
Role of the Atypical MAPK ERK3 in Cancer Growth and Progression

{kind=link}
{kind=link}
{kind=link}
Abstract
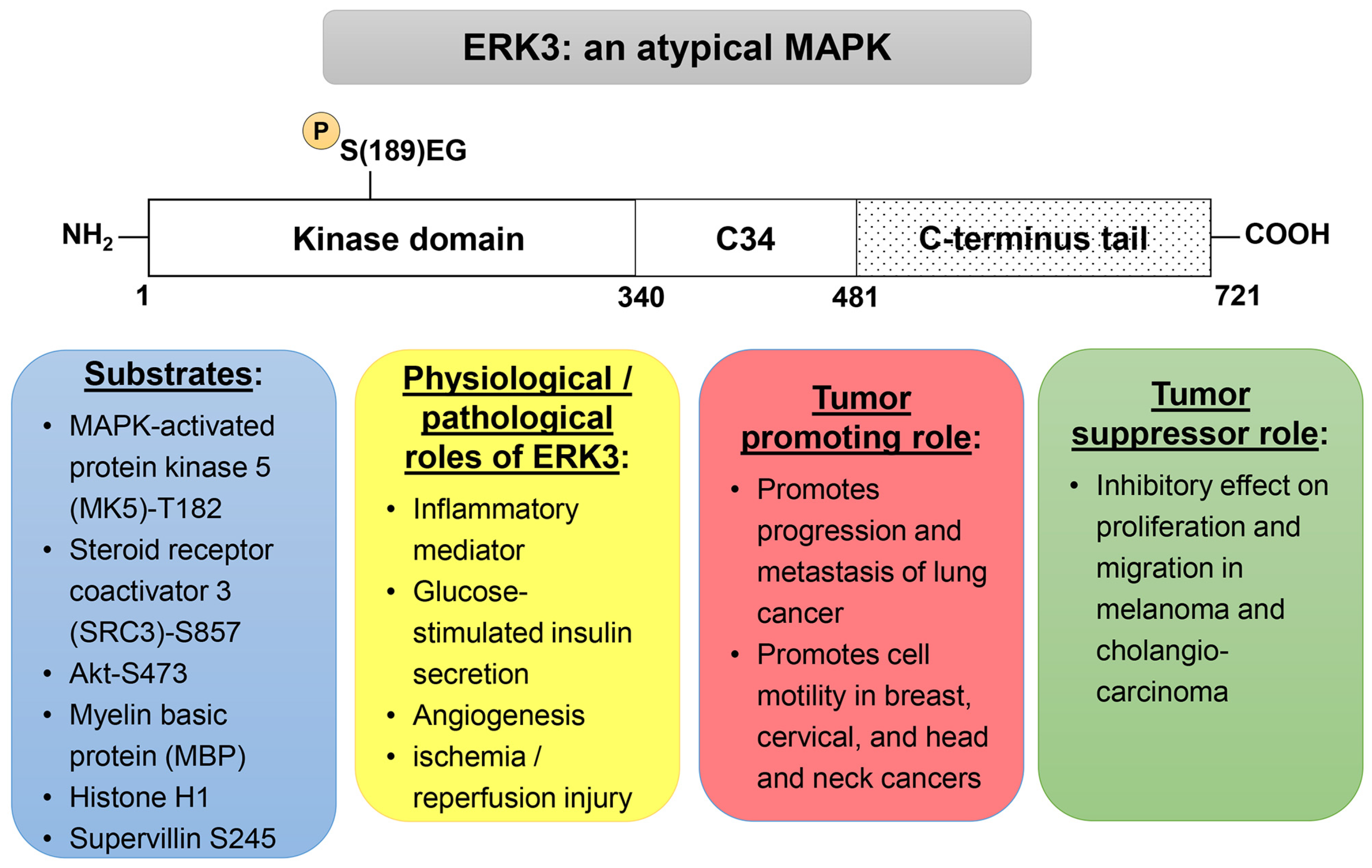
:Simple Summary
Abstract
1. Introduction
2. Physiological Functions of ERK3
3. Roles of ERK3 in Cancer
3.1. Expression and Mutations of ERK3 in Cancers
3.2. Tumor-Promoting Roles of ERK3
3.2.1. Cancer Cell Proliferation and Tumor Growth
3.2.2. Cancer Cell Migration, Invasion and Metastasis
3.2.3. ERK3 Confers Chemoresistance on Cancer Cells
3.3. Tumor-Suppressing Roles of ERK3
4. Regulation of ERK3 in Cancers
4.1. Regulation of ERK3 Kinase Activity
4.2. Molecular Regulation of ERK3 Expression
4.3. Regulation of ERK3 Stability
4.4. Regulation of ERK3 Localization
5. Conclusions and Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Raman, M.; Chen, W.; Cobb, M.H. Differential Regulation and Properties of MAPKs. Oncogene 2007, 26, 3100–3112. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, P.; Meloche, S. Atypical Mitogen-Activated Protein Kinases: Structure, Regulation and Functions. Biochim. Biophys. Acta 2007, 1773, 1376–1387. [Google Scholar] [CrossRef] [PubMed]
- Boulton, T.G.; Nye, S.H.; Robbins, D.J.; Ip, N.Y.; Radziejewska, E.; Morgenbesser, S.D.; DePinho, R.A.; Panayotatos, N.; Cobb, M.H.; Yancopoulos, G.D. ERKs: A Family of Protein-Serine/Threonine Kinases That Are Activated and Tyrosine Phosphorylated in Response to Insulin and NGF. Cell 1991, 65, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Zhao, Y.; Moller, D.E.; Flier, J.S. Cloning and Characterization of P97MAPK, a Novel Human Homolog of Rat ERK-3. Mol. Cell. Biol. 1994, 14, 8202–8211. [Google Scholar] [CrossRef] [PubMed]
- Meloche, S.; Beatty, B.G.; Pellerin, J. Primary Structure, Expression and Chromosomal Locus of a Human Homolog of Rat ERK3. Oncogene 1996, 13, 1575–1579. [Google Scholar] [PubMed]
- Turgeon, B.; Saba-El-Leil, M.K.; Meloche, S. Cloning and Characterization of Mouse Extracellular-Signal-Regulated Protein Kinase 3 as a Unique Gene Product of 100 KDa. Biochem. J. 2000, 346 Pt 1, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Boulton, T.G.; Cobb, M.H. ERK3 Is a Constitutively Nuclear Protein Kinase. J. Biol. Chem. 1996, 271, 8951–8958. [Google Scholar] [CrossRef]
- Brand, F.; Schumacher, S.; Kant, S.; Menon, M.B.; Simon, R.; Turgeon, B.; Britsch, S.; Meloche, S.; Gaestel, M.; Kotlyarov, A. The Extracellular Signal-Regulated Kinase 3 (Mitogen-Activated Protein Kinase 6 [MAPK6])-MAPK-Activated Protein Kinase 5 Signaling Complex Regulates Septin Function and Dendrite Morphology. Mol. Cell. Biol. 2012, 32, 2467–2478. [Google Scholar] [CrossRef]
- Seternes, O.-M.; Mikalsen, T.; Johansen, B.; Michaelsen, E.; Armstrong, C.G.; Morrice, N.A.; Turgeon, B.; Meloche, S.; Moens, U.; Keyse, S.M. Activation of MK5/PRAK by the Atypical MAP Kinase ERK3 Defines a Novel Signal Transduction Pathway. EMBO J. 2004, 23, 4780–4791. [Google Scholar] [CrossRef]
- Schumacher, S.; Laass, K.; Kant, S.; Shi, Y.; Visel, A.; Gruber, A.D.; Kotlyarov, A.; Gaestel, M. Scaffolding by ERK3 Regulates MK5 in Development. EMBO J. 2004, 23, 4770–4779. [Google Scholar] [CrossRef] [PubMed]
