
History of Developing Acute Promyelocytic Leukemia Treatment and Role of Promyelocytic Leukemia Bodies

{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
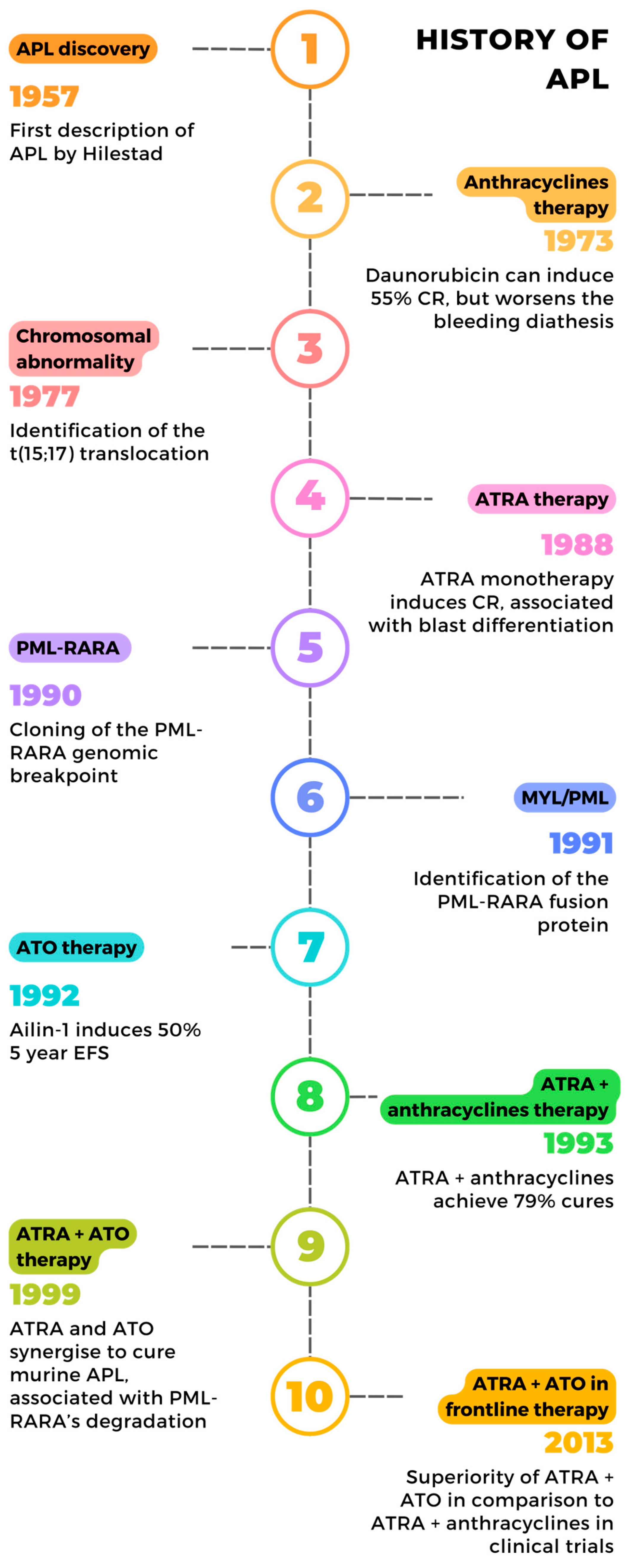
1. Introduction
2. Conventional and Unconventional Therapies
2.1. Chemotherapy’s Early Successes
2.2. ATRA: Toward a Differentiation Therapy
2.3. Arsenic, a Secular Medicine
2.4. The Reintroduction of Arsenic in Modern Medicine
2.5. The ATRA + ATO Synergy
3. Understanding APL
3.1. The Discovery of PML-RARA
3.2. RARA’s Regulation of Myeloid Differentiation
3.3. PML-RARA Impairs the Transcriptional Regulation of Hematopoietic Cells
4. PML’s Contribution to APL Pathogenesis and Response
4.1. PML Nuclear Bodies: Tightly Organized Membrane-Less Compartments
4.2. PML Nuclear Bodies: From Structure to Function
4.3. PML NBs Disruption Block Their Tumor Suppressive Functions
4.4. PML-Mediated Chromatin Regulation
4.5. PML-Mediated PML-RARA Multimerization
4.6. Recruitment of Partner Proteins on PML-RARA
4.7. PML and Leukemia Initiating Cells
5. Understanding APL Cure
5.1. ATO Drives PML-RARA Degradation
5.2. Revisiting ATRA Treatment of APL: Is It a Differentiation Therapy?
6. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kampen, K.R. The Discovery and Early Understanding of Leukemia. Leuk. Res. 2012, 36, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Bernard, J. Polyglobulies et Leucémies Provoquées Par Des Injections Intramédullaires de Goudron. Ph.D. Thesis, Université de Strasbourg, Strasbourg, France, 1936. [Google Scholar]
- Lederman, M. The Early History of Radiotherapy: 1895–1939. Int. J. Radiat. Oncol. Biol. Phys. 1981, 7, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Bessis, M.; Bernard, J. Remarkable results of the exsanguino-transfusion treatment of a case of acute leukemia. Bull. Mem. Soc. Med. Hop. Paris 1947, 63, 871–877. [Google Scholar]
- Hillestad, L.K. Acute Promyelocytic Leukemia. Acta Med. Scand. 1957, 159, 189–194. [Google Scholar] [CrossRef]
- Bernard, J.; Mathe, G.; Boulay, J.; Ceoard, B.; Chome, J. Acute promyelocytic leukemia: A study made on 20 cases. Schweiz. Med. Wochenschr. 1959, 89, 604–608. [Google Scholar] [PubMed]
- Sant, M.; Allemani, C.; Tereanu, C.; De Angelis, R.; Capocaccia, R.; Visser, O.; Marcos-Gragera, R.; Maynadié, M.; Simonetti, A.; Lutz, J.-M.; et al. Incidence of Hematologic Malignancies in Europe by Morphologic Subtype: Results of the HAEMACARE Project. Blood 2010, 116, 3724–3734. [Google Scholar] [CrossRef] [PubMed]
- Rickles, F.R.; Falanga, A.; Montesinos, P.; Sanz, M.A.; Brenner, B.; Barbui, T. Bleeding and Thrombosis in Acute Leukemia: What Does the Future of Therapy Look Like? Thromb. Res. 2007, 120 (Suppl. S2), S99–S106. [Google Scholar] [CrossRef] [PubMed]
- Krumbhaar, E.B.; Krumbhaar, H.D. The Blood and Bone Marrow in Yelloe Cross Gas (Mustard Gas) Poisoning: Changes Produced in the Bone Marrow of Fatal Cases. J. Med. Res. 1919, 40, 497–508.3. [Google Scholar] [PubMed]
- Goodman, L.S. Nitrogen Mustard Therapy: Use of Methyl-Bis(Beta-Chloroethyl)Amine Hydrochloride and Tris(Beta-Chloroethyl)Amine Hydrochloride for Hodgkin’s Disease, Lymphosarcoma, Leukemia and Certain Allied and Miscellaneous Disorders. JAMA 1946, 132, 126. [Google Scholar] [CrossRef]
- Gilman, A. Therapeutic Applications of Chemical Warfare Agents. Fed. Proc. 1946, 5, 285–292. [Google Scholar]
- Gilman, A.; Philips, F.S. The Biological Actions and Therapeutic Applications of the B-Chloroethyl Amines and Sulfides. Science 1946, 103, 409–436. [Google Scholar] [CrossRef]
- Grein, A.; Spalla, C.; di Marco, A.; Canevazzi, G. Descrizione e Classificazione Di Un Attinomicete (Streptomyces Peucetius Sp. Nova). Produttore Di Una Sostanza Ad Attivit Antitumorale: La Daunomicina. Giorn. Microbiol. 1963, 11, 109–118. (In Italian) [Google Scholar]
- Tan, C.; Tasaka, H.; Yu, K.-P.; Murphy, M.L.; Karnofsky, D.A. Daunomycin, an Antitumor Antibiotic, in the Treatment of Neoplastic Disease.Clinical Evaluation with Special Reference to Childhood Leukemia. Cancer 1967, 20, 333–353. [Google Scholar] [CrossRef]
- Bernard, J.; Weil, M.; Boiron, M.; Jacquillat, C.; Flandrin, G.; Gemon, M.F. Acute Promyelocytic Leukemia: Results of Treatment by Daunorubicin. Blood 1973, 41, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Mandelli, F.; Labopin, M.; Granena, A.; Iriondo, A.; Prentice, G.; Bacigalupo, A.; Sierra, J.; Meloni, G.; Frassoni, F.; Goldman, J. European Survey of Bone Marrow Transplantation in Acute Promyelocytic Leukemia (M3). Working Party on Acute Leukemia of the European Cooperative Group for Bone Marrow Transplantation (EMBT). Bone Marrow Transpl. 1994, 14, 293–298. [Google Scholar]
- Fenaux, P.; Le Deley, M.C.; Castaigne, S.; Archimbaud, E.; Chomienne, C.; Link, H.; Guerci, A.; Duarte, M.; Daniel, M.T.; Bowen, D. Effect of All Transretinoic Acid in Newly Diagnosed Acute Promyelocytic Leukemia. Results of a Multicenter Randomized Trial. European APL 91 Group. Blood 1993, 82, 3241–3249. [Google Scholar] [CrossRef]
- Bollag, W. Retinoids and Cancer. Cancer Chemother. Pharmacol. 1979, 3, 209–215. [Google Scholar] [CrossRef]
- Liang, C.; Qiao, G.; Liu, Y.; Tian, L.; Hui, N.; Li, J.; Ma, Y.; Li, H.; Zhao, Q.; Cao, W.; et al. Overview of All-Trans-Retinoic Acid (ATRA) and Its Analogues: Structures, Activities, and Mechanisms in Acute Promyelocytic Leukaemia. Eur. J. Med. Chem. 2021, 220, 113451. [Google Scholar] [CrossRef] [PubMed]
- Daenen, S.; Vellenga, E.; van Dobbenburgh, O.A.; Halie, M.R. Retinoic Acid as Antileukemic Therapy in a Patient with Acute Promyelocytic Leukemia and Aspergillus Pneumonia. Blood 1986, 67, 559–561. [Google Scholar] [CrossRef]
- Flynn, P.J.; Miller, W.J.; Weisdorf, D.J.; Arthur, D.C.; Brunning, R.; Branda, R.F. Retinoic Acid Treatment of Acute Promyelocytic Leukemia: In Vitro and in Vivo Observations. Blood 1983, 62, 1211–1217. [Google Scholar] [CrossRef]
- Fontana, J.A.; Rogers, J.S.; Durham, J.P. The Role of 13 Cis-Retinoic Acid in the Remission Induction of a Patient with Acute Promyelocytic Leukemia. Cancer 1986, 57, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, B. Probable in Vivo Induction of Differentiation by Retinoic Acid of Promyelocytes in Acute Promyelocytic Leukaemia. Br. J. Haematol. 1984, 57, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Sampi, K.; Honma, Y.; Hozumi, M.; Sakurai, M. Discrepancy between In-Vitro and in-Vivo Inductions of Differentiation by Retinoids of Human Acute Promyelocytic Leukemia Cells in Relapse. Leuk. Res. 1985, 9, 1475–1478. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Ye, Y.; Chen, S.; Chai, J.; Lu, J.; Zhoa, L.; Gu, L.; Wang, Z. Use of All-Trans Retinoic Acid in the Treatment of Acute Promyelocytic Leukemia. Blood 1988, 72, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Degos, L.; Chomienne, C.; Daniel, M.T.; Berger, R.; Dombret, H.; Fenaux, P.; Castaigne, S. Treatment of First Relapse in Acute Promyelocytic Leukaemia with All-Trans Retinoic Acid. Lancet 1990, 336, 1440–1441. [Google Scholar] [CrossRef]
- Warrell, R.P.; Frankel, S.R.; Miller, W.H.; Scheinberg, D.A.; Itri, L.M.; Hittelman, W.N.; Vyas, R.; Andreeff, M.; Tafuri, A.; Jakubowski, A.; et al. Differentiation Therapy of Acute Promyelocytic Leukemia with Tretinoin (All-Trans-Retinoic Acid). N. Engl. J. Med. 1991, 324, 1385–1393. [Google Scholar] [CrossRef]
