siudakowska, K. Optimizing the therapeutic potential of tyrosine kinase inhibitors in chemo-immunotherapy of B-cell acute lymphoblastic leukemia involving rituximab. Cancer Res. 2022, 82, 3324. [Google Scholar] [CrossRef]
- Fielding, A.K.; Rowe, J.M.; Buck, G.; Foroni, L.; Gerrard, G.; Litzow, M.R.; Lazarus, H.; Luger, S.M.; Marks, D.I.; McMillan, A.K. UKALLXII/ECOG2993: Addition of imatinib to a standard treatment regimen enhances long-term outcomes in Philadelphia positive acute lymphoblastic leukemia. Blood 2014, 123, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; O’Brien, S.; Smith, T.L.; Cortes, J.; Giles, F.J.; Beran, M.; Pierce, S.; Huh, Y.; Andreeff, M.; Koller, C. Results of treatment with hyper-CVAD, a dose-intensive regimen, in adult acute lymphocytic leukemia. J. Clin. Oncol. 2000, 18, 547. [Google Scholar] [CrossRef] [PubMed]
- King, A.C.; Pappacena, J.J.; Tallman, M.S.; Park, J.H.; Geyer, M.B. Blinatumomab administered concurrently with oral tyrosine kinase inhibitor therapy is a well-tolerated consolidation strategy and eradicates measurable residual disease in adults with Philadelphia chromosome positive acute lymphoblastic leukemia. Leuk. Res. 2019, 79, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Zerbit, J.; Tamburini, J.; Goldwirt, L.; Decroocq, J.; Cayuela, J.M.; Chapuis, N.; Contejean, A.; Batista, R.; Bouscary, D.; Willems, L. Asciminib and ponatinib combination in Philadelphia chromosome-positive acute lymphoblastic leukemia. Leuk. Lymphoma 2021, 62, 3558–3560. [Google Scholar] [CrossRef] [PubMed]
- Foà, R.; Bassan, R.; Vitale, A.; Elia, L.; Piciocchi, A.; Puzzolo, M.-C.; Canichella, M.; Viero, P.; Ferrara, F.; Lunghi, M. Dasatinib–blinatumomab for Ph-positive acute lymphoblastic leukemia in adults. N. Engl. J. Med. 2020, 383, 1613–1623. [Google Scholar] [CrossRef] [PubMed]
- Advani, A.S.; Moseley, A.; O’Dwyer, K.M.; Wood, B.L.; Park, J.; Wieduwilt, M.; Jeyakumar, D.; Yaghmour, G.; Atallah, E.L.; Gerds, A.T. Dasatinib/prednisone induction followed by blinatumomab/dasatinib in Ph+ acute lymphoblastic leukemia. Blood Adv. 2023, 7, 1279–1285. [Google Scholar] [CrossRef]
- Maury, S.; Chevret, S.; Thomas, X.; Heim, D.; Leguay, T.; Huguet, F.; Chevallier, P.; Hunault, M.; Boissel, N.; Escoffre-Barbe, M. Rituximab in B-lineage adult acute lymphoblastic leukemia. N. Engl. J. Med. 2016, 375, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; O’Brien, S.; Faderl, S.; Garcia-Manero, G.; Ferrajoli, A.; Wierda, W.; Ravandi, F.; Verstovsek, S.; Jorgensen, J.L.; Bueso-Ramos, C. Chemoimmunotherapy with a modified hyper-CVAD and rituximab regimen improves outcome in de novo Philadelphia chromosome–negative precursor B-lineage acute lymphoblastic leukemia. J. Clin. Oncol. 2010, 28, 3880. [Google Scholar] [CrossRef]
- Klisovic, R.B.; Leung, W.H.; Brugger, W.; Dirnberger-Hertweck, M.; Winderlich, M.; Ambarkhane, S.V.; Jabbour, E.J. A phase 2a, single-arm, open-label study of tafasitamab, a humanized, Fc-modified, anti-CD19 antibody, in patients with relapsed/refractory B-precursor cell acute lymphoblastic leukemia. Cancer 2021, 127, 4190–4197. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Song, Z.; Zheng, G.; Nicolazzi, C.; Fromm, J.R.; Shehu, E.; Srinivasan, S.; Chen, X.; Zhu, C.; Blondel, M.C. Evaluation of preclinical activity of isatuximab in patients with acute lymphoblastic leukemia. Mol. Cancer Ther. 2021, 20, 1916–1925. [Google Scholar] [CrossRef] [PubMed]
