RNA m6a Methylation Regulator Expression in Castration-Resistant Prostate Cancer Progression and Its Genetic Associations

 ,
, 
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. RNA Isolation and Quantitative PCR Analysis of the Formalin-Fixed, Paraffin-Embedded (FFPE) Tissues and PCa Cell Lines
2.3. Sequential Window Acquisition of All THeoretical Mass Spectra (SWATH-MS) Analysis
2.4. RNAseq Analysis for the PCa Cell Lines and PCa Clinical Tissues
2.5. RNA Expression Analysis Using Published Transcriptomic Data
2.6. Survival Analysis
2.7. Cis-Expression Quantitative Trait Loci (eQTL) Analysis
3. Results
3.1. Differential Expression of m6A Methylation Regulators in PCa Cell Lines
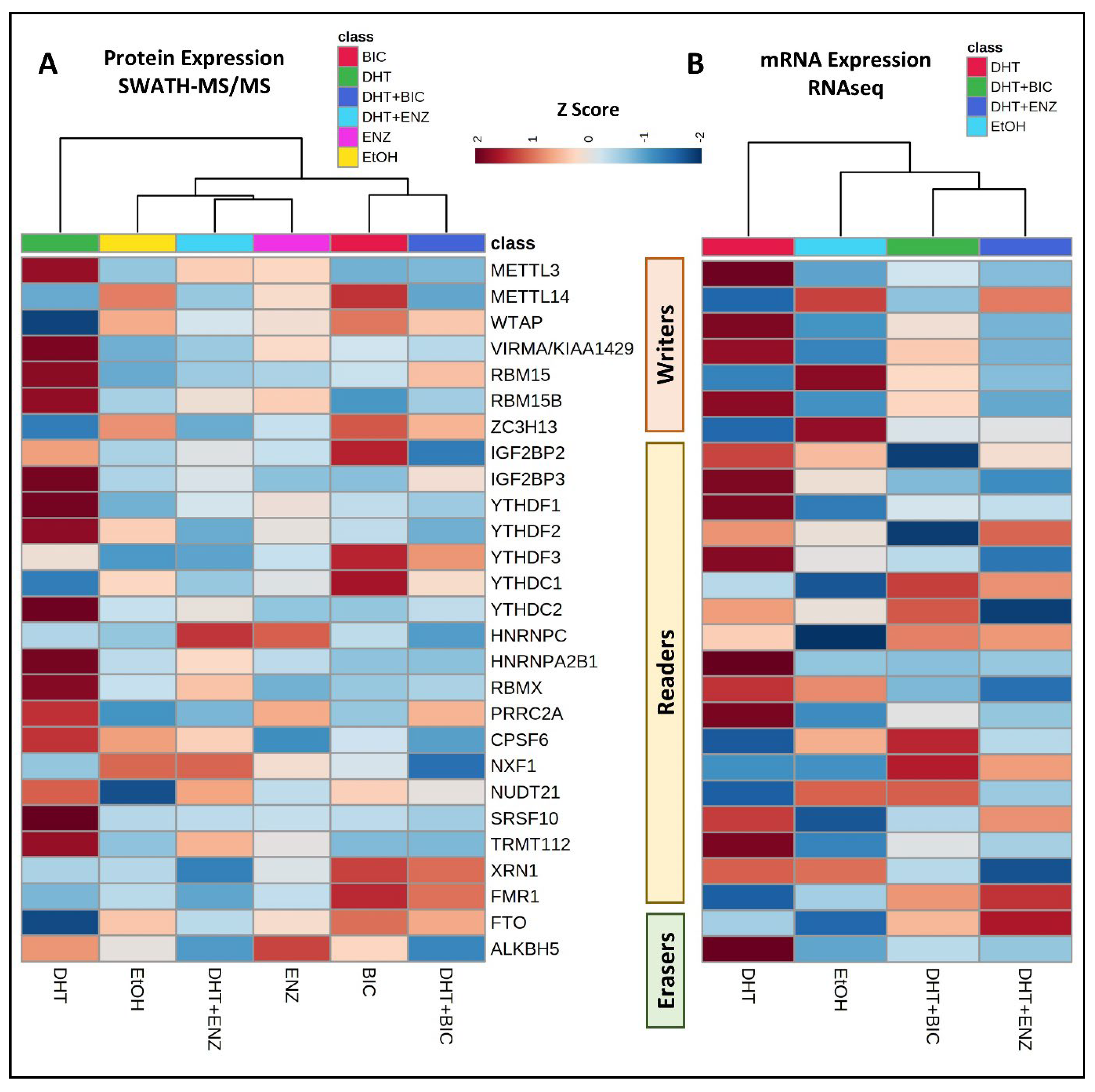
3.2. Effects of Androgen and Anti-Androgen Treatment on the Expression of m6A Methylation Regulators in PCa
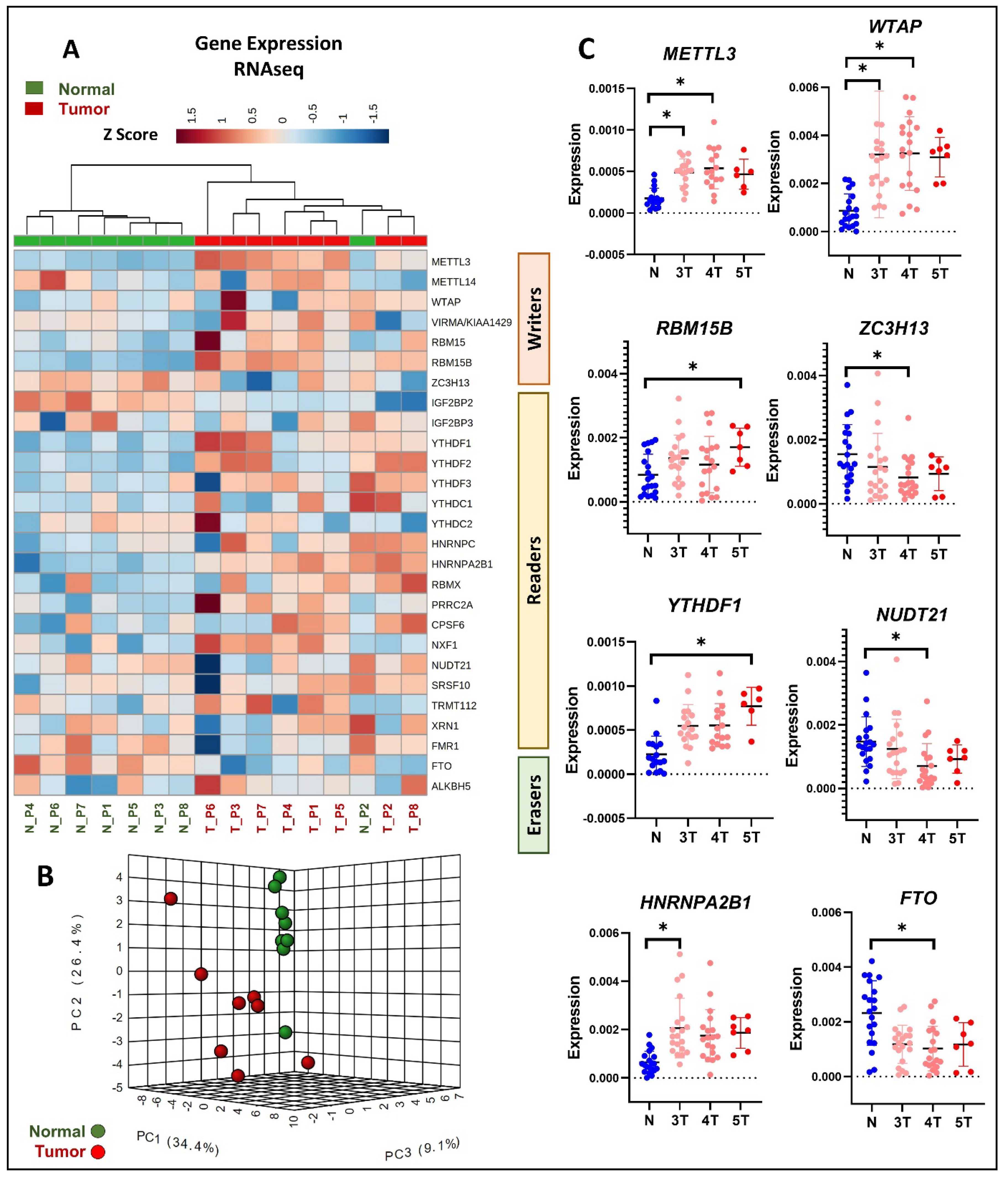
3.3. Differential Expression of m6A Methylation Regulators in Primary PCa Tissue Using RNAseq
3.4. Differential Expression of m6A Methylation Regulators in Response to Disease Progression
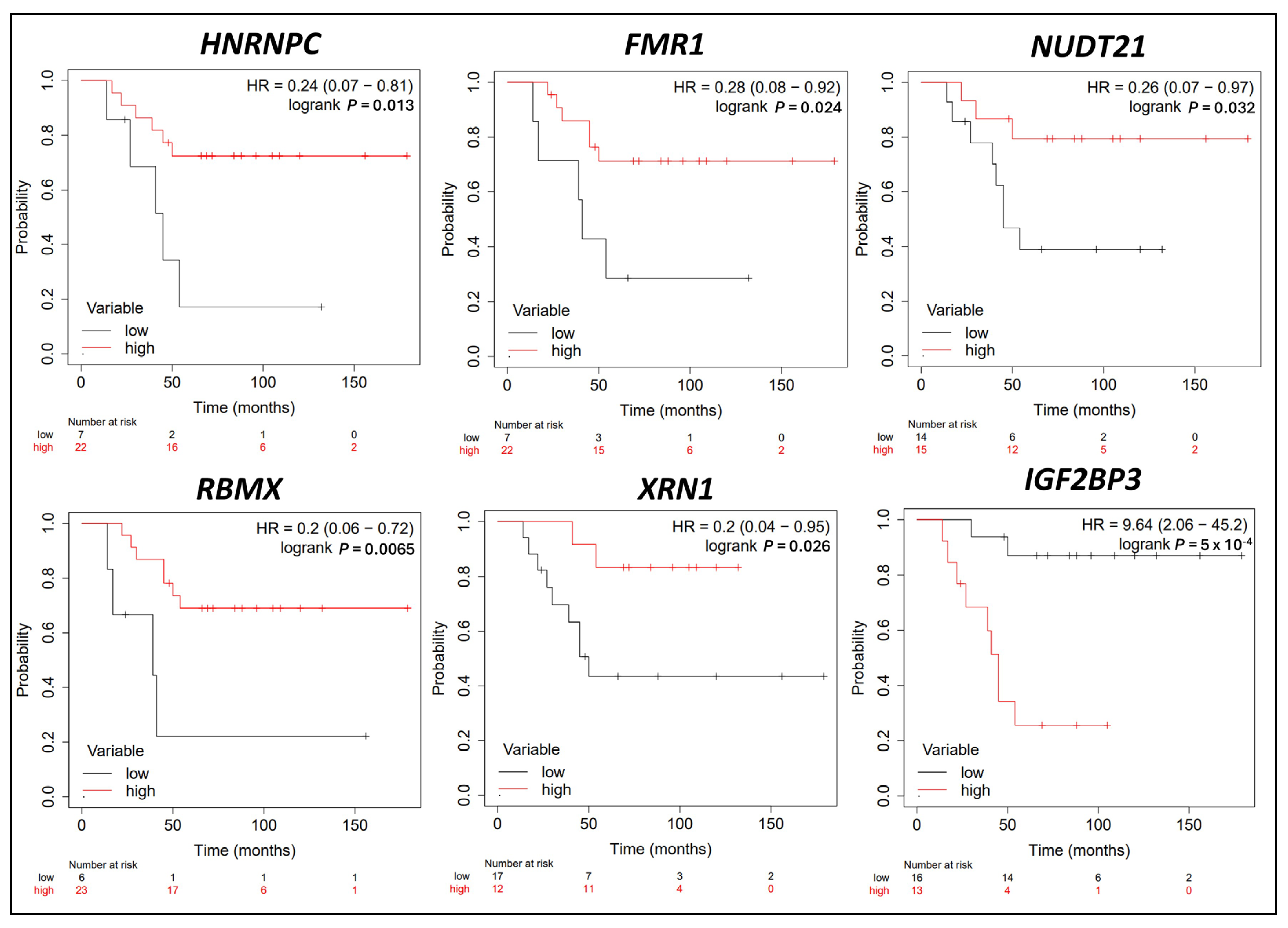
3.5. Survival Analysis in CRPC Patients Correlating with m6A Methylation Regulator Expression Levels
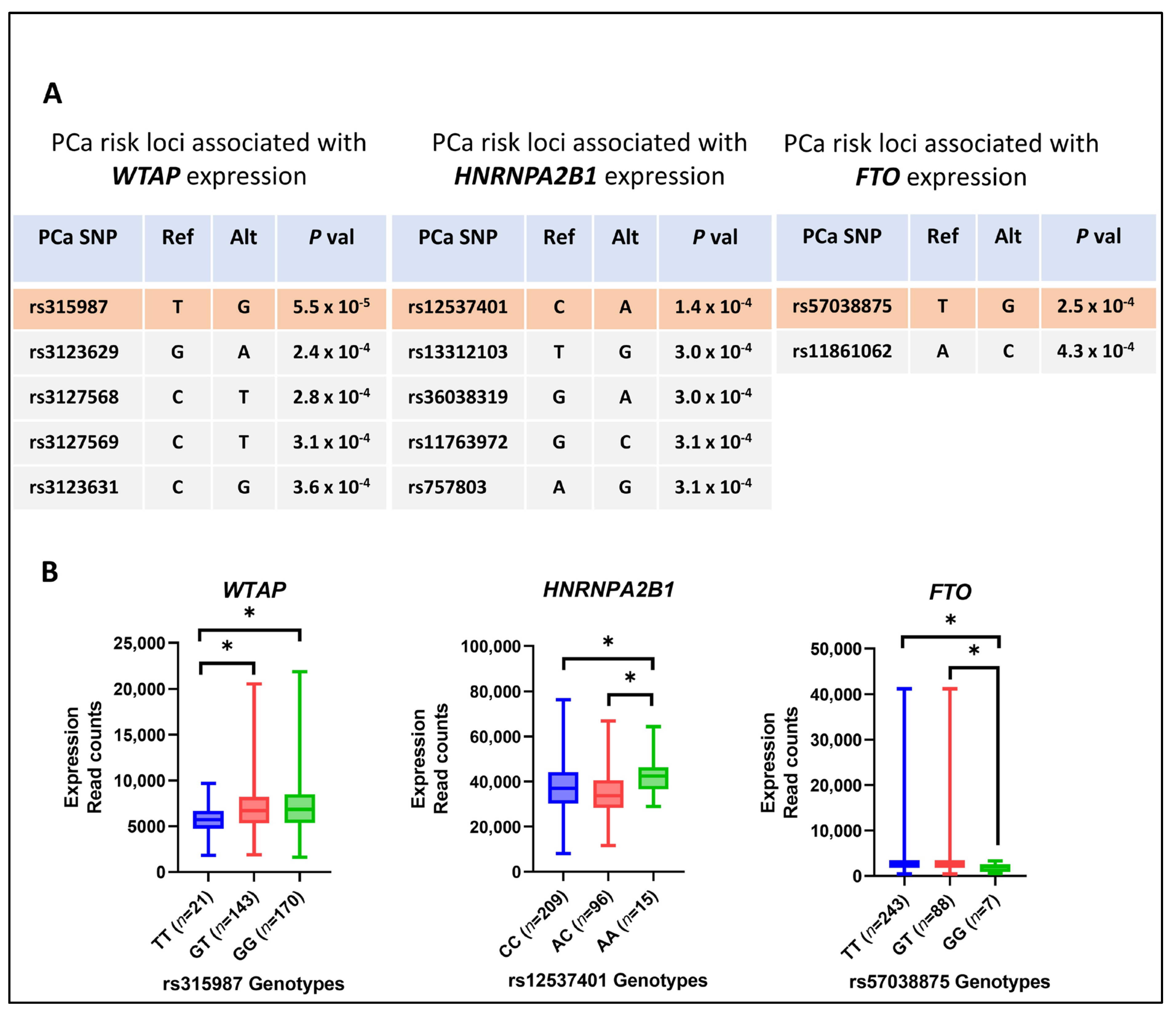
3.6. PCa-Risk-Associated Single-Nucleotide Polymorphisms (SNPs) and the Expression of m6A Methylation Regulators
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Culp, M.B.; Soerjomataram, I.; Efstathiou, J.A.; Bray, F.; Jemal, A. Recent Global Patterns in Prostate Cancer Incidence and Mortality Rates. Eur. Urol. 2020, 77, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.N.; Ferraldeschi, R.; Attard, G.; de Bono, J. Evolution of androgen receptor targeted therapy for advanced prostate cancer. Nat. Rev. Clin. Oncol. 