- Castaigne, S.; Chomienne, C.; Daniel, M.T.; Ballerini, P.; Berger, R.; Fenaux, P.; Degos, L. All-Trans Retinoic Acid as a Differentiation Therapy for Acute Promyelocytic Leukemia. I. Clinical Results. Blood 1990, 76, 1704–1709. [Google Scholar] [CrossRef]
- Chen, Z.X.; Xue, Y.Q.; Zhang, R.; Tao, R.F.; Xia, X.M.; Li, C.; Wang, W.; Zu, W.Y.; Yao, X.Z.; Ling, B.J. A Clinical and Experimental Study on All-Trans Retinoic Acid-Treated Acute Promyelocytic Leukemia Patients. Blood 1991, 78, 1413–1419. [Google Scholar] [CrossRef]
- Fenaux, P.; Castaigne, S.; Dombret, H.; Archimbaud, E.; Duarte, M.; Morel, P.; Lamy, T.; Tilly, H.; Guerci, A.; Maloisel, F. All-Transretinoic Acid Followed by Intensive Chemotherapy Gives a High Complete Remission Rate and May Prolong Remissions in Newly Diagnosed Acute Promyelocytic Leukemia: A Pilot Study on 26 Cases. Blood 1992, 80, 2176–2181. [Google Scholar] [CrossRef]
- Tallman, M.S.; Andersen, J.W.; Schiffer, C.A.; Appelbaum, F.R.; Feusner, J.H.; Ogden, A.; Shepherd, L.; Willman, C.; Bloomfield, C.D.; Rowe, J.M.; et al. All- Trans -Retinoic Acid in Acute Promyelocytic Leukemia. N. Engl. J. Med. 1997, 337, 1021–1028. [Google Scholar] [CrossRef]
- Fenaux, P.; Chastang, C.; Chevret, S.; Sanz, M.; Dombret, H.; Archimbaud, E.; Fey, M.; Rayon, C.; Huguet, F.; Sotto, J.J.; et al. A Randomized Comparison of All Transretinoic Acid (ATRA) Followed by Chemotherapy and ATRA plus Chemotherapy and the Role of Maintenance Therapy in Newly Diagnosed Acute Promyelocytic Leukemia. The European APL Group. Blood 1999, 94, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Degos, L.; Dombret, H.; Chomienne, C.; Daniel, M.; Miclea, J.; Chastang, C.; Castaigne, S.; Fenaux, P. All-Trans-Retinoic Acid as a Differentiating Agent in the Treatment of Acute Promyelocytic Leukemia. Blood 1995, 85, 2643–2653. [Google Scholar] [CrossRef] [PubMed]
- Lo-Coco, F.; Avvisati, G.; Vignetti, M.; Thiede, C.; Orlando, S.M.; Iacobelli, S.; Ferrara, F.; Fazi, P.; Cicconi, L.; Di Bona, E.; et al. Retinoic Acid and Arsenic Trioxide for Acute Promyelocytic Leukemia. N. Engl. J. Med. 2013, 369, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Frankel, S.R.; Eardley, A.; Lauwers, G.; Weiss, M.; Warrell, R.P. The “Retinoic Acid Syndrome” in Acute Promyelocytic Leukemia. Ann. Intern. Med. 1992, 117, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.A.; Montesinos, P. How We Prevent and Treat Differentiation Syndrome in Patients with Acute Promyelocytic Leukemia. Blood 2014, 123, 2777–2782. [Google Scholar] [CrossRef] [PubMed]
- Reyhanoglu, G.; Hughes, B.; King, K.E.; Cambridge, R. Differentiation Syndrome, a Side Effect from the Therapy of Acute Promyelocytic Leukemia. Cureus 2020, 12, e12042. [Google Scholar] [CrossRef] [PubMed]
- Warrell, R.P.; de The, H.; Wang, Z.-Y.; Degos, L. Acute Promyelocytic Leukemia. N. Engl. J. Med. 1993, 329, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Haller, J.S. Therapeutic Mule: The Use of Arsenic in the Nineteenth Century Materia Medica. Pharm. Hist. 1975, 17, 87–100. [Google Scholar] [PubMed]
- Zhu, J.; Chen, Z.; Lallemand-Breitenbach, V.; de Thé, H. How Acute Promyelocytic Leukaemia Revived Arsenic. Nat. Rev. Cancer 2002, 2, 705–714. [Google Scholar] [CrossRef]
- Degos, L. The History of Acute Promyelocytic Leukaemia. Br. J. Haematol. 2003, 122, 539–553. [Google Scholar] [CrossRef]
- Sun, H.D.; Ma, L.; Hu, X.C.; Zhang, T.D. Ai-Lin I Treated 32 Cases of Acute Promyelocytic Leukemia. Chin. J Integrat. Chin. West Med. 1992, 2, 170–172. [Google Scholar]
- Shen, Z.-X.; Chen, G.-Q.; Ni, J.-H.; Li, X.-S.; Xiong, S.-M.; Qiu, Q.-Y.; Zhu, J.; Tang, W.; Sun, G.-L.; Yang, K.-Q.; et al. Use of Arsenic Trioxide (As2O3) in the Treatment of Acute Promyelocytic Leukemia (APL): II. Clinical Efficacy and Pharmacokinetics in Relapsed Patients. Blood 1997, 89, 3354–3360. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Q.; Shi, X.G.; Tang, W.; Xiong, S.M.; Zhu, J.; Cai, X.; Han, Z.G.; Ni, J.H.; Shi, G.Y.; Jia, P.M.; et al. Use of Arsenic Trioxide (As2O3) in the Treatment of Acute Promyelocytic Leukemia (APL): I. As2O3 Exerts Dose-Dependent Dual Effects on APL Cells. Blood 1997, 89, 3345–3353. [Google Scholar] [PubMed]
- Niu, C.; Yan, H.; Yu, T.; Sun, H.P.; Liu, J.X.; Li, X.S.; Wu, W.; Zhang, F.Q.; Chen, Y.; Zhou, L.; et al. Studies on Treatment of Acute Promyelocytic Leukemia with Arsenic Trioxide: Remission Induction, Follow-up, and Molecular Monitoring in 11 Newly Diagnosed and 47 Relapsed Acute Promyelocytic Leukemia Patients. Blood 1999, 94, 3315–3324. [Google Scholar] [CrossRef] [PubMed]
- Soignet, S.L.; Maslak, P.; Wang, Z.-G.; Jhanwar, S.; Calleja, E.; Dardashti, L.J.; Corso, D.; DeBlasio, A.; Gabrilove, J.; Scheinberg, D.A.; et al. Complete Remission after Treatment of Acute Promyelocytic Leukemia with Arsenic Trioxide. N. Engl. J. Med. 1998, 339, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Mathews, V.; George, B.; Chendamarai, E.; Lakshmi, K.M.; Desire, S.; Balasubramanian, P.; Viswabandya, A.; Thirugnanam, R.; Abraham, A.; Shaji, R.V.; et al. Single-Agent Arsenic Trioxide in the Treatment of Newly Diagnosed Acute Promyelocytic Leukemia: Long-Term Follow-Up Data. J. Clin. Oncol. 2010, 28, 3866–3871. [Google Scholar] [CrossRef]
- Ghavamzadeh, A.; Alimoghaddam, K.; Rostami, S.; Ghaffari, S.H.; Jahani, M.; Iravani, M.; Mousavi, S.A.; Bahar, B.; Jalili, M. Phase II Study of Single-Agent Arsenic Trioxide for the Front-Line Therapy of Acute Promyelocytic Leukemia. J. Clin. Oncol. 2011, 29, 2753–2757. [Google Scholar] [CrossRef]
- Chen, G.; Zhu, J.; Shi, X.; Ni, J.; Zhong, H.; Si, G.; Jin, X.; Tang, W.; Li, X.; Xong, S.; et al. In Vitro Studies on Cellular and Molecular Mechanisms of Arsenic Trioxide (As2O3) in the Treatment of Acute Promyelocytic Leukemia: As2O3 Induces NB4 Cell Apoptosis with Downregulation of Bcl-2 Expression and Modulation of PML-RAR Alpha/PML Proteins. Blood 1996, 88, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Fanelli, M.; Ferrara, F.F.; Riccioni, R.; Rosenauer, A.; Davison, K.; Lamph, W.W.; Waxman, S.; Pelicci, P.G.; Lo Coco, F.; et al. Arsenic Trioxide as an Inducer of Apoptosis and Loss of PML/RARα Protein in Acute Promyelocytic Leukemia Cells. JNCI J. Natl. Cancer Inst. 1998, 90, 124–133. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; Guillemin, M.-C.; Janin, A.; Daniel, M.-T.; Degos, L.; Kogan, S.C.; Michael Bishop, J.; de Thé, H. Retinoic Acid and Arsenic Synergize to Eradicate Leukemic Cells in a Mouse Model of Acute Promyelocytic Leukemia. J. Exp. Med. 1999, 189, 1043–1052. [Google Scholar] [CrossRef]
- Rego, E.M.; He, L.-Z.; Warrell, R.P.; Wang, Z.-G.; Pandolfi, P.P. Retinoic Acid (RA) and As2O3 Treatment in Transgenic Models of Acute Promyelocytic Leukemia (APL) Unravel the Distinct Nature of the Leukemogenic Process Induced by the PML-RARα and PLZF-RARα Oncoproteins. Proc. Natl. Acad. Sci. USA 2000, 97, 10173–10178. [Google Scholar] [CrossRef] [PubMed]