- Raetz, E.A.; Cairo, M.S.; Borowitz, M.J.; Lu, X.; Devidas, M.; Reid, J.M.; Goldenberg, D.M.; Wegener, W.A.; Zeng, H.; Whitlock, J.A. Re-induction chemoimmunotherapy with epratuzumab in relapsed acute lymphoblastic leukemia (ALL): Phase II results from Children’s Oncology Group (COG) study ADVL04P2. Pediatr. Blood Cancer 2015, 62, 1171–1175. [Google Scholar] [CrossRef] [PubMed]
- Salih, J.; Hilpert, J.; Placke, T.; Grünebach, F.; Steinle, A.; Salih, H.R.; Krusch, M. The BCR/ABL-inhibitors imatinib, nilotinib and dasatinib differentially affect NK cell reactivity. Int. J. Cancer 2010, 127, 2119–2128. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Rusakiewicz, S.; Routy, B.; Ayyoub, M.; Kroemer, G. Immunological off-target effects of imatinib. Nat. Rev. Clin. Oncol. 2016, 13, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Luskin, M.; Murakami, M.A.; Stevenson, K.E.; Wadleigh, M.; McMasters, M.; Winter, P.; Weinstock, D.M.; Stone, R.; DeAngelo, D.J. A phase I study of asciminib (ABL001) in combination with dasatinib and prednisone for untreated BCR-ABL1-positive ALL in older adults. Blood 2019, 134, 3879. [Google Scholar] [CrossRef]
- Kantarjian, H.; Giles, F.; Wunderle, L.; Bhalla, K.; O’Brien, S.; Wassmann, B.; Tanaka, C.; Manley, P.; Rae, P.; Mietlowski, W. Nilotinib in imatinib-resistant CML and Philadelphia chromosome–positive ALL. N. Engl. J. Med. 2006, 354, 2542–2551. [Google Scholar] [CrossRef] [PubMed]
- Eide, C.A.; Zabriskie, M.S.; Stevens, S.L.S.; Antelope, O.; Vellore, N.A.; Than, H.; Schultz, A.R.; Clair, P.; Bowler, A.D.; Pomicter, A.D. Combining the allosteric inhibitor asciminib with ponatinib suppresses emergence of and restores efficacy against highly resistant BCR-ABL1 mutants. Cancer Cell 2019, 36, 431–443.e435. [Google Scholar] [CrossRef] [PubMed]
- Abbas, R.; Hsyu, P.-H. Clinical pharmacokinetics and pharmacodynamics of bosutinib. Clin. Pharmacokinet. 2016, 55, 1191–1204. [Google Scholar] [CrossRef] [PubMed]
- Golas, J.M.; Arndt, K.; Etienne, C.; Lucas, J.; Nardin, D.; Gibbons, J.; Frost, P.; Ye, F.; Boschelli, D.H.; Boschelli, F. SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res. 2003, 63, 375–381. [Google Scholar] [PubMed]
- Hughes, T.P.; Mauro, M.J.; Cortes, J.E.; Minami, H.; Rea, D.; DeAngelo, D.J.; Breccia, M.; Goh, Y.-T.; Talpaz, M.; Hochhaus, A. Asciminib in chronic myeloid leukemia after ABL kinase inhibitor failure. N. Engl. J. Med. 2019, 381, 2315–2326. [Google Scholar] [CrossRef] [PubMed]
- Rix, U.; Hantschel, O.; Dürnberger, G.; Remsing Rix, L.L.; Planyavsky, M.; Fernbach, N.V.; Kaupe, I.; Bennett, K.L.; Valent, P.; Colinge, J. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood J. Am. Soc. Hematol. 2007, 110, 4055–4063. [Google Scholar] [CrossRef] [PubMed]
- Menna, P.; De Grazia, U.; Marchesi, F.; Minotti, G.; Salvatorelli, E. Further analytical, pharmacokinetic, and clinical observations on low-dose ponatinib in patients with philadelphia chromosome-positive acute lymphoblastic leukemia. Chemotherapy 2020, 65, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Hayes, M.; Resta, D.; Racine-Poon, A.; Druker, B.J.; Talpaz, M.; Sawyers, C.L.; Rosamilia, M.; Ford, J.; Lloyd, P. Pharmacokinetics and pharmacodynamics of imatinib in a phase I trial with chronic myeloid leukemia patients. J. Clin. Oncol. 2004, 22, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, A.; Réa, D.; Boquimpani, C.; Minami, Y.; Cortes, J.E.; Hughes, T.P.; Apperley, J.F.; Lomaia, E.; Voloshin, S.; Turkina, A. Asciminib vs bosutinib in chronic-phase chronic myeloid leukemia previously treated with at least two tyrosine kinase inhibitors: Longer-term follow-up of ASCEMBL. Leukemia 2023, 37, 617–626. [Google Scholar] [CrossRef]