- Aberg, E.; Torgersen, K.M.; Johansen, B.; Keyse, S.M.; Perander, M.; Seternes, O.-M. Docking of PRAK/MK5 to the Atypical MAPKs ERK3 and ERK4 Defines a Novel MAPK Interaction Motif. J. Biol. Chem. 2009, 284, 19392–19401. [Google Scholar] [CrossRef] [PubMed]
- Déléris, P.; Rousseau, J.; Coulombe, P.; Rodier, G.; Tanguay, P.-L.; Meloche, S. Activation Loop Phosphorylation of the Atypical MAP Kinases ERK3 and ERK4 Is Required for Binding, Activation and Cytoplasmic Relocalization of MK5. J. Cell. Physiol. 2008, 217, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Soulez, M.; Tanguay, P.-L.; Dô, F.; Dort, J.; Crist, C.; Kotlyarov, A.; Gaestel, M.; Ferron, M.; Dumont, N.A.; Meloche, S. ERK3-MK5 Signaling Regulates Myogenic Differentiation and Muscle Regeneration by Promoting FoxO3 Degradation. J. Cell. Physiol. 2022, 237, 2271–2287. [Google Scholar] [CrossRef] [PubMed]
- Ronkina, N.; Schuster-Gossler, K.; Hansmann, F.; Kunze-Schumacher, H.; Sandrock, I.; Yakovleva, T.; Lafera, J.; Baumgärtner, W.; Krueger, A.; Prinz, I.; et al. Germ Line Deletion Reveals a Nonessential Role of Atypical Mitogen-Activated Protein Kinase 6/Extracellular Signal-Regulated Kinase 3. Mol. Cell. Biol. 2019, 39, e00516-18. [Google Scholar] [CrossRef] [PubMed]
- Klinger, S.; Turgeon, B.; Lévesque, K.; Wood, G.A.; Aagaard-Tillery, K.M.; Meloche, S. Loss of Erk3 Function in Mice Leads to Intrauterine Growth Restriction, Pulmonary Immaturity, and Neonatal Lethality. Proc. Natl. Acad. Sci. USA 2009, 106, 16710–16715. [Google Scholar] [CrossRef] [PubMed]
- Marquis, M.; Boulet, S.; Mathien, S.; Rousseau, J.; Thébault, P.; Daudelin, J.-F.; Rooney, J.; Turgeon, B.; Beauchamp, C.; Meloche, S.; et al. The Non-Classical MAP Kinase ERK3 Controls T Cell Activation. PLoS ONE 2014, 9, e86681. [Google Scholar] [CrossRef] [PubMed]
- Marquis, M.; Daudelin, J.-F.; Boulet, S.; Sirois, J.; Crain, K.; Mathien, S.; Turgeon, B.; Rousseau, J.; Meloche, S.; Labrecque, N. The Catalytic Activity of the Mitogen-Activated Protein Kinase Extracellular Signal-Regulated Kinase 3 Is Required to Sustain CD4+ CD8+ Thymocyte Survival. Mol. Cell. Biol. 2014, 34, 3374–3387. [Google Scholar] [CrossRef] [PubMed]
- Sirois, J.; Daudelin, J.-F.; Boulet, S.; Marquis, M.; Meloche, S.; Labrecque, N. The Atypical MAPK ERK3 Controls Positive Selection of Thymocytes. Immunology 2015, 145, 161–169. [Google Scholar] [CrossRef]
- Soulez, M.; Saba-El-Leil, M.K.; Turgeon, B.; Mathien, S.; Coulombe, P.; Klinger, S.; Rousseau, J.; Lévesque, K.; Meloche, S. Reevaluation of the Role of Extracellular Signal-Regulated Kinase 3 in Perinatal Survival and Postnatal Growth Using New Genetically Engineered Mouse Models. Mol. Cell. Biol. 2019, 39, e00527. [Google Scholar] [CrossRef]