- Estey, E.; Garcia-Manero, G.; Ferrajoli, A.; Faderl, S.; Verstovsek, S.; Jones, D.; Kantarjian, H. Use of All-Trans Retinoic Acid plus Arsenic Trioxide as an Alternative to Chemotherapy in Untreated Acute Promyelocytic Leukemia. Blood 2006, 107, 3469–3473. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, X.; Wang, B.; Rong, Z.; Qi, H.; Chen, H. The Efficacy and Safety of Arsenic Trioxide with or without All-Trans Retinoic Acid for the Treatment of Acute Promyelocytic Leukemia: A Meta-Analysis. Leuk. Res. 2011, 35, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.A.; Fenaux, P.; Tallman, M.S.; Estey, E.H.; Löwenberg, B.; Naoe, T.; Lengfelder, E.; Döhner, H.; Burnett, A.K.; Chen, S.-J.; et al. Management of Acute Promyelocytic Leukemia: Updated Recommendations from an Expert Panel of the European LeukemiaNet. Blood 2019, 133, 1630–1643. [Google Scholar] [CrossRef] [PubMed]
- Long, Z.-J.; Hu, Y.; Li, X.-D.; He, Y.; Xiao, R.-Z.; Fang, Z.-G.; Wang, D.-N.; Liu, J.-J.; Yan, J.-S.; Huang, R.-W.; et al. ATO/ATRA/Anthracycline-Chemotherapy Sequential Consolidation Achieves Long-Term Efficacy in Primary Acute Promyelocytic Leukemia. PLoS ONE 2014, 9, e104610. [Google Scholar] [CrossRef]
- Golomb, H.M.; Rowley, J.; Vardiman, J.; Baron, J.; Locker, G.; Krasnow, S. Partial Deletion of Long Arm of Chromosome 17: A Specific Abnormality in Acute Promyelocytic Leukemia? Arch. Intern. Med. 1976, 136, 825–828. [Google Scholar] [CrossRef]
- Rowley, J.D.; Golomb, H.M.; Vardiman, J.; Fukuhara, S.; Dougherty, C.; Potter, D. Further Evidence for a Non-Random Chromosomal Abnormality in Acute Promyelocytic Leukemia. Int. J. Cancer 1977, 20, 869–872. [Google Scholar] [CrossRef]
- Rowley, J.D.; Golomb, H.M.; Dougherty, C. 15/17 Translocation, a Consistent Chromosomal Change in Acute Promyelocytic Leukaemia. Lancet 1977, 309, 549–550. [Google Scholar] [CrossRef]
- Larson, R.A.; Kondo, K.; Vardiman, J.W.; Butler, A.E.; Golomb, H.M.; Rowley, J.D. Evidence for a 15; 17 Translocation in Every Patient with Acute Promyelocytic Leukemia. Am. J. Med. 1984, 76, 827–841. [Google Scholar] [CrossRef]
- Mattei, M.-G.; Petkovich, M.; Mattei, J.-F.; Brand, N.; Chambon, P. Mapping of the Human Retinoic Acid Receptor to the Q21 Band of Chromosome 17. Hum. Genet. 1988, 80, 186–188. [Google Scholar] [CrossRef]
- Chomienne, C.; Ballerini, P.; Balitrand, N.; Huang, M.E.; Krawice, I.; Castaigne, S.; Fenaux, P.; Tiollais, P.; Dejean, A.; Degos, L.; et al. The Retinoic Acid Receptor Alpha Gene Is Rearranged in Retinoic Acid-Sensitive Promyelocytic Leukemias. Leukemia 1990, 4, 802–807. [Google Scholar] [PubMed]
- de Thé, H.; Chomienne, C.; Lanotte, M.; Degos, L.; Dejean, A. The t(15;17) Translocation of Acute Promyelocytic Leukaemia Fuses the Retinoic Acid Receptor α Gene to a Novel Transcribed Locus. Nature 1990, 347, 558–561. [Google Scholar] [CrossRef]
- Borrow, J.; Goddard, A.; Sheer, D.; Solomon, E. Molecular Analysis of Acute Promyelocytic Leukemia Breakpoint Cluster Region on Chromosome 17. Science 1990, 249, 1577–1580. [Google Scholar] [CrossRef] [PubMed]
- Longo, L.; Pandolfi, P.P.; Biondi, A.; Rambaldi, A.; Mencarelli, A.; Lo Coco, F.; Diverio, D.; Pegoraro, L.; Avanzi, G.; Tabilio, A. Rearrangements and Aberrant Expression of the Retinoic Acid Receptor Alpha Gene in Acute Promyelocytic Leukemias. J. Exp. Med. 1990, 172, 1571–1575. [Google Scholar] [CrossRef] [PubMed]
- de Thé, H.; Lavau, C.; Marchio, A.; Chomienne, C.; Degos, L.; Dejean, A. The PML-RARα Fusion mRNA Generated by the t(15;17) Translocation in Acute Promyelocytic Leukemia Encodes a Functionally Altered RAR. Cell 1991, 66, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Goddard, A.D.; Borrow, J.; Freemont, P.S.; Solomon, E. Characterization of a Zinc Finger Gene Disrupted by the t(15;17) in Acute Promyelocytic Leukemia. Science 1991, 254, 1371–1374. [Google Scholar] [CrossRef] [PubMed]
- Kakizuka, A.; Miller, W.H.; Umesono, K.; Warrell, R.P.; Frankel, S.R.; Murty, V.V.V.S.; Dmitrovsky, E.; Evans, R.M. Chromosomal Translocation t(15;17) in Human Acute Promyelocytic Leukemia Fuses RARα with a Novel Putative Transcription Factor, PML. Cell 1991, 66, 663–674. [Google Scholar] [CrossRef]
- Rousselot, P.; Hardas, B.; Patel, A.; Guidez, F.; Gäken, J.; Castaigne, S.; Dejean, A.; de Thé, H.; Degos, L.; Farzaneh, F. The PML-RAR Alpha Gene Product of the t(15;17) Translocation Inhibits Retinoic Acid-Induced Granulocytic Differentiation and Mediated Transactivation in Human Myeloid Cells. Oncogene 1994, 9, 545–551. [Google Scholar] [PubMed]
- Kastner, P.; Lawrence, H.J.; Waltzinger, C.; Ghyselinck, N.B.; Chambon, P.; Chan, S. Positive and Negative Regulation of Granulopoiesis by Endogenous RARα. Blood 2001, 97, 1314–1320. [Google Scholar] [CrossRef]
- Collins, S. The Role of Retinoids and Retinoic Acid Receptors in Normal Hematopoiesis. Leukemia 2002, 16, 1896–1905. [Google Scholar] [CrossRef]
- Du, C.; Redner, R.L.; Cooke, M.P.; Lavau, C. Overexpression of Wild-Type Retinoic Acid Receptor Alpha (RARalpha) Recapitulates Retinoic Acid-Sensitive Transformation of Primary Myeloid Progenitors by Acute Promyelocytic Leukemia RARalpha-Fusion Genes. Blood 1999, 94, 793–802. [Google Scholar] [CrossRef]
- Astolfi, A.; Masetti, R.; Indio, V.; Bertuccio, S.N.; Messelodi, D.; Rampelli, S.; Leardini, D.; Carella, M.; Serravalle, S.; Libri, V.; et al. Torque Teno Mini Virus as a Cause of Childhood Acute Promyelocytic Leukemia Lacking PML/RARA Fusion. Blood 2021, 138, 1773–1777. [Google Scholar] [CrossRef]
- Chen, X.; Wang, F.; Zhou, X.; Zhang, Y.; Cao, P.; Ma, X.; Yuan, L.; Fang, J.; Liu, M.; Liu, M.; et al. Torque Teno Mini Virus Driven Childhood Acute Promyelocytic Leukemia: The Third Case Report and Sequence Analysis. Front. Oncol. 2022, 12, 1074913. [Google Scholar] [CrossRef] [PubMed]
- Privalsky, M.L. The Role of Corepressors in Transcriptional Regulation by Nuclear Hormone Receptors. Annu. Rev. Physiol. 2004, 66, 315–360. [Google Scholar] [CrossRef]
- Huang, P.; Chandra, V.; Rastinejad, F. Retinoic Acid Actions through Mammalian Nuclear Receptors. Chem. Rev. 2014, 114, 233–254. [Google Scholar] [CrossRef]
- le Maire, A.; Teyssier, C.; Balaguer, P.; Bourguet, W.; Germain, P. Regulation of RXR-RAR Heterodimers by RXR- and RAR-Specific Ligands and Their Combinations. Cells 2019, 8, 1392. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.; Kogan, S.; Lagasse, E.; Weissman, I.; Alcalay, M.; Pelicci, P.G.; Atwater, S.; Bishop, J.M. A PMLRARα Transgene Initiates Murine Acute Promyelocytic Leukemia. Proc. Natl. Acad. Sci. USA 1997, 94, 2551–2556. [Google Scholar] [CrossRef] [PubMed]
- Grisolano, J.L.; Wesselschmidt, R.L.; Pelicci, P.G.; Ley, T.J. Altered Myeloid Development and Acute Leukemia in Transgenic Mice Expressing PML-RAR Alpha under Control of Cathepsin G Regulatory Sequences. Blood 1997, 89, 376–387. [Google Scholar] [CrossRef]
- Lanotte, M.; Martin-Thouvenin, V.; Najman, S.; Balerini, P.; Valensi, F.; Berger, R. NB4, a Maturation Inducible Cell Line with t(15;17) Marker Isolated from a Human Acute Promyelocytic Leukemia (M3). Blood 1991, 77, 1080–1086. [Google Scholar] [CrossRef]
- Collins, S.J.; Robertson, K.A.; Mueller, L. Retinoic Acid-Induced Granulocytic Differentiation of HL-60 Myeloid Leukemia Cells Is Mediated Directly through the Retinoic Acid Receptor (RAR-α). Mol. Cell. Biol. 1990, 10, 2154–2163. [Google Scholar] [CrossRef]
- Kogan, S.C. Mouse Models of Acute Promyelocytic Leukemia. In Acute Promyelocytic Leukemia; Pandolfi, P.P., Vogt, P.K., Eds.; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2007; Volume 313, pp. 3–29. ISBN 978-3-540-34592-3. [Google Scholar]