2014, 11, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.; Conteduca, V.; Zoubeidi, A.; Beltran, H. Biological Evolution of Castration-resistant Prostate Cancer. Eur. Urol. Focus 2019, 5, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Kirby, M.; Hirst, C.; Crawford, E.D. Characterising the castration-resistant prostate cancer population: A systematic review. Int. J. Clin. Pract. 2011, 65, 1180–1192. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Chu, Q.; Zeng, Y.; Yuan, X.; Wang, J.; Zhang, Y.; Xue, C.; Li, L. Non-coding RNA methylation modifications in hepatocellular carcinoma: Interactions and potential implications. Cell Commun. Signal. 2023, 21, 359. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhu, H.; Luo, C.; Yan, Z.; Zheng, G.; Zou, X.; Zou, J.; Zhang, G. The role of RNA modification in urological cancers: Mechanisms and clinical potential. Discov. Oncol. 2023, 14, 235. [Google Scholar] [CrossRef]
- Gu, C.; Shi, X.; Dai, C.; Shen, F.; Rocco, G.; Chen, J.; Huang, Z.; Chen, C.; He, C.; Huang, T.; et al. RNA m6A Modification in Cancers: Molecular Mechanisms and Potential Clinical Applications. Innovation 2020, 1, 100066. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Kong, S.; Tao, M.; Ju, S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol. Cancer 2020, 19, 88. [Google Scholar] [CrossRef]
- Boccaletto, P.; Baginski, B. MODOMICS: An Operational Guide to the Use of the RNA Modification Pathways Database. Methods Mol. Biol. 2021, 2284, 481–505. [Google Scholar] [CrossRef]
- Wilkinson, E.; Cui, Y.H.; He, Y.Y. Roles of RNA Modifications in Diverse Cellular Functions. Front. Cell Dev. Biol. 2022, 10, 828683. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Ji, X.; Guo, X.; Ji, S. Regulatory Role of N6-methyladenosine (m6A) Methylation in RNA Processing and Human Diseases. J. Cell. Biochem. 2017, 118, 2534–2543. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.; Li, T.; Xu, Y.; Zhang, Y.; Pan, H.; Wang, Y. The role of m6A epigenetic modifications in tumor coding and non-coding RNA processing. Cell Commun. Signal. 2023, 21, 355. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Du, Y.; Wang, L.; Liu, X. The M6A methyltransferase METTL3 promotes the development and progression of prostate carcinoma via mediating MYC methylation. J. Cancer 2020, 11, 3588–3595. [Google Scholar] [CrossRef] [PubMed]
- Barros-Silva, D.; Lobo, J.; Guimaraes-Teixeira, C.; Carneiro, I.; Oliveira, J.; Martens-Uzunova, E.S.; Henrique, R.; Jeronimo, C. VIRMA-Dependent N6-Methyladenosine Modifications Regulate the Expression of Long Non-Coding RNAs CCAT1 and CCAT2 in Prostate Cancer. Cancers 2020, 12, 771. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Meng, S.; Xu, M.; Wang, S.; He, L.; Xu, X.; Wang, X.; Xie, L. Downregulation of N6-methyladenosine binding YTHDF2 protein mediated by miR-493-3p suppresses prostate cancer by elevating N6-methyladenosine levels. Oncotarget 2018, 9, 3752–3764. [Google Scholar] [CrossRef]
- Nie, Q.; Wu, X.; Huang, Y.; Guo, T.; Kuang, J.; Du, C. RNA N6-methyladenosine-modified-binding protein YTHDF1 promotes prostate cancer progression by regulating androgen function-related gene TRIM68. Eur. J. Med. Res. 2023, 28, 552. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Li, Y.; Xu, Y. The FTO m6A demethylase inhibits the invasion and migration of prostate cancer cells by regulating total m6A levels. Life Sci. 2021, 271, 119180. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, J.; Zhu, X.; Yang, B.; He, Z.; Yao, X. AZGP1P2/UBA1/RBM15 Cascade Mediates the Fate Determinations of Prostate Cancer Stem Cells and Promotes Therapeutic Effect of Docetaxel in Castration-Resistant Prostate Cancer via TPM1 m6A Modification. Research 2023, 6, 0252. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, C.; Malik, A.; Abeysinghe, P.; Clements, J.; Batra, J. SWATH-MS Based Proteomic Profiling of Prostate Cancer Cells Reveals Adaptive Molecular Mechanisms in Response to Anti-Androgen Therapy. Cancers 2021, 13, 715. [Google Scholar] [CrossRef] [PubMed]
- Fernando, A.; Liyanage, C.; Moradi, A.; Janaththani, P.; Batra, J. Identification and Characterization of Alternatively Spliced Transcript Isoforms of IRX4 in Prostate Cancer. Genes 2021, 12, 615. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Moya, L.; An, J.; Hoffman, A.; Srinivasan, S.; Panchadsaram, J.; Walpole, C.; Perry-Keene, J.L.; Chambers, S.; Australian Prostate Cancer, B.; et al. A microsatellite repeat in PCA3 long non-coding RNA is associated with prostate cancer risk and aggressiveness. Sci. Rep. 2017, 7, 16862. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; An, J.; Nelson, C.C.; Lehman, M.L.; Batra, J.; Clements, J.A. Analysis of androgen and anti-androgen regulation of KLK-related peptidase 2, 3, and 4 alternative transcripts in prostate cancer. Biol. Chem. 2014, 395, 1127–1132. [Google Scholar] [CrossRef]
- Rich, J.T.; Neely, J.G.; Paniello, R.C.; Voelker, C.C.; Nussenbaum, B.; Wang, E.W. A practical guide to understanding Kaplan-Meier curves. Otolaryngol. Head Neck Surg. 2010, 143, 331–336. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Sampson, N.; Neuwirt, H.; Puhr, M.; Klocker, H.; Eder, I.E. In vitro model systems to study androgen receptor signaling in prostate cancer. Endocr. Relat. Cancer 2013, 20, R49–R64. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, N.; Granata, I.; Capaia, M.; Piccirillo, M.; Guarracino, M.R.; Vene, R.; Brizzolara, A.; Petretto, A.; Inglese, E.; Morini, M.; et al. Adaptive phenotype drives resistance to androgen deprivation therapy in prostate cancer. Cell Commun. Signal. 2017, 15, 51. [Google Scholar] [CrossRef] [PubMed]