- Wang, W.; Bian, K.; Vallabhaneni, S.; Zhang, B.; Wu, R.-C.; O’Malley, B.W.; Long, W. ERK3 Promotes Endothelial Cell Functions by Upregulating SRC-3/SP1-Mediated VEGFR2 Expression. J. Cell. Physiol. 2014, 229, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Bogucka, K.; Pompaiah, M.; Marini, F.; Binder, H.; Harms, G.; Kaulich, M.; Klein, M.; Michel, C.; Radsak, M.P.; Rosigkeit, S.; et al. ERK3/MAPK6 Controls IL-8 Production and Chemotaxis. eLife 2020, 9, e52511. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Meng, J.; Liao, X.; Liu, Y.; Zhou, Q.; Xu, Z.; Yin, S.; Cao, Q.; Su, G.; He, S.; et al. A de Novo Missense Mutation in MPP2 Confers an Increased Risk of Vogt-Koyanagi-Harada Disease as Shown by Trio-Based Whole-Exome Sequencing. Cell. Mol. Immunol. 2023, 20, 1379–1392. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-Y.; Lin, C.-F. Annexin A2: Its Molecular Regulation and Cellular Expression in Cancer Development. Dis. Markers 2014, 2014, 308976. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.-Q.; Xu, W.; Wu, J.-L.; Lu, X.; Chen, X.-M. MicroRNA-374a Protects against Myocardial Ischemia-Reperfusion Injury in Mice by Targeting the MAPK6 Pathway. Life Sci. 2019, 232, 116619. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Sun, Q.; Zhao, H.; Tao, J.; Yan, D. Long Noncoding RNA NEAT1 Sponges MiR-495-3p to Enhance Myocardial Ischemia-Reperfusion Injury via MAPK6 Activation. J. Cell. Physiol. 2020, 235, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Zhao, Z.-H.; Meng, Q.-T.; Tie, M.-E.; Lei, S.-Q.; Xia, Z.-Y. Propofol Protects against Hepatic Ischemia/Reperfusion Injury via MiR-133a-5p Regulating the Expression of MAPK6. Cell Biol. Int. 2017, 41, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Junhua, W.; Long, L.; Jun, Y.; Yang, X. Exogenous Hydrogen Sulfide Protects SH-SY5Y Cells from OGD/RInduced Injury. Curr. Mol. Med. 2017, 17, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Anhê, G.F.; Torrão, A.S.; Nogueira, T.C.A.; Caperuto, L.C.; Amaral, M.E.C.; Medina, M.C.; Azevedo-Martins, A.K.; Carpinelli, A.R.; Carvalho, C.R.O.; Curi, R.; et al. ERK3 Associates with MAP2 and Is Involved in Glucose-Induced Insulin Secretion. Mol. Cell. Endocrinol. 2006, 251, 33–41. [Google Scholar] [CrossRef]
- El-Merahbi, R.; Viera, J.T.; Valdes, A.L.; Kolczynska, K.; Reuter, S.; Löffler, M.C.; Erk, M.; Ade, C.P.; Karwen, T.; Mayer, A.E.; et al. The Adrenergic-Induced ERK3 Pathway Drives Lipolysis and Suppresses Energy Dissipation. Genes Dev. 2020, 34, 495–510. [Google Scholar] [CrossRef]
- Loza-Valdes, A.; El-Merahbi, R.; Kassouf, T.; Demczuk, A.; Reuter, S.; Viera, J.T.; Karwen, T.; Noh, M.; Löffler, M.C.; Romero-Becerra, R.; et al. Targeting ERK3/MK5 Complex for Treatment of Obesity and Diabetes. Biochem. Biophys. Res. Commun. 2022, 612, 119–125. [Google Scholar] [CrossRef]
- Xie, Y.; Vessey, J.P.; Konecna, A.; Dahm, R.; Macchi, P.; Kiebler, M.A. The GTP-Binding Protein Septin 7 Is Critical for Dendrite Branching and Dendritic-Spine Morphology. Curr. Biol. 2007, 17, 1746–1751. [Google Scholar] [CrossRef]
- Zhang, C.; Zhu, M.; Wang, W.; Chen, D.; Chen, S.; Zheng, H. TNF-α Promotes Tumor Lymph Angiogenesis in Head and Neck Squamous Cell Carcinoma through Regulation of ERK3. Transl. Cancer Res. 2019, 8, 2439–2448. [Google Scholar] [CrossRef]
- Coulombe, P.; Rodier, G.; Pelletier, S.; Pellerin, J.; Meloche, S. Rapid Turnover of Extracellular Signal-Regulated Kinase 3 by the Ubiquitin-Proteasome Pathway Defines a Novel Paradigm of Mitogen-Activated Protein Kinase Regulation during Cellular Differentiation. Mol. Cell. Biol. 2003, 23, 4542–4558. [Google Scholar] [CrossRef]