- Welch, J.S.; Yuan, W.; Ley, T.J. Expression of PML-RARα by the Murine PML Locus Leads to Myeloid Self-Renewal, Clonal Expansion and Morphologic Promyelocytic Leukemia. Blood 2008, 112, 932. [Google Scholar] [CrossRef]
- Lehmann-Che, J.; Bally, C.; Letouzé, E.; Berthier, C.; Yuan, H.; Jollivet, F.; Ades, L.; Cassinat, B.; Hirsch, P.; Pigneux, A.; et al. Dual Origin of Relapses in Retinoic-Acid Resistant Acute Promyelocytic Leukemia. Nat. Commun. 2018, 9, 2047. [Google Scholar] [CrossRef] [PubMed]
- Vickers, M.; Jackson, G.; Taylor, P. The Incidence of Acute Promyelocytic Leukemia Appears Constant over Most of a Human Lifespan, Implying Only One Rate Limiting Mutation. Leukemia 2000, 14, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Shyamsunder, P.; Han, L.; Mayakonda, A.; Nagata, Y.; Sundaresan, J.; Kanojia, D.; Yoshida, K.; Ganesan, S.; Hattori, N.; et al. Comprehensive Mutational Analysis of Primary and Relapse Acute Promyelocytic Leukemia. Leukemia 2016, 30, 1672–1681. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.S.; Greenblatt, S.M.; Shirley, C.M.; Duffield, A.S.; Bruner, J.K.; Li, L.; Nguyen, B.; Jung, E.; Aplan, P.D.; Ghiaur, G.; et al. All-Trans Retinoic Acid Synergizes with FLT3 Inhibition to Eliminate FLT3/ITD+ Leukemia Stem Cells in Vitro and in Vivo. Blood 2016, 127, 2867–2878. [Google Scholar] [CrossRef] [PubMed]
- Esnault, C.; Rahmé, R.; Rice, K.L.; Berthier, C.; Gaillard, C.; Quentin, S.; Maubert, A.-L.; Kogan, S.; De Thé, H. FLT3-ITD Impedes Retinoic Acid, but Not Arsenic, Responses in Murine Acute Promyelocytic Leukemias. Blood 2019, 133, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.J.; Evans, R.M. Acquisition of Oncogenic Potential by RAR Chimeras in Acute Promyelocytic Leukemia through Formation of Homodimers. Mol. Cell 2000, 5, 821–830. [Google Scholar] [CrossRef]
- Ablain, J.; de Thé, H. Retinoic Acid Signaling in Cancer: The Parable of Acute Promyelocytic Leukemia: Retinoic Acid Signaling in Cancer. Int. J. Cancer 2014, 135, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Nasr, R.; Pérès, L.; Riaucoux-Lormière, F.; Honoré, N.; Berthier, C.; Kamashev, D.; Zhou, J.; Vitoux, D.; Lavau, C.; et al. RXR Is an Essential Component of the Oncogenic PML/RARA Complex In Vivo. Cancer Cell 2007, 12, 23–35. [Google Scholar] [CrossRef]
- Martens, J.H.A.; Brinkman, A.B.; Simmer, F.; Francoijs, K.-J.; Nebbioso, A.; Ferrara, F.; Altucci, L.; Stunnenberg, H.G. PML-RARα/RXR Alters the Epigenetic Landscape in Acute Promyelocytic Leukemia. Cancer Cell 2010, 17, 173–185. [Google Scholar] [CrossRef]
- He, L.-Z.; Guidez, F.; Tribioli, C.; Peruzzi, D.; Ruthardt, M.; Zelent, A.; Pandolfi, P.P. Distinct Interactions of PML-RARα and PLZF-RARα with Co-Repressors Determine Differential Responses to RA in APL. Nat. Genet. 1998, 18, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Kamashev, D.; Vitoux, D.; De Thé, H. PML–RARA-RXR Oligomers Mediate Retinoid and Rexinoid/cAMP Cross-Talk in Acute Promyelocytic Leukemia Cell Differentiation. J. Exp. Med. 2004, 199, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, P.; Shi, J.; Zhu, X.; He, M.; Jia, X.; Yang, X.; Qiu, F.; Jin, W.; Qian, M.; et al. PML/RARα Targets Promoter Regions Containing PU.1 Consensus and RARE Half Sites in Acute Promyelocytic Leukemia. Cancer Cell 2010, 17, 186–197. [Google Scholar] [CrossRef]
- Tan, Y.; Wang, X.; Song, H.; Zhang, Y.; Zhang, R.; Li, S.; Jin, W.; Chen, S.; Fang, H.; Chen, Z.; et al. A PML/RARα Direct Target Atlas Redefines Transcriptional Deregulation in Acute Promyelocytic Leukemia. Blood 2021, 137, 1503–1516. [Google Scholar] [CrossRef]
- Villiers, W.; Kelly, A.; He, X.; Kaufman-Cook, J.; Elbasir, A.; Bensmail, H.; Lavender, P.; Dillon, R.; Mifsud, B.; Osborne, C.S. Multi-Omics and Machine Learning Reveal Context-Specific Gene Regulatory Activities of PML::RARA in Acute Promyelocytic Leukemia. Nat. Commun. 2023, 14, 724. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, M.-C.; de Thé, H. Classic and Variants APLs, as Viewed from a Therapy Response. Cancers 2020, 12, 967. [Google Scholar] [CrossRef] [PubMed]
- Liquori, A.; Ibañez, M.; Sargas, C.; Sanz, M.; Barragán, E.; Cervera, J. Acute Promyelocytic Leukemia: A Constellation of Molecular Events around a Single PML-RARA Fusion Gene. Cancers 2020, 12, 624. [Google Scholar] [CrossRef]
- Kastner, P.; Perez, A.; Lutz, Y.; Rochette-Egly, C.; Gaub, M.P.; Durand, B.; Lanotte, M.; Berger, R.; Chambon, P. Structure, Localization and Transcriptional Properties of Two Classes of Retinoic Acid Receptor Alpha Fusion Proteins in Acute Promyelocytic Leukemia (APL): Structural Similarities with a New Family of Oncoproteins. EMBO J. 1992, 11, 629–642. [Google Scholar] [CrossRef]
- Daniel, M.T.; Koken, M.; Romagné, O.; Barbey, S.; Bazarbachi, A.; Stadler, M.; Guillemin, M.C.; Degos, L.; Chomienne, C.; de Thé, H. PML Protein Expression in Hematopoietic and Acute Promyelocytic Leukemia Cells. Blood 1993, 82, 1858–1867. [Google Scholar] [CrossRef]
- De Thé, G.; Riviere, M.; Bernhard, W. Examen Au Microscope Électronique de La Tumeur VX2 Du Lapin Domestique Dérivée Du Papillome de Shope. Bull. Cancer 1960, 47, 570–584. [Google Scholar]
- Fagioli, M.; Alcalay, M.; Pandolfi, P.P.; Venturini, L.; Mencarelli, A.; Simeone, A.; Acampora, D.; Grignani, F.; Pelicci, P.G. Alternative Splicing of PML Transcripts Predicts Coexpression of Several Carboxy-Terminally Different Protein Isoforms. Oncogene 1992, 7, 1083–1091. [Google Scholar] [PubMed]
- Koken, M.H.; Puvion-Dutilleul, F.; Guillemin, M.C.; Viron, A.; Linares-Cruz, G.; Stuurman, N.; De Jong, L.; Szostecki, C.; Calvo, F.; Chomienne, C. The t(15;17) Translocation Alters a Nuclear Body in a Retinoic Acid-Reversible Fashion. EMBO J. 1994, 13, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, F.-M.; Hendzel, M.J.; Bazett-Jones, D.P. Promyelocytic Leukemia (Pml) Nuclear Bodies Are Protein Structures That Do Not Accumulate RNA. J. Cell Biol. 2000, 148, 283–292. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; De Thé, H. PML Nuclear Bodies. Cold Spring Harb. Perspect. Biol. 2010, 2, a000661. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.; Pandolfi, P.P. Structure, Dynamics and Functions of Promyelocytic Leukaemia Nuclear Bodies. Nat. Rev. Mol. Cell Biol. 2007, 8, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Reymond, A. The Tripartite Motif Family Identifies Cell Compartments. EMBO J. 2001, 20, 2140–2151. [Google Scholar] [CrossRef] [PubMed]
- Meroni, G.; Desagher, S. Cellular Function of TRIM E3 Ubiquitin Ligases in Health and Disease. Cells 2022, 11, 250. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Benhenda, S.; Wu, H.; Lallemand-Breitenbach, V.; Zhen, T.; Jollivet, F.; Peres, L.; Li, Y.; Chen, S.-J.; Chen, Z.; et al. RING Tetramerization Is Required for Nuclear Body Biogenesis and PML Sumoylation. Nat. Commun. 2018, 9, 1277. [Google Scholar] [CrossRef]
- Kamitani, T.; Kito, K.; Nguyen, H.P.; Wada, H.; Fukuda-Kamitani, T.; Yeh, E.T.H. Identification of Three Major Sentrinization Sites in PML. J. Biol. Chem. 1998, 273, 26675–26682. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; Zhu, J.; Puvion, F.; Koken, M.; Honoré, N.; Doubeikovsky, A.; Duprez, E.; Pandolfi, P.P.; Puvion, E.; Freemont, P.; et al. Role of Promyelocytic Leukemia (Pml) Sumolation in Nuclear Body Formation, 11s Proteasome Recruitment, and As2O3-Induced Pml or Pml/Retinoic Acid Receptor α Degradation. J. Exp. Med. 2001, 193, 1361–1372. [Google Scholar] [CrossRef]