- Ji, G.; Huang, C.; He, S.; Gong, Y.; Song, G.; Li, X.; Zhou, L. Comprehensive analysis of m6A regulators prognostic value in prostate cancer. Aging 2020, 12, 14863–14884. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhang, X.; Ma, C.; Hu, J. m6A Methylation Regulators Are Predictive Biomarkers for Tumour Metastasis in Prostate Cancer. Cancers 2022, 14, 4035. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Wang, Y.; Li, H. RNA m6A Methylation Regulators Multi-Omics Analysis in Prostate Cancer. Front. Genet. 2021, 12, 768041. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, H.; Zheng, J.; Shao, C. Analysis of RNA m6A methylation regulators and tumour immune cell infiltration characterization in prostate cancer. Artif. Cells Nanomed. Biotechnol. 2021, 49, 407–435. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, Z.; He, L.; Li, K.; Hu, C.; Chen, J.; Zhou, F.; Wang, J.; Li, Y.; Xiao, H. Molecular Characterization and Clinical Relevance of N6-Methyladenosine Regulators in Metastatic Prostate Cancer. Front. Oncol. 2022, 12, 914692. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Wei, B.; Wang, X.; Kang, R. METTL3 enhances cell adhesion through stabilizing integrin beta1 mRNA via an m6A-HuR-dependent mechanism in prostatic carcinoma. Am. J. Cancer Res. 2020, 10, 1012–1025. [Google Scholar] [PubMed]
- Wang, Z.; Sun, H.; Zhu, H.; Gu, D.; Chen, X.; Pan, Y.; Zheng, B.; Yang, D. Demethylase FTO inhibits the development of prostate cancer by upregulating EGR2 expression in an m6A manner. Turk. J. Biol. 2022, 46, 426–438. [Google Scholar] [CrossRef]
- Ma, X.X.; Cao, Z.G.; Zhao, S.L. m6A methyltransferase METTL3 promotes the progression of prostate cancer via m6A-modified LEF1. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3565–3571. [Google Scholar] [CrossRef] [PubMed]