- Mathien, S.; Déléris, P.; Soulez, M.; Voisin, L.; Meloche, S. Deubiquitinating Enzyme USP20 Regulates Extracellular Signal-Regulated Kinase 3 Stability and Biological Activity. Mol. Cell. Biol. 2017, 37, e00432-16. [Google Scholar] [CrossRef]
- Long, W.; Foulds, C.E.; Qin, J.; Liu, J.; Ding, C.; Lonard, D.M.; Solis, L.M.; Wistuba, I.I.; Qin, J.; Tsai, S.Y.; et al. ERK3 Signals through SRC-3 Coactivator to Promote Human Lung Cancer Cell Invasion. J. Clin. Investig. 2012, 122, 1869–1880. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Mahale, A.; Saranath, D. Molecular Cloning, Isolation and Characterisation of ERK3 Gene from Chewing-Tobacco Induced Oral Squamous Cell Carcinoma. Oral Oncol. 2004, 40, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Wang, S.; Zhu, X.-G.; Yu, Y.-X.; Cui, Z.-R.; Yu, Y.-Z. Increased Expression of Mitogen-Activated Protein Kinase and Its Upstream Regulating Signal in Human Gastric Cancer. World J. Gastroenterol. 2005, 11, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, T.; Li, X.; Li, T.; Ma, X.; Zhao, D.; Zhao, X. Increased Methylation of ZNF671 Suppresses Tumor Progression by Promoting MAPK6 Transcription in Laryngeal Carcinoma. Int. J. Biol. Sci. 2023, 19, 2443–2457. [Google Scholar] [CrossRef]
- Alshammari, E.S.; Aljagthmi, A.A.; Stacy, A.J.; Bottomley, M.; Shamma, H.N.; Kadakia, M.P.; Long, W. ERK3 Is Transcriptionally Upregulated by ∆Np63α and Mediates the Role of ∆Np63α in Suppressing Cell Migration in Non-Melanoma Skin Cancers. BMC Cancer 2021, 21, 155. [Google Scholar] [CrossRef]
- Vallabhaneni, S.; Liu, J.; Morel, M.; Wang, J.; DeMayo, F.J.; Long, W. Conditional ERK3 Overexpression Cooperates with PTEN Deletion to Promote Lung Adenocarcinoma Formation in Mice. Mol. Oncol. 2022, 16, 1184–1199. [Google Scholar] [CrossRef] [PubMed]
- Kostenko, S.; Dumitriu, G.; Moens, U. Tumour Promoting and Suppressing Roles of the Atypical MAP Kinase Signalling Pathway ERK3/4-MK5. J. Mol. Signal. 2012, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Zhou, W.; Wang, W.; Dong, B.; Han, D.; Shen, T.; Creighton, C.J.; Moore, D.D.; Yang, F. MAPK6-AKT Signaling Promotes Tumor Growth and Resistance to MTOR Kinase Blockade. Sci. Adv. 2021, 7, eabi6439. [Google Scholar] [CrossRef] [PubMed]
- Bogucka, K.; Marini, F.; Rosigkeit, S.; Schloeder, J.; Jonuleit, H.; David, K.; Schlackow, M.; Rajalingam, K. ERK3/MAPK6 Is Required for KRAS-Mediated NSCLC Tumorigenesis. Cancer Gene Ther. 2021, 28, 359–374. [Google Scholar] [CrossRef]
- Tanguay, P.-L.; Rodier, G.; Meloche, S. C-Terminal Domain Phosphorylation of ERK3 Controlled by Cdk1 and Cdc14 Regulates Its Stability in Mitosis. Biochem. J. 2010, 428, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Kling, D.E.; Brandon, K.L.; Sollinger, C.A.; Cavicchio, A.J.; Ge, Q.; Kinane, T.B.; Donahoe, P.K.; Schnitzer, J.J. Distribution of ERK1/2 and ERK3 during Normal Rat Fetal Lung Development. Anat. Embryol. 2006, 211, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Javary, J.; Goupil, E.; Soulez, M.; Kanshin, E.; Bouchard, A.; Seternes, O.-M.; Thibault, P.; Labbé, J.-C.; Meloche, S. Phosphoproteomic Analysis Identifies Supervillin as an ERK3 Substrate Regulating Cytokinesis and Cell Ploidy. J. Cell. Physiol. 2022, 239, e30938. [Google Scholar] [CrossRef] [PubMed]