- Sahin, U.; de Thé, H.; Lallemand-Breitenbach, V. PML Nuclear Bodies: Assembly and Oxidative Stress-Sensitive Sumoylation. Nucleus 2014, 5, 499–507. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; Jeanne, M.; Benhenda, S.; Nasr, R.; Lei, M.; Peres, L.; Zhou, J.; Zhu, J.; Raught, B.; de Thé, H. Arsenic Degrades PML or PML–RARα through a SUMO-Triggered RNF4/Ubiquitin-Mediated Pathway. Nat. Cell Biol. 2008, 10, 547–555. [Google Scholar] [CrossRef]
- Van Damme, E.; Laukens, K.; Dang, T.H.; Van Ostade, X. A Manually Curated Network of the PML Nuclear Body Interactome Reveals an Important Role for PML-NBs in SUMOylation Dynamics. Int. J. Biol. Sci. 2010, 6, 51–67. [Google Scholar] [CrossRef]
- Sahin, U.; Ferhi, O.; Jeanne, M.; Benhenda, S.; Berthier, C.; Jollivet, F.; Niwa-Kawakita, M.; Faklaris, O.; Setterblad, N.; de Thé, H.; et al. Oxidative Stress–Induced Assembly of PML Nuclear Bodies Controls Sumoylation of Partner Proteins. J. Cell Biol. 2014, 204, 931–945. [Google Scholar] [CrossRef]
- Müller, S.; Matunis, M.J.; Dejean, A. Conjugation with the Ubiquitin-Related Modifier SUMO-1 Regulates the Partitioning of PML within the Nucleus. EMBO J. 1998, 17, 61–70. [Google Scholar] [CrossRef]
- Shen, T.H.; Lin, H.-K.; Scaglioni, P.P.; Yung, T.M.; Pandolfi, P.P. The Mechanisms of PML-Nuclear Body Formation. Mol. Cell 2006, 24, 331–339. [Google Scholar] [CrossRef]
- Barroso-Gomila, O.; Trulsson, F.; Muratore, V.; Canosa, I.; Merino-Cacho, L.; Cortazar, A.R.; Pérez, C.; Azkargorta, M.; Iloro, I.; Carracedo, A.; et al. Identification of Proximal SUMO-Dependent Interactors Using SUMO-ID. Nat. Commun. 2021, 12, 6671. [Google Scholar] [CrossRef]
- Hirano, S.; Udagawa, O. SUMOylation Regulates the Number and Size of Promyelocytic Leukemia-Nuclear Bodies (PML-NBs) and Arsenic Perturbs SUMO Dynamics on PML by Insolubilizing PML in THP-1 Cells. Arch. Toxicol. 2022, 96, 545–558. [Google Scholar] [CrossRef]
- Antolini, F.; Bello, M.L.; Sette, M. Purified Promyelocytic Leukemia Coiled-Coil Aggregates as a Tetramer Displaying Low α-Helical Content. Protein Expr. Purif. 2003, 29, 94–102. [Google Scholar] [CrossRef]
- Li, Y.; Ma, X.; Chen, Z.; Wu, H.; Wang, P.; Wu, W.; Cheng, N.; Zeng, L.; Zhang, H.; Cai, X.; et al. B1 Oligomerization Regulates PML Nuclear Body Biogenesis and Leukemogenesis. Nat. Commun. 2019, 10, 3789. [Google Scholar] [CrossRef]
- Bercier, P.; Wang, Q.Q.; Zang, N.; Zhang, J.; Yang, C.; Maimaitiyiming, Y.; Abou-Ghali, M.; Berthier, C.; Wu, C.; Niwa-Kawakita, M.; et al. Structural Basis of PML/RARA Oncoprotein Targeting by Arsenic Unravels a Cysteine Rheostat Controlling PML Body Assembly and Function. Cancer Discov. 2023, 13, 2548–2565. [Google Scholar] [CrossRef]
- Guan, D.; Kao, H.-Y. The Function, Regulation and Therapeutic Implications of the Tumor Suppressor Protein, PML. Cell Biosci. 2015, 5, 60. [Google Scholar] [CrossRef]
- Weidtkamp-Peters, S.; Lenser, T.; Negorev, D.; Gerstner, N.; Hofmann, T.G.; Schwanitz, G.; Hoischen, C.; Maul, G.; Dittrich, P.; Hemmerich, P. Dynamics of Component Exchange at PML Nuclear Bodies. J. Cell Sci. 2008, 121, 2731–2743. [Google Scholar] [CrossRef]
- Tessier, S.; Ferhi, O.; Geoffroy, M.-C.; González-Prieto, R.; Canat, A.; Quentin, S.; Pla, M.; Niwa-Kawakita, M.; Bercier, P.; Rérolle, D.; et al. Exploration of Nuclear Body-Enhanced Sumoylation Reveals That PML Represses 2-Cell Features of Embryonic Stem Cells. Nat. Commun. 2022, 13, 5726. [Google Scholar] [CrossRef]
- Cossec, J.-C.; Theurillat, I.; Chica, C.; Búa Aguín, S.; Gaume, X.; Andrieux, A.; Iturbide, A.; Jouvion, G.; Li, H.; Bossis, G.; et al. SUMO Safeguards Somatic and Pluripotent Cell Identities by Enforcing Distinct Chromatin States. Cell Stem Cell 2018, 23, 742–757.e8. [Google Scholar] [CrossRef]
- Hsu, K.-S.; Kao, H.-Y. PML: Regulation and Multifaceted Function beyond Tumor Suppression. Cell Biosci. 2018, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-K.; Bergmann, S.; Pandolfi, P.P. Cytoplasmic PML Function in TGF-β Signalling. Nature 2004, 431, 205–211. [Google Scholar] [CrossRef]
- Wang, Z.-G.; Ruggero, D.; Ronchetti, S.; Zhong, S.; Gaboli, M.; Rivi, R.; Pandolfi, P.P. Pml Is Essential for Multiple Apoptotic Pathways. Nat. Genet. 1998, 20, 266–272. [Google Scholar] [CrossRef]
- Wang, Z.G.; Delva, L.; Gaboli, M.; Rivi, R.; Giorgio, M.; Cordon-Cardo, C.; Grosveld, F.; Pandolfi, P.P. Role of PML in Cell Growth and the Retinoic Acid Pathway. Sci. New Ser. 1998, 279, 1547–1551. [Google Scholar] [CrossRef]
- Koken, M.H.; Linares-Cruz, G.; Quignon, F.; Viron, A.; Chelbi-Alix, M.K.; Sobczak-Thépot, J.; Juhlin, L.; Degos, L.; Calvo, F.; de Thé, H. The PML Growth-Suppressor Has an Altered Expression in Human Oncogenesis. Oncogene 1995, 10, 1315–1324. [Google Scholar]
- Scaglioni, P.P.; Yung, T.M.; Cai, L.F.; Erdjument-Bromage, H.; Kaufman, A.J.; Singh, B.; Teruya-Feldstein, J.; Tempst, P.; Pandolfi, P.P. A CK2-Dependent Mechanism for Degradation of the PML Tumor Suppressor. Cell 2006, 126, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Trotman, L.C.; Alimonti, A.; Scaglioni, P.P.; Koutcher, J.A.; Cordon-Cardo, C.; Pandolfi, P.P. Identification of a Tumour Suppressor Network Opposing Nuclear Akt Function. Nature 2006, 441, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Wolyniec, K.; Shortt, J.; de Stanchina, E.; Levav-Cohen, Y.; Alsheich-Bartok, O.; Louria-Hayon, I.; Corneille, V.; Kumar, B.; Woods, S.J.; Opat, S.; et al. E6AP Ubiquitin Ligase Regulates PML-Induced Senescence in Myc-Driven Lymphomagenesis. Blood 2012, 120, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Mitchell, C.; Corneille, V.; Shortt, J.; Fox, S.; Pandolfi, P.P.; Castillo-Martin, M.; Bonal, D.; Cordon-Carlo, C.; Lozano, G.; et al. Loss of PML Cooperates with Mutant P53 to Drive More Aggressive Cancers in a Gender-Dependent Manner. Cell Cycle 2013, 12, 1722–1731. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Chin, W.; Chow, L.T.; Chan, A.S.; Yim, A.P.; Leung, S.F.; Mok, T.S.; Chang, K.S.; Johnson, P.J.; Chan, J.Y. Lack of Expression for the Suppressor PML in Human Small Cell Lung Carcinoma. Int. J. Cancer 2000, 85, 599–605. [Google Scholar] [CrossRef]
- Gurrieri, C.; Capodieci, P.; Bernardi, R.; Scaglioni, P.P.; Nafa, K.; Rush, L.J.; Verbel, D.A.; Cordon-Cardo, C.; Pandolfi, P.P. Loss of the Tumor Suppressor PML in Human Cancers of Multiple Histologic Origins. JNCI J. Natl. Cancer Inst. 2004, 96, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Datta, N.; Islam, S.; Chatterjee, U.; Chatterjee, S.; Panda, C.K.; Ghosh, M.K. Promyelocytic Leukemia (PML) Gene Regulation: Implication towards Curbing Oncogenesis. Cell Death Dis. 2019, 10, 656. [Google Scholar] [CrossRef] [PubMed]
- Rego, E.M.; Wang, Z.G.; Peruzzi, D.; He, L.Z.; Cordon-Cardo, C.; Pandolfi, P.P. Role of Promyelocytic Leukemia (PML) Protein in Tumor Suppression. J. Exp. Med. 2001, 193, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Voisset, E.; Moravcsik, E.; Stratford, E.W.; Jaye, A.; Palgrave, C.J.; Hills, R.K.; Salomoni, P.; Kogan, S.C.; Solomon, E.; Grimwade, D. Pml Nuclear Body Disruption Cooperates in APL Pathogenesis and Impairs DNA Damage Repair Pathways in Mice. Blood 2018, 131, 636–648. [Google Scholar] [CrossRef]
