
The Exacerbating Effects of the Tumor Necrosis Factor in Cardiovascular Stenosis: Intimal Hyperplasia

Abstract
Simple Summary
Abstract
1. Introduction
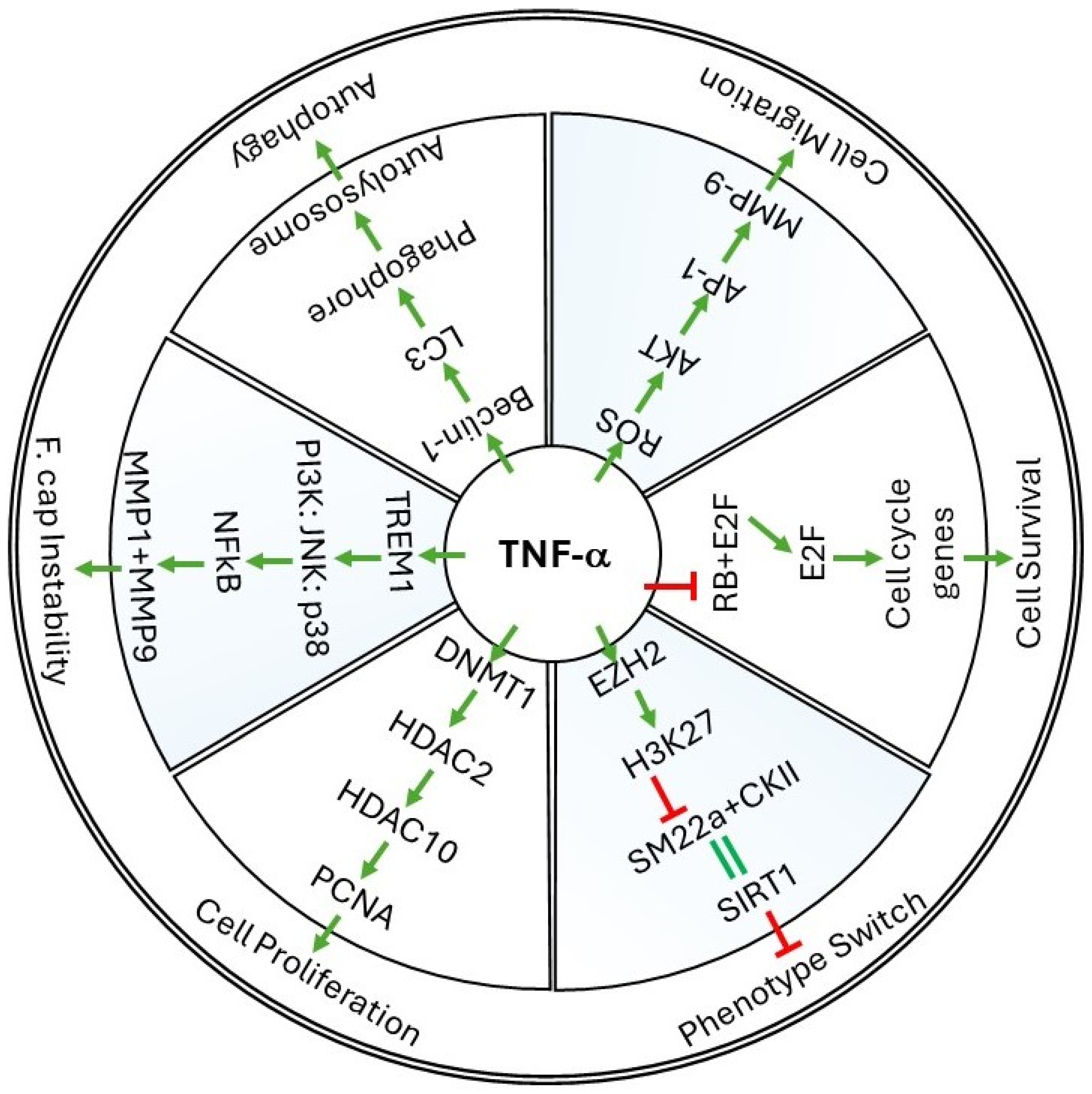
2. Cellular Events Regulated by TNF-α in Vessel Walls
2.1. Cell Survival
2.2. Cell Proliferation and Migration
2.3. Phenotype Switch and Cell Differentiation
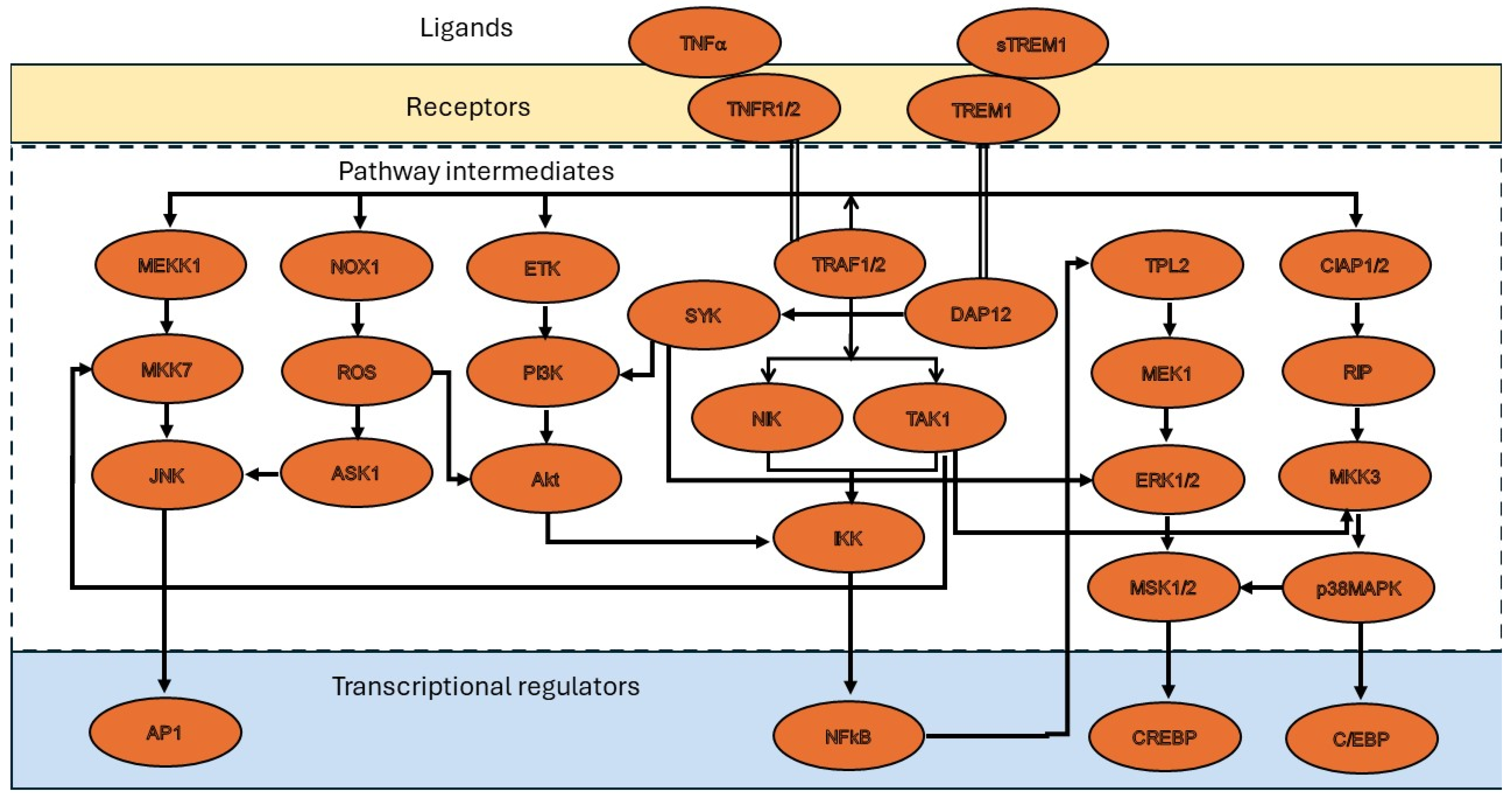
3. Molecular Mechanisms of TNF-α in Restenosis
3.1. ROS Production by TNF-α
3.2. TNF-α-Induced Epigenetic Changes during Restenosis
3.3. TNF-α-Induced Autophagy Mechanisms
4. Role of TNF Superfamily Proteins in Restenosis
5. Stenosis Consequent to Interventional Procedures
5.1. Vascular Stenosis Induced Due to Balloon Angioplasty
5.2. Vein Graft Stenosis in Coronary Artery Bypass Grafts
6. Indications and Contraindications of Using TNF-α Inhibitors to Treat Stenosis

{kind=link}
{kind=link}
| Drug Name | Target(s) | Mechanism | Indications/Implications | References | |
|---|---|---|---|---|---|
| 1 | Benpyrine racemate | TNF-α | Targets TNF-α and attenuates TNF-α-induced inflammation | Liver and lung injury | [82,83,84] |
| 2 | Clodronic acid | TNF-α, IL-1β, and IL-6 | Inhibits secretion of inflammatory cytokines by macrophages | Osteoporosis, vertebral fractures, hyperparathyroidism, hypercalcemia in malignancy, multiple myeloma, pain | [85] |
| 3 | CPI-1189 | TNF-α | Inhibits release of TNF-α release | Sciatica and postherpetic neuralgia, and AIDS dementia complex (ADC) | [86,87] |
| 4 | Hispidol Ataquimast | COX2, TNF-α, IL-5, IL-4, Leukotrienes and GM-CSF | Inhibits the secretion of Leukotrienes, TNF-α, and GM-CSF | Chronic obstructive bronchopneumopathies | [88,89,90] |
| 5 | Hispidol C87 | TNF-α | Inhibits TNF-α mediated activity by modulating TNF-TNFR interaction by affinity binding | Liver damage | [91] |
| 6 | Hispidol CDC801 | TNF-α and PDE4 | A potent, orally active, and dual inhibitor | Autoimmune diseases, Crohn’s disease | [92] |
| 7 | Hispidol Hemay007 | TNF-α | Inhibits TNF-α induced adhesion of monocytes to colon epithelial cells | Inflammatory Bowel Disease and rheumatoid arthritis | [93,94] |
| 8 | IA-14069 | TNF-α | Directly targets TNF-α | Collagen-induced arthritis | [95,96] |