- An, H.-J.; Lee, C.-J.; Lee, G.-E.; Choi, Y.; Jeung, D.; Chen, W.; Lee, H.S.; Kang, H.C.; Lee, J.Y.; Kim, D.J.; et al. FBXW7-Mediated ERK3 Degradation Regulates the Proliferation of Lung Cancer Cells. Exp. Mol. Med. 2022, 54, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Myers, A.K.; Morel, M.; Gee, S.H.; Hoffmann, K.A.; Long, W. ERK3 and DGKζ Interact to Modulate Cell Motility in Lung Cancer Cells. Front. Cell Dev. Biol. 2023, 11, 1192221. [Google Scholar] [CrossRef]
- Elkhadragy, L.; Alsaran, H.; Morel, M.; Long, W. Activation Loop Phosphorylation of ERK3 Is Important for Its Kinase Activity and Ability to Promote Lung Cancer Cell Invasiveness. J. Biol. Chem. 2018, 293, 16193–16205. [Google Scholar] [CrossRef]
- Elkhadragy, L.; Alsaran, H.; Long, W. The C-Terminus Tail Regulates ERK3 Kinase Activity and Its Ability in Promoting Cancer Cell Migration and Invasion. Int. J. Mol. Sci. 2020, 21, 4044. [Google Scholar] [CrossRef] [PubMed]
- Elkhadragy, L.; Chen, M.; Miller, K.; Yang, M.-H.; Long, W. A Regulatory BMI1/Let-7i/ERK3 Pathway Controls the Motility of Head and Neck Cancer Cells. Mol. Oncol. 2017, 11, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Alsaran, H.; Elkhadragy, L.; Shakya, A.; Long, W. L290P/V Mutations Increase ERK3′s Cytoplasmic Localization and Migration/Invasion-Promoting Capability in Cancer Cells. Sci. Rep. 2017, 7, 14979. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, R.; Han, X.; Hou, X.; Tian, Y.; Zhang, W. Rab31 Promotes the Invasion and Metastasis of Cervical Cancer Cells by Inhibiting MAPK6 Degradation. Int. J. Biol. Sci. 2022, 18, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Al-Mahdi, R.; Babteen, N.; Thillai, K.; Holt, M.; Johansen, B.; Wetting, H.L.; Seternes, O.-M.; Wells, C.M. A Novel Role for Atypical MAPK Kinase ERK3 in Regulating Breast Cancer Cell Morphology and Migration. Cell Adh. Migr. 2015, 9, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Ryu, K.-J.; Hong, K.-S.; Kim, H.; Han, H.; Kim, M.; Kim, T.; Ok, D.W.; Yang, J.W.; Hwangbo, C.; et al. ERK3 Increases Snail Protein Stability by Inhibiting FBXO11-Mediated Snail Ubiquitination. Cancers 2023, 16, 105. [Google Scholar] [CrossRef] [PubMed]
- Bogucka-Janczi, K.; Harms, G.; Coissieux, M.-M.; Bentires-Alj, M.; Thiede, B.; Rajalingam, K. ERK3/MAPK6 Dictates CDC42/RAC1 Activity and ARP2/3-Dependent Actin Polymerization. eLife 2023, 12, e85167. [Google Scholar] [CrossRef] [PubMed]
- Torres-Arzayus, M.I.; Font de Mora, J.; Yuan, J.; Vazquez, F.; Bronson, R.; Rue, M.; Sellers, W.R.; Brown, M. High Tumor Incidence and Activation of the PI3K/AKT Pathway in Transgenic Mice Define AIB1 as an Oncogene. Cancer Cell 2004, 6, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Erdem, H.; Li, R.; Cai, Y.; Ayala, G.; Ittmann, M.; Yu-Lee, L.; Tsai, S.Y.; Tsai, M.-J. Steroid Receptor Coactivator-3/AIB1 Promotes Cell Migration and Invasiveness through Focal Adhesion Turnover and Matrix Metalloproteinase Expression. Cancer Res. 2008, 68, 5460–5468. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.T.; Murray, G.I. Current Mechanistic Insights into the Roles of Matrix Metalloproteinases in Tumour Invasion and Metastasis. J. Pathol. 2015, 237, 273–281. [Google Scholar] [CrossRef]