- Ferbeyre, G.; de Stanchina, E.; Querido, E.; Baptiste, N.; Prives, C.; Lowe, S.W. PML Is Induced by Oncogenic Ras and Promotes Premature Senescence. Genes Dev. 2000, 14, 2015–2027. [Google Scholar] [CrossRef]
- Pearson, M.; Carbone, R.; Sebastiani, C.; Cioce, M.; Fagioli, M.; Saito, S.; Higashimoto, Y.; Appella, E.; Minucci, S.; Pandolfi, P.P.; et al. PML Regulates P53 Acetylation and Premature Senescence Induced by Oncogenic Ras. Nature 2000, 406, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Bischof, O. Deconstructing PML-Induced Premature Senescence. EMBO J. 2002, 21, 3358–3369. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.; Papa, A.; Pandolfi, P.P. Regulation of Apoptosis by PML and the PML-NBs. Oncogene 2008, 27, 6299–6312. [Google Scholar] [CrossRef]
- Vernier, M.; Bourdeau, V.; Gaumont-Leclerc, M.-F.; Moiseeva, O.; Begin, V.; Saad, F.; Mes-Masson, A.-M.; Ferbeyre, G. Regulation of E2Fs and Senescence by PML Nuclear Bodies. Genes Dev. 2011, 25, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Talluri, S.; Dick, F.A. The Retinoblastoma Protein and PML Collaborate to Organize Heterochromatin and Silence E2F-Responsive Genes during Senescence. Cell Cycle 2014, 13, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Ablain, J.; Rice, K.; Soilihi, H.; de Reynies, A.; Minucci, S.; de Thé, H. Activation of a Promyelocytic Leukemia–Tumor Protein 53 Axis Underlies Acute Promyelocytic Leukemia Cure. Nat. Med. 2014, 20, 167–174. [Google Scholar] [CrossRef] [PubMed]
- de Thé, H.; Pandolfi, P.P.; Chen, Z. Acute Promyelocytic Leukemia: A Paradigm for Oncoprotein-Targeted Cure. Cancer Cell 2017, 32, 552–560. [Google Scholar] [CrossRef]
- Liebl, M.C.; Hofmann, T.G. Regulating the P53 Tumor Suppressor Network at PML Biomolecular Condensates. Cancers 2022, 14, 4549. [Google Scholar] [CrossRef]
- Ivanschitz, L.; Takahashi, Y.; Jollivet, F.; Ayrault, O.; Le Bras, M.; de Thé, H. PML IV/ARF Interaction Enhances P53 SUMO-1 Conjugation, Activation, and Senescence. Proc. Natl. Acad. Sci. USA 2015, 112, 14278–14283. [Google Scholar] [CrossRef]
- Dellaire, G. The Number of PML Nuclear Bodies Increases in Early S Phase by a Fission Mechanism. J. Cell Sci. 2006, 119, 1026–1033. [Google Scholar] [CrossRef]
- Corpet, A.; Kleijwegt, C.; Roubille, S.; Juillard, F.; Jacquet, K.; Texier, P.; Lomonte, P. PML Nuclear Bodies and Chromatin Dynamics: Catch Me If You Can! Nucleic Acids Res. 2020, 48, 11890–11912. [Google Scholar] [CrossRef] [PubMed]
- Delbarre, E.; Ivanauskiene, K.; Spirkoski, J.; Shah, A.; Vekterud, K.; Moskaug, J.Ø.; Bøe, S.O.; Wong, L.H.; Küntziger, T.; Collas, P. PML Protein Organizes Heterochromatin Domains Where It Regulates Histone H3.3 Deposition by ATRX/DAXX. Genome Res. 2017, 27, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Fracassi, C.; Ugge’, M.; Abdelhalim, M.; Zapparoli, E.; Simoni, M.; Magliulo, D.; Mazza, D.; Lazarevic, D.; Morelli, M.J.; Collas, P.; et al. PML Modulates Epigenetic Composition of Chromatin to Regulate Expression of Pro-Metastatic Genes in Triple-Negative Breast Cancer. Nucleic Acids Res. 2023, 51, 11024–11039. [Google Scholar] [CrossRef] [PubMed]
- Ishov, A.M.; Sotnikov, A.G.; Negorev, D.; Vladimirova, O.V.; Neff, N.; Kamitani, T.; Yeh, E.T.H.; Strauss, J.F.; Maul, G.G. Pml Is Critical for Nd10 Formation and Recruits the Pml-Interacting Protein Daxx to This Nuclear Structure When Modified by Sumo-1. J. Cell Biol. 1999, 147, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, F.-M.; Kruhlak, M.J.; Box, A.K.; Hendzel, M.J.; Bazett-Jones, D.P. The Transcription Coactivator Cbp Is a Dynamic Component of the Promyelocytic Leukemia Nuclear Body. J. Cell Biol. 2001, 152, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- D’Orazi, G.; Cecchinelli, B.; Bruno, T.; Manni, I.; Higashimoto, Y.; Saito, S.; Gostissa, M.; Coen, S.; Marchetti, A.; Del Sal, G.; et al. Homeodomain-Interacting Protein Kinase-2 Phosphorylates P53 at Ser 46 and Mediates Apoptosis. Nat. Cell Biol. 2002, 4, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T.G.; Möller, A.; Sirma, H.; Zentgraf, H.; Taya, Y.; Dröge, W.; Will, H.; Schmitz, M.L. Regulation of P53 Activity by Its Interaction with Homeodomain-Interacting Protein Kinase-2. Nat. Cell Biol. 2002, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Liang, W.-W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.-W.; Reynolds, S.M.; Wyczalkowski, M.A.; Yao, L.; et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 2018, 23, 227–238.e3. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Shirnekhi, H.K.; Gorman, S.D.; Chandra, B.; Baggett, D.W.; Park, C.-G.; Somjee, R.; Lang, B.; Hosseini, S.M.H.; Pioso, B.J.; et al. Defining the Condensate Landscape of Fusion Oncoproteins. Nat. Commun. 2023, 14, 6008. [Google Scholar] [CrossRef]
- Cambiaghi, V.; Giuliani, V.; Lombardi, S.; Marinelli, C.; Toffalorio, F.; Pelicci, P.G. TRIM Proteins in Cancer. In TRIM/RBCC Proteins; Meroni, G., Ed.; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2012; Volume 770, pp. 77–91. ISBN 978-1-4614-5397-0. [Google Scholar]
- Crawford, L.J.; Johnston, C.K.; Irvine, A.E. TRIM Proteins in Blood Cancers. J. Cell Commun. Signal. 2018, 12, 21–29. [Google Scholar] [CrossRef]
- Occhionorelli, M.; Santoro, F.; Pallavicini, I.; Gruszka, A.; Moretti, S.; Bossi, D.; Viale, A.; Shing, D.; Ronzoni, S.; Muradore, I.; et al. The Self-Association Coiled-Coil Domain of PML Is Sufficient for the Oncogenic Conversion of the Retinoic Acid Receptor (RAR) Alpha. Leukemia 2011, 25, 814–820. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, X.; Wu, W.; Chen, Z.; Meng, G. PML Nuclear Body Biogenesis, Carcinogenesis, and Targeted Therapy. Trends Cancer 2020, 6, 889–906. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.; Zeisig, B.B.; Dong, S.; So, C.W.E. Forced Homo-Oligomerization of RARα Leads to Transformation of Primary Hematopoietic Cells. Cancer Cell 2006, 9, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Sternsdorf, T.; Phan, V.T.; Maunakea, M.L.; Ocampo, C.B.; Sohal, J.; Silletto, A.; Galimi, F.; Le Beau, M.M.; Evans, R.M.; Kogan, S.C. Forced Retinoic Acid Receptor α Homodimers Prime Mice for APL-like Leukemia. Cancer Cell 2006, 9, 81–94. [Google Scholar] [CrossRef]
- Zhu, J.; Zhou, J.; Peres, L.; Riaucoux, F.; Honoré, N.; Kogan, S.; de Thé, H. A Sumoylation Site in PML/RARA Is Essential for Leukemic Transformation. Cancer Cell 2005, 7, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Vitaliano-Prunier, A.; Halftermeyer, J.; Ablain, J.; de Reynies, A.; Peres, L.; Le Bras, M.; Metzger, D.; de The, H. Clearance of PML/RARA-Bound Promoters Suffice to Initiate APL Differentiation. Blood 2014, 124, 3772–3780. [Google Scholar] [CrossRef]
- Grignani, F.; De Matteis, S.; Nervi, C.; Tomassoni, L.; Gelmetti, V.; Cioce, M.; Fanelli, M.; Ruthardt, M.; Ferrara, F.F.; Zamir, I.; et al. Fusion Proteins of the Retinoic Acid Receptor-α Recruit Histone Deacetylase in Promyelocytic Leukaemia. Nature 1998, 391, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Tiefenbach, J.; Novac, N.; Ducasse, M.; Eck, M.; Melchior, F.; Heinzel, T. SUMOylation of the Corepressor N-CoR Modulates Its Capacity to Repress Transcription. Mol. Biol. Cell 2006, 17, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.J.; Nagy, L.; Inoue, S.; Shao, W.; Miller, W.H.; Evans, R.M. Role of the Histone Deacetylase Complex in Acute Promyelocytic Leukaemia. Nature 1998, 391, 811–814. [Google Scholar] [CrossRef]