| 9 | Opinercept | TNF-α | When combined with disease-modifying anti-rheumatic drugs (DMARDs) it shows superior efficacy compared to DMARDs alone | Rheumatoid arthritis | [97] |
| 10 | OPS-2071 | TNF-α | Suppresses TNF-α production from T cells | Crohn’s disease, IBD | [98,99] |
| 11 | Pomalidomide | TNF-α, IL-12, IL-16, IL-1β, MIP-1α and MCP-1 | Inhibits production of TNF-α | Multiple myeloma | [100] |
| 12 | Roquinimex (Linomide) | TNF-α | Reduces the secretion of TNF-α by tumor-associated macrophages. Enhance activity of T cells, NK cells, and macrophages | Antineoplastic activity | [101] |
| 13 | Tanfanercept | TNF-α | Neutralizes TNF-α activity | Dry eye disease | [102] |
| 14 | TAPI-1 | TNF-α | Prevents release of the soluble forms of TNF-α, by inhibiting TACE | Arthritis | [103] |
| 15 | UTL-5G | TNF-α | Inhibits TNF-α and other factors | Reduces cisplatin-induced hepatotoxicity, nephrotoxicity, and bone marrow toxicity | [104] |
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhao, S.; Jiang, J.; Jing, Y.; Liu, W.; Yang, X.; Hou, X.; Gao, L.; Wei, L. The Concentration of Tumor Necrosis Factor-α Determines Its Protective or Damaging Effect on Liver Injury by Regulating Yap Activity. Cell Death Dis. 2020, 11, 70. [Google Scholar] [CrossRef] [PubMed]
- Jang, D.; Lee, A.-H.; Shin, H.-Y.; Song, H.-R.; Park, J.-H.; Kang, T.-B.; Lee, S.-R.; Yang, S.-H. The Role of Tumor Necrosis Factor Alpha (TNF-α) in Autoimmune Disease and Current TNF-α Inhibitors in Therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Otsuka, S.; Wong, N.; Abbasi, A.; Gaida, M.M.; Fan, Y.; Meerzaman, D.; Ashwell, J.D. TNF Plays a Crucial Role in Inflammation by Signaling via T Cell TNFR2. Proc. Natl. Acad. Sci. USA 2021, 118, e2109972118. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, D.R. Tumor Necrosis Factor in the Heart. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1998, 274, R577–R595. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zheng, Y.; Chen, X. Drugs for Autoimmune Inflammatory Diseases: From Small Molecule Compounds to Anti-TNF Biologics. Front. Pharmacol. 2017, 8, 460. [Google Scholar] [CrossRef] [PubMed]