- Yang, J.Y.; Ha, S.-A.; Yang, Y.-S.; Kim, J.W. P-Glycoprotein ABCB5 and YB-1 Expression Plays a Role in Increased Heterogeneity of Breast Cancer Cells: Correlations with Cell Fusion and Doxorubicin Resistance. BMC Cancer 2010, 10, 388. [Google Scholar] [CrossRef] [PubMed]
- Bian, K.; Muppani, N.R.; Elkhadragy, L.; Wang, W.; Zhang, C.; Chen, T.; Jung, S.; Seternes, O.M.; Long, W. ERK3 Regulates TDP2-Mediated DNA Damage Response and Chemoresistance in Lung Cancer Cells. Oncotarget 2016, 7, 6665–6675. [Google Scholar] [CrossRef] [PubMed]
- Martincic, D.; Hande, K.R. Topoisomerase II Inhibitors. Cancer Chemother. Biol. Response Modif. 2005, 22, 101–121. [Google Scholar] [CrossRef] [PubMed]
- Cortes Ledesma, F.; El Khamisy, S.F.; Zuma, M.C.; Osborn, K.; Caldecott, K.W. A Human 5′-Tyrosyl DNA Phosphodiesterase That Repairs Topoisomerase-Mediated DNA Damage. Nature 2009, 461, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zhao, G.; Bian, Y.; Bi, G.; Sui, Q.; Zhang, H.; Shi, H.; Shan, G.; Huang, Y.; Chen, Z.; et al. HNF4G Increases Cisplatin Resistance in Lung Adenocarcinoma via the MAPK6/Akt Pathway. PeerJ 2023, 11, e14996. [Google Scholar] [CrossRef] [PubMed]
- Crowe, D.L. Induction of P97MAPK Expression Regulates Collagen Mediated Inhibition of Proliferation and Migration in Human Squamous Cell Carcinoma Lines. Int. J. Oncol. 2004, 24, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Myers, A.K.; Markey, M.P.; Long, W. The Atypical MAPK ERK3 Potently Suppresses Melanoma Cell Growth and Invasiveness. J. Cell. Physiol. 2019, 234, 13220–13232. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Xie, H.; Yang, F.; Shan, Q.; Dai, H.; Zhuo, J.; Wei, X.; Song, P.; Zhou, L.; Xu, X.; et al. Metformin Potentiates the Effect of Arsenic Trioxide Suppressing Intrahepatic Cholangiocarcinoma: Roles of P38 MAPK, ERK3, and MTORC1. J. Hematol. Oncol. 2017, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Wang, S.; Xiang, Y. Up-Regulated MicroRNA499a by Hepatitis B Virus Induced Hepatocellular Carcinogenesis via Targeting MAPK6. PLoS ONE 2014, 9, e111410. [Google Scholar] [CrossRef]
- Adams, J.A. Activation Loop Phosphorylation and Catalysis in Protein Kinases: Is There Functional Evidence for the Autoinhibitor Model? Biochemistry 2003, 42, 601–607. [Google Scholar] [CrossRef]
- Canagarajah, B.J.; Khokhlatchev, A.; Cobb, M.H.; Goldsmith, E.J. Activation Mechanism of the MAP Kinase ERK2 by Dual Phosphorylation. Cell 1997, 90, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Zhen, E.; Robinson, M.J.; Ebert, D.; Goldsmith, E.; Cobb, M.H. Characterization of a Protein Kinase That Phosphorylates Serine 189 of the Mitogen-Activated Protein Kinase Homolog ERK3. J. Biol. Chem. 1996, 271, 12057–12062. [Google Scholar] [CrossRef] [PubMed]