- Wang, P.; Tang, Z.; Lee, B.; Zhu, J.J.; Cai, L.; Szalaj, P.; Tian, S.Z.; Zheng, M.; Plewczynski, D.; Ruan, X.; et al. Chromatin Topology Reorganization and Transcription Repression by PML-RARα in Acute Promyeloid Leukemia. Genome Biol. 2020, 21, 110. [Google Scholar] [CrossRef]
- Iaccarino, L.; Ottone, T.; Divona, M.; Cicconi, L.; Cairoli, R.; Voso, M.T.; Lo-Coco, F. Mutations Affecting Both the Rearranged and the Unrearranged PML Alleles in Refractory Acute Promyelocytic Leukaemia. Br. J. Haematol. 2016, 172, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Hadjimichael, C.; Chanoumidou, K.; Nikolaou, C.; Klonizakis, A.; Theodosi, G.-I.; Makatounakis, T.; Papamatheakis, J.; Kretsovali, A. Promyelocytic Leukemia Protein Is an Essential Regulator of Stem Cell Pluripotency and Somatic Cell Reprogramming. Stem Cell Rep. 2017, 8, 1366–1378. [Google Scholar] [CrossRef]
- Amodeo, V.; Deli, A.; Betts, J.; Bartesaghi, S.; Zhang, Y.; Richard-Londt, A.; Ellis, M.; Roshani, R.; Vouri, M.; Galavotti, S.; et al. A PML/Slit Axis Controls Physiological Cell Migration and Cancer Invasion in the CNS. Cell Rep. 2017, 20, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A Cell Initiating Human Acute Myeloid Leukaemia after Transplantation into SCID Mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human Acute Myeloid Leukemia Is Organized as a Hierarchy That Originates from a Primitive Hematopoietic Cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Shlush, L.I.; Mitchell, A.; Heisler, L.; Abelson, S.; Ng, S.W.K.; Trotman-Grant, A.; Medeiros, J.J.F.; Rao-Bhatia, A.; Jaciw-Zurakowsky, I.; Marke, R.; et al. Tracing the Origins of Relapse in Acute Myeloid Leukaemia to Stem Cells. Nature 2017, 547, 104–108. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.; Zeinabad, H.A.; Szegezdi, E. Hematopoietic versus Leukemic Stem Cell Quiescence: Challenges and Therapeutic Opportunities. Blood Rev. 2021, 50, 100850. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Bernardi, R.; Morotti, A.; Matsuoka, S.; Saglio, G.; Ikeda, Y.; Rosenblatt, J.; Avigan, D.E.; Teruya-Feldstein, J.; Pandolfi, P.P. PML Targeting Eradicates Quiescent Leukaemia-Initiating Cells. Nature 2008, 453, 1072–1078. [Google Scholar] [CrossRef]
- Ito, K.; Carracedo, A.; Weiss, D.; Arai, F.; Ala, U.; Avigan, D.E.; Schafer, Z.T.; Evans, R.M.; Suda, T.; Lee, C.-H.; et al. A PML–PPAR-δ Pathway for Fatty Acid Oxidation Regulates Hematopoietic Stem Cell Maintenance. Nat. Med. 2012, 18, 1350–1358. [Google Scholar] [CrossRef]
- Carracedo, A.; Weiss, D.; Leliaert, A.K.; Bhasin, M.; de Boer, V.C.J.; Laurent, G.; Adams, A.C.; Sundvall, M.; Song, S.J.; Ito, K.; et al. A Metabolic Prosurvival Role for PML in Breast Cancer. J. Clin. Investig. 2012, 122, 3088–3100. [Google Scholar] [CrossRef]
- Martín-Martín, N.; Piva, M.; Urosevic, J.; Aldaz, P.; Sutherland, J.D.; Fernández-Ruiz, S.; Arreal, L.; Torrano, V.; Cortazar, A.R.; Planet, E.; et al. Stratification and Therapeutic Potential of PML in Metastatic Breast Cancer. Nat. Commun. 2016, 7, 12595. [Google Scholar] [CrossRef]
- Gentric, G.; Kieffer, Y.; Mieulet, V.; Goundiam, O.; Bonneau, C.; Nemati, F.; Hurbain, I.; Raposo, G.; Popova, T.; Stern, M.-H.; et al. PML-Regulated Mitochondrial Metabolism Enhances Chemosensitivity in Human Ovarian Cancers. Cell Metab. 2019, 29, 156–173.e10. [Google Scholar] [CrossRef]
- Arreal, L.; Piva, M.; Fernández, S.; Revandkar, A.; Schaub- Clerigué, A.; Villanueva, J.; Zabala-Letona, A.; Pujana, M.; Astobiza, I.; Cortazar, A.R.; et al. Targeting PML in Triple Negative Breast Cancer Elicits Growth Suppression and Senescence. Cell Death Differ. 2020, 27, 1186–1199. [Google Scholar] [CrossRef]
- Zhu, J.; Koken, M.H.M.; Quignon, F.; Chelbi-Alix, M.K.; Degos, L.; Wang, Z.Y.; Chen, Z.; de The, H. Arsenic-Induced PML Targeting onto Nuclear Bodies: Implications for the Treatment of Acute Promyelocytic Leukemia. Proc. Natl. Acad. Sci. USA 1997, 94, 3978–3983. [Google Scholar] [CrossRef]
- Islam, K.; Wang, Q.Q.; Naranmandura, H. Molecular Mechanisms of Arsenic Toxicity. In Advances in Molecular Toxicology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 9, pp. 77–107. ISBN 978-0-12-802229-0. [Google Scholar]
- Jeanne, M.; Lallemand-Breitenbach, V.; Ferhi, O.; Koken, M.; Le Bras, M.; Duffort, S.; Peres, L.; Berthier, C.; Soilihi, H.; Raught, B.; et al. PML/RARA Oxidation and Arsenic Binding Initiate the Antileukemia Response of As2O3. Cancer Cell 2010, 18, 88–98. [Google Scholar] [CrossRef]
- Niwa-Kawakita, M.; Ferhi, O.; Soilihi, H.; Le Bras, M.; Lallemand-Breitenbach, V.; de Thé, H. PML Is a ROS Sensor Activating P53 upon Oxidative Stress. J. Exp. Med. 2017, 214, 3197–3206. [Google Scholar] [CrossRef]
- Tatham, M.H.; Geoffroy, M.-C.; Shen, L.; Plechanovova, A.; Hattersley, N.; Jaffray, E.G.; Palvimo, J.J.; Hay, R.T. RNF4 Is a Poly-SUMO-Specific E3 Ubiquitin Ligase Required for Arsenic-Induced PML Degradation. Nat. Cell Biol. 2008, 10, 538–546. [Google Scholar] [CrossRef]
- Erker, Y.; Neyret-Kahn, H.; Seeler, J.S.; Dejean, A.; Atfi, A.; Levy, L. Arkadia, a Novel SUMO-Targeted Ubiquitin Ligase Involved in PML Degradation. Mol. Cell. Biol. 2013, 33, 2163–2177. [Google Scholar] [CrossRef]
- Tsai, J.M.; Aguirre, J.D.; Li, Y.-D.; Brown, J.; Focht, V.; Kater, L.; Kempf, G.; Sandoval, B.; Schmitt, S.; Rutter, J.C.; et al. UBR5 Forms Ligand-Dependent Complexes on Chromatin to Regulate Nuclear Hormone Receptor Stability. Mol. Cell 2023, 83, 2753–2767.e10. [Google Scholar] [CrossRef]
- Jaffray, E.G.; Tatham, M.H.; Mojsa, B.; Liczmanska, M.; Rojas-Fernandez, A.; Yin, Y.; Ball, G.; Hay, R.T. The P97/VCP Segregase Is Essential for Arsenic-Induced Degradation of PML and PML-RARA. J. Cell Biol. 2023, 222, e202201027. [Google Scholar] [CrossRef]
- de Thé, H. Differentiation Therapy Revisited. Nat. Rev. Cancer 2018, 18, 117–127. [Google Scholar] [CrossRef]
- Yilmaz, M.; Kantarjian, H.; Ravandi, F. Acute Promyelocytic Leukemia Current Treatment Algorithms. Blood Cancer J. 2021, 11, 123. [Google Scholar] [CrossRef]
- Gallagher, R.E.; Moser, B.K.; Racevskis, J.; Poiré, X.; Bloomfield, C.D.; Carroll, A.J.; Ketterling, R.P.; Roulston, D.; Schachter-Tokarz, E.; Zhou, D.-C.; et al. Treatment-Influenced Associations of PML-RARα Mutations, FLT3 Mutations, and Additional Chromosome Abnormalities in Relapsed Acute Promyelocytic Leukemia. Blood 2012, 120, 2098–2108. [Google Scholar] [CrossRef]
- Raelson, J.V.; Nervi, C.; Rosenauer, A.; Benedetti, L.; Monczak, Y.; Pearson, M.; Pelicci, P.G.; Miller, W.H. The PML/RAR Alpha Oncoprotein Is a Direct Molecular Target of Retinoic Acid in Acute Promyelocytic Leukemia Cells. Blood 1996, 88, 2826–2832. [Google Scholar] [CrossRef]
- Zhu, J.; Gianni, M.; Kopf, E.; Honore, N.; Chelbi-Alix, M.; Koken, M.; Quignon, F.; Rochette-Egly, C.; de The, H. Retinoic Acid Induces Proteasome-Dependent Degradation of Retinoic Acid Receptor Alpha (RARalpha) and Oncogenic RARalpha Fusion Proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 14807–14812. [Google Scholar] [CrossRef]