- Boosani, C.S.; Agrawal, D.K. Methylation and microRNA-Mediated Epigenetic Regulation of SOCS3. Mol. Biol. Rep. 2015, 42, 853–872. [Google Scholar] [CrossRef] [PubMed]
- Boosani, C.S.; Dhar, K.; Agrawal, D.K. Down-Regulation of Hsa-miR-1264 Contributes to DNMT1-Mediated Silencing of SOCS3. Mol. Biol. Rep. 2015, 42, 1365–1376. [Google Scholar] [CrossRef] [PubMed]
- Boosani, C.S.; Gunasekar, P.; Block, M.; Jiang, W.; Zhang, Z.; Radwan, M.M.; Agrawal, D.K. Inhibition of DNA Methyltransferase-1 Instigates the Expression of DNA Methyltransferase-3a in Angioplasty-Induced Restenosis. Can. J. Physiol. Pharmacol. 2018, 96, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, R.; Altmann, C.J.; Andres-Hernando, A.; Webb, R.G.; Okamura, K.; Yang, Y.; Falk, S.; Schmidt, E.P.; Faubel, S. Acute Lung Injury and Acute Kidney Injury Are Established by Four Hours in Experimental Sepsis and Are Improved with Pre, but Not Post, Sepsis Administration of TNF-α Antibodies. PLoS ONE 2013, 8, e79037. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.; Gaines, G.C.; Rosenberg, J.; Minter, R.; Bahjat, F.R.; Rectenwald, J.; MacKay, S.L.D.; Edwards, C.K.; Moldawer, L.L. LPS-Induced Liver Injury Ind-Galactosamine-Sensitized Mice Requires Secreted TNF-α and the TNF-P55 Receptor. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 278, R1202–R1209. [Google Scholar] [CrossRef]
- Wang, F.; Li, C.; Ding, F.H.; Shen, Y.; Gao, J.; Liu, Z.H.; Chen, J.W.; Zhang, R.Y.; Shen, W.F.; Wang, X.Q.; et al. Increased Serum TREM-1 Level Is Associated with in-Stent Restenosis, and Activation of TREM-1 Promotes Inflammation, Proliferation and Migration in Vascular Smooth Muscle Cells. Atherosclerosis 2017, 267, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, S.; Rizwani, W.; Joshi, B.; Kunigal, S.; Chellappan, S.P. TNF-α Response of Vascular Endothelial and Vascular Smooth Muscle Cells Involve Differential Utilization of ASK1 Kinase and P73. Cell Death Differ. 