- Hoch, M.; Sengupta, T.; Hourcade-Potelleret, F. Pharmacokinetic drug interactions of asciminib with the sensitive cytochrome P450 probe substrates midazolam, warfarin, and repaglinide in healthy participants. Clin. Transl. Sci. 2022, 15, 1406–1416. [Google Scholar] [CrossRef] [PubMed]
- Eadie, L.N.; Schutz, C.E.; Page, E.C.; Conlin, T.S.; Heatley, S.L.; Lagonik, E.; Rehn, J.A.; Yeung, D.T.; Hughes, T.P.; White, D.L. Asciminib Is Effective Against ABL1 Gene Fusions in Acute Lymphoblastic Leukemia but Only When the ABL1 SH3 Domain Is Present. Blood 2023, 142, 1602. [Google Scholar] [CrossRef]
- Maury, S.; Huguet, F.; Leguay, T.; Lacombe, F.; Maynadié, M.; Girard, S.; de Labarthe, A.; Kuhlein, E.; Raffoux, E.; Thomas, X. Adverse prognostic significance of CD20 expression in adults with Philadelphia chromosome-negative B-cell precursor acute lymphoblastic leukemia. Haematologica 2010, 95, 324. [Google Scholar] [CrossRef] [PubMed]
- Marks, D.I.; Kirkwood, A.A.; Rowntree, C.J.; Aguiar, M.; Bailey, K.E.; Beaton, B.; Cahalin, P.; Castleton, A.Z.; Clifton-Hadley, L.; Copland, M. Addition of four doses of rituximab to standard induction chemotherapy in adult patients with precursor B-cell acute lymphoblastic leukaemia (UKALL14): A phase 3, multicentre, randomised controlled trial. Lancet Haematol. 2022, 9, e262–e275. [Google Scholar] [CrossRef] [PubMed]
- Bieerkehazhi, S.; Chen, Z.; Zhao, Y.; Yu, Y.; Zhang, H.; Vasudevan, S.A.; Woodfield, S.E.; Tao, L.; Yi, J.S.; Muscal, J.A.; et al. Novel Src/Abl tyrosine kinase inhibitor bosutinib suppresses neuroblastoma growth via inhibiting Src/Abl signaling. Oncotarget 2017, 8, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Griffith, D.J.; Druker, B.J.; Wait, C.L.; Ott, K.A.; Zigler, A.J. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood 2000, 96, 925–932. [Google Scholar] [CrossRef]
- Ballinger, M.L.; Osman, N.; Hashimura, K.; de Haan, J.B.; Jandeleit-Dahm, K.; Allen, T.; Tannock, L.R.; Rutledge, J.C.; Little, P.J. Imatinib inhibits vascular smooth muscle proteoglycan synthesis and reduces LDL binding in vitro and aortic lipid deposition in vivo. J. Cell Mol. Med. 2010, 14, 1408–1418. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, T.; Pollock, R.; Stoffregen, E.P.; Keats, J.A.; Abdullah, O.M.; Moseson, E.M.; Rivera, V.M.; Tang, H.; Metcalf, C.A., 3rd; Bohacek, R.S.; et al. Inhibition of wild-type and mutant Bcr-Abl by AP23464, a potent ATP-based oncogenic protein kinase inhibitor: Implications for CML. Blood 2004, 104, 2532–2539. [Google Scholar] [CrossRef] [PubMed]