- Ablain, J.; Leiva, M.; Peres, L.; Fonsart, J.; Anthony, E.; de Thé, H. Uncoupling RARA Transcriptional Activation and Degradation Clarifies the Bases for APL Response to Therapies. J. Exp. Med. 2013, 210, 647–653. [Google Scholar] [CrossRef]
- Nasr, R.; Guillemin, M.-C.; Ferhi, O.; Soilihi, H.; Peres, L.; Berthier, C.; Rousselot, P.; Robledo-Sarmiento, M.; Lallemand-Breitenbach, V.; Gourmel, B.; et al. Eradication of Acute Promyelocytic Leukemia-Initiating Cells through PML-RARA Degradation. Nat. Med. 2008, 14, 1333–1342. [Google Scholar] [CrossRef]
- McKenzie, M.D.; Ghisi, M.; Oxley, E.P.; Ngo, S.; Cimmino, L.; Esnault, C.; Liu, R.; Salmon, J.M.; Bell, C.C.; Ahmed, N.; et al. Interconversion between Tumorigenic and Differentiated States in Acute Myeloid Leukemia. Cell Stem Cell 2019, 25, 258–272.e9. [Google Scholar] [CrossRef]
- Douer, D.; Estey, E.; Santillana, S.; Bennett, J.M.; Lopez-Bernstein, G.; Boehm, K.; Williams, T. Treatment of Newly Diagnosed and Relapsed Acute Promyelocytic Leukemia with Intravenous Liposomal All-Transretinoic Acid. Blood 2001, 97, 73–80. [Google Scholar] [CrossRef]
- Tsimberidou, A.-M.; Tirado-Gomez, M.; Andreeff, M.; O’Brien, S.; Kantarjian, H.; Keating, M.; Lopez-Berestein, G.; Estey, E. Single-Agent Liposomal All-Trans Retinoic Acid Can Cure Some Patients with Untreated Acute Promyelocytic Leukemia: An Update of The University of Texas M. D. Anderson Cancer Center Series. Leuk. Lymphoma 2006, 47, 1062–1068. [Google Scholar] [CrossRef]
- Rabellino, A.; Scaglioni, P.P. PML Degradation: Multiple Ways to Eliminate PML. Front. Oncol. 2013, 3, 60. [Google Scholar] [CrossRef]
- Kchour, G.; Tarhini, M.; Kooshyar, M.-M.; El Hajj, H.; Wattel, E.; Mahmoudi, M.; Hatoum, H.; Rahimi, H.; Maleki, M.; Rafatpanah, H.; et al. Phase 2 Study of the Efficacy and Safety of the Combination of Arsenic Trioxide, Interferon Alpha, and Zidovudine in Newly Diagnosed Chronic Adult T-Cell Leukemia/Lymphoma (ATL). Blood 2009, 113, 6528–6532. [Google Scholar] [CrossRef]
- Dassouki, Z.; Sahin, U.; El Hajj, H.; Jollivet, F.; Kfoury, Y.; Lallemand-Breitenbach, V.; Hermine, O.; Bazarbachi, A. ATL Response to Arsenic/Interferon Therapy Is Triggered by SUMO/PML/RNF4-Dependent Tax Degradation. Blood 2015, 125, 474–482. [Google Scholar] [CrossRef]
- Marçais, A.; Cook, L.; Witkover, A.; Asnafi, V.; Avettand-Fenoel, V.; Delarue, R.; Cheminant, M.; Sibon, D.; Frenzel, L.; de Thé, H.; et al. Arsenic Trioxide (As2O3) as a Maintenance Therapy for Adult T Cell Leukemia/Lymphoma. Retrovirology 2020, 17, 5. [Google Scholar] [CrossRef]
- Hleihel, R.; El Hajj, H.; Wu, H.-C.; Berthier, C.; Zhu, H.-H.; Massoud, R.; Chakhachiro, Z.; El Sabban, M.; De The, H.; Bazarbachi, A. A Pin1/PML/P53 Axis Activated by Retinoic Acid in NPM-1c Acute Myeloid Leukemia. Haematologica 2021, 106, 3090–3099. [Google Scholar] [CrossRef]
- Schlenk, R.F.; Dohner, K.; Kneba, M.; Gotze, K.; Hartmann, F.; del Valle, F.; Kirchen, H.; Koller, E.; Fischer, J.T.; Bullinger, L.; et al. Gene Mutations and Response to Treatment with All-Trans Retinoic Acid in Elderly Patients with Acute Myeloid Leukemia. Results from the AMLSG Trial AML HD98B. Haematologica 2009, 94, 54–60. [Google Scholar] [CrossRef]
- Wu, H.-C.; Rérolle, D.; Berthier, C.; Hleihel, R.; Sakamoto, T.; Quentin, S.; Benhenda, S.; Morganti, C.; Wu, C.; Conte, L.; et al. Actinomycin D Targets NPM1c-Primed Mitochondria to Restore PML-Driven Senescence in AML Therapy. Cancer Discov. 2021, 11, 3198–3213. [Google Scholar] [CrossRef]
- Okazaki, T.; Kageji, T.; Kuwayama, K.; Kitazato, K.T.; Mure, H.; Hara, K.; Morigaki, R.; Mizobuchi, Y.; Matsuzaki, K.; Nagahiro, S. Up-Regulation of Endogenous PML Induced by a Combination of Interferon-Beta and Temozolomide Enhances P73/YAP-Mediated Apoptosis in Glioblastoma. Cancer Lett. 2012, 323, 199–207. [Google Scholar] [CrossRef]
- Dagher, T.; Maslah, N.; Edmond, V.; Cassinat, B.; Vainchenker, W.; Giraudier, S.; Pasquier, F.; Verger, E.; Niwa-Kawakita, M.; Lallemand-Breitenbach, V.; et al. JAK2V617F Myeloproliferative Neoplasm Eradication by a Novel Interferon/Arsenic Therapy Involves PML. J. Exp. Med. 2021, 218, e20201268. [Google Scholar] [CrossRef]
- Zhou, W.; Cheng, L.; Shi, Y.; Ke, S.Q.; Huang, Z.; Fang, X.; Chu, C.; Xie, Q.; Bian, X.; Rich, J.N.; et al. Arsenic Trioxide Disrupts Glioma Stem Cells via Promoting PML Degradation to Inhibit Tumor Growth. Oncotarget 2015, 6, 37300–37315. [Google Scholar] [CrossRef]
- Voon, H.P.J.; Hii, L.; Garvie, A.; Udugama, M.; Krug, B.; Russo, C.; Chüeh, A.C.; Daly, R.J.; Morey, A.; Bell, T.D.M.; et al. Pediatric Glioma Histone H3.3 K27M/G34R Mutations Drive Abnormalities in PML Nuclear Bodies. Genome Biol. 2023, 24, 284. [Google Scholar] [CrossRef]
- Khetchoumian, K.; Teletin, M.; Tisserand, J.; Mark, M.; Herquel, B.; Ignat, M.; Zucman-Rossi, J.; Cammas, F.; Lerouge, T.; Thibault, C.; et al. Loss of Trim24 (Tif1α) Gene Function Confers Oncogenic Activity to Retinoic Acid Receptor Alpha. Nat. Genet. 2007, 39, 1500–1506. [Google Scholar] [CrossRef]
- Tan, J.; Ong, C.K.; Lim, W.K.; Ng, C.C.Y.; Thike, A.A.; Ng, L.M.; Rajasegaran, V.; Myint, S.S.; Nagarajan, S.; Thangaraju, S.; et al. Genomic Landscapes of Breast Fibroepithelial Tumors. Nat. Genet. 2015, 47, 1341–1345. [Google Scholar] [CrossRef]
- McKeown, M.R.; Corces, M.R.; Eaton, M.L.; Fiore, C.; Lee, E.; Lopez, J.T.; Chen, M.W.; Smith, D.; Chan, S.M.; Koenig, J.L.; et al. Superenhancer Analysis Defines Novel Epigenomic Subtypes of Non-APL AML, Including an RARα Dependency Targetable by SY-1425, a Potent and Selective RARα Agonist. Cancer Discov. 2017, 7, 1136–1153. [Google Scholar] [CrossRef]
- Lübbert, M.; Grishina, O.; Schmoor, C.; Schlenk, R.F.; Jost, E.; Crysandt, M.; Heuser, M.; Thol, F.; Salih, H.R.; Schittenhelm, M.M.; et al. Valproate and Retinoic Acid in Combination With Decitabine in Elderly Nonfit Patients With Acute Myeloid Leukemia: Results of a Multicenter, Randomized, 2 × 2, Phase II Trial. J. Clin. Oncol. 2020, 38, 257–270. [Google Scholar] [CrossRef]
- De Botton, S.; Cluzeau, T.; Vigil, C.; Cook, R.J.; Rousselot, P.; Rizzieri, D.A.; Liesveld, J.L.; Fenaux, P.; Braun, T.; Banos, A.; et al. Targeting RARA Overexpression with Tamibarotene, a Potent and Selective RARα Agonist, Is a Novel Approach in AML. Blood Adv. 2023, 7, 1858–1870. [Google Scholar] [CrossRef]
- Zhao, J.; Liang, J.-W.; Xue, H.-L.; Shen, S.-H.; Chen, J.; Tang, Y.-J.; Yu, L.-S.; Liang, H.-H.; Gu, L.-J.; Tang, J.-Y.; et al. The Genetics and Clinical Characteristics of Children Morphologically Diagnosed as Acute Promyelocytic Leukemia. Leukemia 2019, 33, 1387–1399. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bercier, P.; de Thé, H. History of Developing Acute Promyelocytic Leukemia Treatment and Role of Promyelocytic Leukemia Bodies. Cancers 2024, 16, 1351. https://doi.org/10.3390/cancers16071351
Bercier P, de Thé H. History of Developing Acute Promyelocytic Leukemia Treatment and Role of Promyelocytic Leukemia Bodies. Cancers. 2024; 16(7):1351. https://doi.org/10.3390/cancers16071351
Chicago/Turabian StyleBercier, Pierre, and Hugues de Thé. 2024. "History of Developing Acute Promyelocytic Leukemia Treatment and Role of Promyelocytic Leukemia Bodies" Cancers 16, no. 7: 1351. https://doi.org/10.3390/cancers16071351