2012, 19, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Cheng, G.; Gangahar, D.M.; Agrawal, D.K. Insulin-like Growth Factor-1 and TNF-α Regulate Autophagy through c-Jun N-Terminal Kinase and Akt Pathways in Human Atherosclerotic Vascular Smooth Cells. Immunol. Cell Biol. 2006, 84, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Yoshida, T.; Higashi, Y.; Snarski, P.; Li, Z.; Delafontaine, P.; Danchuk, S.; Sukhanov, S. Abstract 18317: Activation of Autophagy Mediates Insulin-Like Growth Factor I (Igf-1)-Induced Anti-Apoptotic Effect in Vascular Smooth Muscle Cells. Circulation 2016, 134 (Suppl. S1), A18317. [Google Scholar] [CrossRef]
- Rolski, F.; Błyszczuk, P. Complexity of TNF-α Signaling in Heart Disease. J. Clin. Med. 2020, 9, 3267. [Google Scholar] [CrossRef] [PubMed]
- Rectenwald, J.E.; Moldawer, L.L.; Huber, T.S.; Seeger, J.M.; Ozaki, C.K. Direct Evidence for Cytokine Involvement in Neointimal Hyperplasia. Circulation 2000, 102, 1697–1702. [Google Scholar] [CrossRef] [PubMed]
- Dhar, K.; Rakesh, K.; Pankajakshan, D.; Agrawal, D.K. SOCS3 Promotor Hypermethylation and STAT3-NF-κB Interaction Downregulate SOCS3 Expression in Human Coronary Artery Smooth Muscle Cells. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H776–H785. [Google Scholar] [CrossRef] [PubMed]
- Voloshyna, I.; Seshadri, S.; Anwar, K.; Littlefield, M.J.; Belilos, E.; Carsons, S.E.; Reiss, A.B. Infliximab Reverses Suppression of Cholesterol Efflux Proteins by TNF-α: A Possible Mechanism for Modulation of Atherogenesis. BioMed Res. Int. 2014, 2014, e312647. [Google Scholar] [CrossRef]
- Jiang, W.; Block, M.E.; Boosani, C.S. Short Communication: TNF-α and IGF-1 Regulates Epigenetic Mechanisms of HDAC2 and HDAC10. PLoS ONE 2022, 17, e0263190. [Google Scholar] [CrossRef]
- Lamb, F.S.; Choi, H.; Miller, M.R.; Stark, R.J. TNFα and Reactive Oxygen Signaling in Vascular Smooth Muscle Cells in Hypertension and Atherosclerosis. Am. J. Hypertens. 2020, 33, 902–913. [Google Scholar] [CrossRef]
- Goetze, S.; Xi, X.-P.; Kawano, Y.; Kawano, H.; Fleck, E.; Hsueh, W.A.; Law, R.E. TNF-α–Induced Migration of Vascular Smooth Muscle Cells Is MAPK Dependent. Hypertension 1999, 33, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-C.; Wang, C.-P.; Chen, J.-H.; Lin, H.-H. Anti-Atherosclerotic Effect of Hibiscus Leaf Polyphenols against Tumor Necrosis Factor-Alpha-Induced Abnormal Vascular Smooth Muscle Cell Migration and Proliferation. Antioxidants 2019, 8, 620. [Google Scholar] [CrossRef]