- Gozgit, J.M.; Wong, M.J.; Wardwell, S.; Tyner, J.W.; Loriaux, M.M.; Mohemmad, Q.K.; Narasimhan, N.I.; Shakespeare, W.C.; Wang, F.; Druker, B.J.; et al. Potent activity of ponatinib (AP24534) in models of FLT3-driven acute myeloid leukemia and other hematologic malignancies. Mol. Cancer Ther. 2011, 10, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Oykhman, P.; Timm-McCann, M.; Xiang, R.F.; Islam, A.; Li, S.S.; Stack, D.; Huston, S.M.; Ma, L.L.; Mody, C.H. Requirement and redundancy of the Src family kinases Fyn and Lyn in perforin-dependent killing of Cryptococcus neoformans by NK cells. Infect. Immun. 2013, 81, 3912–3922. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Xu, Y.; Wang, Z.; Wang, T.; Du, X.; Song, X.; Guo, X.; Peng, J.; Zhang, J.; Liang, Y.; et al. Tim-3 Hampers Tumor Surveillance of Liver-Resident and Conventional NK Cells by Disrupting PI3K Signaling. Cancer Res. 2020, 80, 1130–1142. [Google Scholar] [CrossRef] [PubMed]
- Romeo, V.; Gierke, S.; Edgar, K.A.; Liu, S.D. Effects of PI3K Inhibition on Afucosylated Antibody-Driven FcgammaRIIIa Events and Phospho-S6 Activity in NK Cells. J. Immunol. 2019, 203, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Wylie, A.A.; Schoepfer, J.; Jahnke, W.; Cowan-Jacob, S.W.; Loo, A.; Furet, P.; Marzinzik, A.L.; Pelle, X.; Donovan, J.; Zhu, W.; et al. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature 2017, 543, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Manley, P.W.; Barys, L.; Cowan-Jacob, S.W. The specificity of asciminib, a potential treatment for chronic myeloid leukemia, as a myristate-pocket binding ABL inhibitor and analysis of its interactions with mutant forms of BCR-ABL1 kinase. Leuk. Res. 2020, 98, 106458. [Google Scholar] [CrossRef]
- Leonard, J.T.; Kosaka, Y.; Malla, P.; LaTocha, D.; Lamble, A.; Hayes-Lattin, B.; Byrd, K.; Druker, B.J.; Tyner, J.W.; Chang, B.H. Concomitant use of a dual Src/ABL kinase inhibitor eliminates the in vitro efficacy of blinatumomab against Ph+ ALL. Blood J. Am. Soc. Hematol. 2021, 137, 939–944. [Google Scholar] [CrossRef]
- Kauer, J.; Märklin, M.; Pflügler, M.; Hörner, S.; Hinterleitner, C.; Tandler, C.; Jung, G.; Salih, H.R.; Heitmann, J.S. BCR:: ABL1 tyrosine kinase inhibitors hamper the therapeutic efficacy of blinatumomab in vitro. J. Cancer Res. Clin. Oncol. 2022, 148, 2759–2771. [Google Scholar] [CrossRef] [PubMed]
- Assi, R.; Kantarjian, H.; Short, N.J.; Daver, N.; Takahashi, K.; Garcia-Manero, G.; DiNardo, C.; Burger, J.; Cortes, J.; Jain, N. Safety and efficacy of blinatumomab in combination with a tyrosine kinase inhibitor for the treatment of relapsed Philadelphia chromosome-positive leukemia. Clin. Lymphoma Myeloma Leuk. 2017, 17, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Häselbarth, L.; Karow, A.; Mentz, K.; Böttcher, M.; Roche-Lancaster, O.; Krumbholz, M.; Jitschin, R.; Mougiakakos, D.; Metzler, M. Effects of the STAMP-inhibitor asciminib on T cell activation and metabolic fitness compared to tyrosine kinase inhibition by imatinib, dasatinib, and nilotinib. Cancer Immunol. Immunother. 2023, 72, 1661–1672. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holzmayer, S.J.; Kauer, J.; Mauermann, J.; Roider, T.; Märklin, M. Asciminib Maintains Antibody-Dependent Cellular Cytotoxicity against Leukemic Blasts. Cancers 2024, 16, 1288. https://doi.org/10.3390/cancers16071288
Holzmayer SJ, Kauer J, Mauermann J, Roider T, Märklin M. Asciminib Maintains Antibody-Dependent Cellular Cytotoxicity against Leukemic Blasts. Cancers. 2024; 16(7):1288. https://doi.org/10.3390/cancers16071288
Chicago/Turabian StyleHolzmayer, Samuel J., Joseph Kauer, Jonas Mauermann, Tobias Roider, and Melanie Märklin. 2024. "Asciminib Maintains Antibody-Dependent Cellular Cytotoxicity against Leukemic Blasts" Cancers 16, no. 7: 1288. https://doi.org/10.3390/cancers16071288