- Déléris, P.; Trost, M.; Topisirovic, I.; Tanguay, P.-L.; Borden, K.L.B.; Thibault, P.; Meloche, S. Activation Loop Phosphorylation of ERK3/ERK4 by Group I P21-Activated Kinases (PAKs) Defines a Novel PAK-ERK3/4-MAPK-Activated Protein Kinase 5 Signaling Pathway. J. Biol. Chem. 2011, 286, 6470–6478. [Google Scholar] [CrossRef] [PubMed]
- De la Mota-Peynado, A.; Chernoff, J.; Beeser, A. Identification of the Atypical MAPK Erk3 as a Novel Substrate for P21-Activated Kinase (Pak) Activity. J. Biol. Chem. 2011, 286, 13603–13611. [Google Scholar] [CrossRef] [PubMed]
- Perander, M.; Al-Mahdi, R.; Jensen, T.C.; Nunn, J.A.L.; Kildalsen, H.; Johansen, B.; Gabrielsen, M.; Keyse, S.M.; Seternes, O.-M. Regulation of Atypical MAP Kinases ERK3 and ERK4 by the Phosphatase DUSP2. Sci. Rep. 2017, 7, 43471. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; Eby, M.T.; Forrest, W.F.; Gray, D.C.; Tien, J.Y.; Stern, H.M.; Murray, L.J.; Davis, D.P.; Modrusan, Z.; Seshagiri, S. Regulation of ERK3/MAPK6 Expression by BRAF. Int. J. Oncol. 2006, 29, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Liu, Q.; Yang, X.; Ding, C.; Wang, Q.; Xiong, Y. LncRNA LINC00649 Recruits TAF15 and Enhances MAPK6 Expression to Promote the Development of Lung Squamous Cell Carcinoma via Activating MAPK Signaling Pathway. Cancer Gene Ther. 2022, 29, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Chen, H.; Deng, Y.; Chen, H.; Xing, L.; Guo, Y.; Wang, M.; Chen, J. CircDNAJC11 Interacts with TAF15 to Promote Breast Cancer Progression via Enhancing MAPK6 Expression and Activating the MAPK Signaling Pathway. J. Transl. Med. 2023, 21, 186. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, K.; Sivasankar, V. MicroRNAs—Biology and Clinical Applications. J. Oral Maxillofac. Pathol. 2014, 18, 229–234. [Google Scholar] [CrossRef]
- Subramanian, M.; Francis, P.; Bilke, S.; Li, X.L.; Hara, T.; Lu, X.; Jones, M.F.; Walker, R.L.; Zhu, Y.; Pineda, M.; et al. A Mutant P53/Let-7i-Axis-Regulated Gene Network Drives Cell Migration, Invasion and Metastasis. Oncogene 2015, 34, 1094–1104. [Google Scholar] [CrossRef]
- Tian, Y.; Hao, S.; Ye, M.; Zhang, A.; Nan, Y.; Wang, G.; Jia, Z.; Yu, K.; Guo, L.; Pu, P.; et al. MicroRNAs Let-7b/i Suppress Human Glioma Cell Invasion and Migration by Targeting IKBKE Directly. Biochem. Biophys. Res. Commun. 2015, 458, 307–312. [Google Scholar] [CrossRef]
- Yang, W.-H.; Lan, H.-Y.; Huang, C.-H.; Tai, S.-K.; Tzeng, C.-H.; Kao, S.-Y.; Wu, K.-J.; Hung, M.-C.; Yang, M.-H. RAC1 Activation Mediates Twist1-Induced Cancer Cell Migration. Nat. Cell. Biol. 2012, 14, 366–374. [Google Scholar] [CrossRef]
- Siddique, H.R.; Saleem, M. Role of BMI1, a Stem Cell Factor, in Cancer Recurrence and Chemoresistance: Preclinical and Clinical Evidences. Stem Cells 2012, 30, 372–378. [Google Scholar] [CrossRef]
- Wu, F.; Mo, Q.; Wan, X.; Dan, J.; Hu, H. NEAT1/Hsa-Mir-98-5p/MAPK6 Axis Is Involved in Non-Small-Cell Lung Cancer Development. J. Cell. Biochem. 2019, 120, 2836–2846. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Huang, S.; Wu, F.; Ding, H. MiR-98 Inhibits Cell Proliferation and Induces Cell Apoptosis by Targeting MAPK6 in HUVECs. Exp. Ther. Med. 2018, 15, 2755–2760. [Google Scholar] [CrossRef]