- Cotter, K.A.; Gallon, J.; Uebersax, N.; Rubin, P.; Meyer, K.D.; Piscuoglio, S.; Jaffrey, S.R.; Rubin, M.A. Mapping of m6A and Its Regulatory Targets in Prostate Cancer Reveals a METTL3-Low Induction of Therapy Resistance. Mol. Cancer Res. 2021, 19, 1398–1411. [Google Scholar] [CrossRef] [PubMed]
- Lothion-Roy, J.; Haigh, D.B.; Harris, A.E.; Metzler, V.M.; Alsaleem, M.; Toss, M.S.; Kariri, Y.; Ntekim, A.; Robinson, B.D.; Khani, F.; et al. Clinical and molecular significance of the RNA m6A methyltransferase complex in prostate cancer. Front. Genet. 2022, 13, 1096071. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Chen, G.; Feng, Z.; Zhu, B.; Zhou, L.; Zhang, Y.; Mai, J.; Jiang, C.; Zeng, J. YTHDF1 promotes the proliferation, migration, and invasion of prostate cancer cells by regulating TRIM44. Genes Genom. 2021, 43, 1413–1421. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Shen, Y.; Jia, G.; Deng, Z.; Shi, F.; Jing, Y.; Xia, S. Activation of the HNRNPA2B1/miR-93-5p/FRMD6 axis facilitates prostate cancer progression in an m6A-dependent manner. J. Cancer 2023, 14, 1242–1256. [Google Scholar] [CrossRef] [PubMed]
- Jafari Najaf Abadi, M.H.; Shafabakhsh, R.; Asemi, Z.; Mirzaei, H.R.; Sahebnasagh, R.; Mirzaei, H.; Hamblin, M.R. CFIm25 and alternative polyadenylation: Conflicting roles in cancer. Cancer Lett. 2019, 459, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Luo, Y.; Zhang, F.; Ma, L. Exosome-derived lncRNA A1BG-AS1 attenuates the progression of prostate cancer depending on ZC3H13-mediated m6A modification. Cell Div. 2024, 19, 5. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lin, H.; Zhou, M.; Xiang, Q.; Deng, Y.; Luo, L.; Liu, Y.; Zhu, Z.; Zhao, Z. The m6A methylation regulator-based signature for predicting the prognosis of prostate cancer. Future Oncol. 2020, 16, 2421–2432. [Google Scholar] [CrossRef] [PubMed]
- Mo, L.; Meng, L.; Huang, Z.; Yi, L.; Yang, N.; Li, G. An analysis of the role of HnRNP C dysregulation in cancers. Biomark. Res. 2022, 10, 19. [Google Scholar] [CrossRef]
- Wang, S.; Xu, G.; Chao, F.; Zhang, C.; Han, D.; Chen, G. HNRNPC Promotes Proliferation, Metastasis and Predicts Prognosis in Prostate Cancer. Cancer Manag. Res. 2021, 13, 7263–7276. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Long, Z.; Yang, T.; Wang, S.; Zhong, W.; Hu, F.; Teoh, J.Y.; Lu, J.; Mao, X. M6A-modified circRBM33 promotes prostate cancer progression via PDHA1-mediated mitochondrial respiration regulation and presents a potential target for ARSI therapy. Int. J. Biol. Sci. 2023, 19, 1543–1563. [Google Scholar] [CrossRef] [PubMed]
- Adamson, B.; Brittain, N.; Walker, L.; Duncan, R.; Luzzi, S.; Rescigno, P.; Smith, G.; McGill, S.; Burchmore, R.J.; Willmore, E.; et al. The catalytic subunit of DNA-PK regulates transcription and splicing of AR in advanced prostate cancer. J. Clin. Investig. 2023, 133, e169200. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Lin, B.; Li, T.; Liu, Y.; Li, Y.; Zhou, X.; Miao, M.; Gu, J.; Pan, H.; Yang, F.; et al. A dual yet opposite growth-regulating function of miR-204 and its target XRN1 in prostate adenocarcinoma cells and neuroendocrine-like prostate cancer cells. Oncotarget 2015, 6, 7686–7700. [Google Scholar] [CrossRef] [PubMed]
- Mancarella, C.; Scotlandi, K. IGF2BP3 From Physiology to Cancer: Novel Discoveries, Unsolved Issues, and Future Perspectives. Front. Cell Dev. Biol. 2019, 7, 363. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Wang, R.; Zheng, Q.; Shen, D.; Wang, H.; Lu, Z.; Luo, W.; Xie, H.; Ren, L.; Jiang, M.; et al. circPDE5A regulates prostate cancer metastasis via controlling WTAP-dependent N6-methyladenisine methylation of EIF3C mRNA. J. Exp. Clin. Cancer Res. 2022, 41, 187. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Chen, W.; Zhou, X.; Yang, T.; Luo, J.; Long, Z.; Wu, J.; Lv, D.; Mao, X.; Cen, S. N6-methyladenosine demethylase FTO suppressed prostate cancer progression by maintaining CLIC4 mRNA stability. Cell Death Discov. 2022, 8, 184. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Li, L.; Wei, X.; Xu, F.; Huang, X.; Qi, F.; Zhang, Y.; Li, X. HNRNPC suppresses tumor immune microenvironment by activating Treg cells promoting the progression of prostate cancer. Cancer Sci. 2023, 114, 1830–1845. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liyanage, C.; Fernando, A.; Chamberlain, A.; Moradi, A.; Batra, J. RNA m6a Methylation Regulator Expression in Castration-Resistant Prostate Cancer Progression and Its Genetic Associations. Cancers 2024, 16, 1303. https://doi.org/10.3390/cancers16071303
Liyanage C, Fernando A, Chamberlain A, Moradi A, Batra J. RNA m6a Methylation Regulator Expression in Castration-Resistant Prostate Cancer Progression and Its Genetic Associations. Cancers. 2024; 16(7):1303. https://doi.org/10.3390/cancers16071303
Chicago/Turabian StyleLiyanage, Chamikara, Achala Fernando, Audrey Chamberlain, Afshin Moradi, and Jyotsna Batra. 2024. "RNA m6a Methylation Regulator Expression in Castration-Resistant Prostate Cancer Progression and Its Genetic Associations" Cancers 16, no. 7: 1303. https://doi.org/10.3390/cancers16071303
APA StyleLiyanage, C., Fernando, A., Chamberlain, A., Moradi, A., & Batra, J. (2024). RNA m6a Methylation Regulator Expression in Castration-Resistant Prostate Cancer Progression and Its Genetic Associations. Cancers, 16(7), 1303. https://doi.org/10.3390/cancers16071303