- Coulombe, P.; Rodier, G.; Bonneil, E.; Thibault, P.; Meloche, S. N-Terminal Ubiquitination of Extracellular Signal-Regulated Kinase 3 and P21 Directs Their Degradation by the Proteasome. Mol. Cell. Biol. 2004, 24, 6140–6150. [Google Scholar] [CrossRef] [PubMed]
- Snyder, N.A.; Silva, G.M. Deubiquitinating Enzymes (DUBs): Regulation, Homeostasis, and Oxidative Stress Response. J. Biol. Chem. 2021, 297, 101077. [Google Scholar] [CrossRef]
- Hansen, C.A.; Bartek, J.; Jensen, S. A Functional Link between the Human Cell Cycle-Regulatory Phosphatase Cdc14A and the Atypical Mitogen-Activated Kinase Erk3. Cell Cycle 2008, 7, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Pilkington, R.; Garcia Munoz, A.; Nguyen, L.K.; Rauch, N.; Kennedy, S.; Monsefi, N.; Herrero, A.; Taylor, C.T.; von Kriegsheim, A. Substrate-Trapped Interactors of PHD3 and FIH Cluster in Distinct Signaling Pathways. Cell Rep. 2016, 14, 2745–2760. [Google Scholar] [CrossRef]
- Jin, Y.; Pan, Y.; Zheng, S.; Liu, Y.; Xu, J.; Peng, Y.; Zhang, Z.; Wang, Y.; Xiong, Y.; Xu, L.; et al. Inactivation of EGLN3 Hydroxylase Facilitates Erk3 Degradation via Autophagy and Impedes Lung Cancer Growth. Oncogene 2022, 41, 1752–1766. [Google Scholar] [CrossRef]
- Bind, E.; Kleyner, Y.; Skowronska-Krawczyk, D.; Bien, E.; Dynlacht, B.D.; Sánchez, I. A Novel Mechanism for Mitogen-Activated Protein Kinase Localization. Mol. Biol. Cell 2004, 15, 4457–4466. [Google Scholar] [CrossRef] [PubMed]
- Julien, C.; Coulombe, P.; Meloche, S. Nuclear Export of ERK3 by a CRM1-Dependent Mechanism Regulates Its Inhibitory Action on Cell Cycle Progression. J. Biol. Chem. 2003, 278, 42615–42624. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.J.; Xu Be, B.; Stippec, S.; Cobb, M.H. Different Domains of the Mitogen-Activated Protein Kinases ERK3 and ERK2 Direct Subcellular Localization and Upstream Specificity in Vivo. J. Biol. Chem. 2002, 277, 5094–5100. [Google Scholar] [CrossRef] [PubMed]
- Aldharee, H. Role of ERK3 in Regulating RhoGDI1-PAKs Signaling Axis. Master’s Thesis, Wright State University, Dayton, OH, USA, 2017. Available online: https://corescholar.libraries.wright.edu/cgi/viewcontent.cgi?article=2923&context=etd_all (accessed on 15 December 2023).
- Grädler, U.; Busch, M.; Leuthner, B.; Raba, M.; Burgdorf, L.; Lehmann, M.; Linde, N.; Esdar, C. Biochemical, Cellular and Structural Characterization of Novel and Selective ERK3 Inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127551. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elkhadragy, L.; Myers, A.; Long, W. Role of the Atypical MAPK ERK3 in Cancer Growth and Progression. Cancers 2024, 16, 1381. https://doi.org/10.3390/cancers16071381
Elkhadragy L, Myers A, Long W. Role of the Atypical MAPK ERK3 in Cancer Growth and Progression. Cancers. 2024; 16(7):1381. https://doi.org/10.3390/cancers16071381
Chicago/Turabian StyleElkhadragy, Lobna, Amanda Myers, and Weiwen Long. 2024. "Role of the Atypical MAPK ERK3 in Cancer Growth and Progression" Cancers 16, no. 7: 1381. https://doi.org/10.3390/cancers16071381