- Hu, Y.; Pan, H.; Peng, J.; He, J.; Tang, M.; Yan, S.; Rong, J.; Li, J.; Zheng, Z.; Wang, H.; et al. Resveratrol Inhibits Necroptosis by Mediating the TNF-α/RIP1/RIP3/MLKL Pathway in Myocardial Hypoxia/Reoxygenation Injury. Acta Biochim. Biophys. Sin. 2021, 53, 430–437. [Google Scholar] [CrossRef]
- Zou, J.; Huang, Y.; Cao, K.; Yang, G.; Yin, H.; Len, J.; Hsieh, T.; Wu, J.M. Effect of Resveratrol on Intimal Hyperplasia after Endothelial Denudation in an Experimental Rabbit Model. Life Sci. 2000, 68, 153–163. [Google Scholar] [CrossRef]
- Wang, Z.; Rao, P.J.; Castresana, M.R.; Newman, W.H. TNF-α Induces Proliferation or Apoptosis in Human Saphenous Vein Smooth Muscle Cells Depending on Phenotype. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H293–H301. [Google Scholar] [CrossRef]
- Lim, J.; Ehsanipour, A.; Hsu, J.J.; Lu, J.; Pedego, T.; Wu, A.; Walthers, C.M.; Demer, L.L.; Seidlits, S.K.; Tintut, Y. Inflammation Drives Retraction, Stiffening, and Nodule Formation via Cytoskeletal Machinery in a Three-Dimensional Culture Model of Aortic Stenosis. Am. J. Pathol. 2016, 186, 2378–2389. [Google Scholar] [CrossRef]
- Cho, M.; Park, J.-K. Modular 3D In Vitro Artery-Mimicking Multichannel System for Recapitulating Vascular Stenosis and Inflammation. Micromachines 2021, 12, 1528. [Google Scholar] [CrossRef]
- Yang, Z.; Oemar, B.S.; Carrel, T.; Kipfer, B.; Julmy, F.; Lüscher, T.F. Different Proliferative Properties of Smooth Muscle Cells of Human Arterial and Venous Bypass Vessels. Circulation 1998, 97, 181–187. [Google Scholar] [CrossRef]
- Peppel, K.; Zhang, L.; Orman, E.S.; Hagen, P.-O.; Amalfitano, A.; Brian, L.; Freedman, N.J. Activation of Vascular Smooth Muscle Cells by TNF and PDGF: Overlapping and Complementary Signal Transduction Mechanisms. Cardiovasc. Res. 2005, 65, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Castresana, M.R.; Newman, W.H. NF-κB Is Required for TNF-α-directed Smooth Muscle Cell Migration. FEBS Lett. 2001, 508, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Monraats, P.S.; Pires, N.M.M.; Schepers, A.; Agema, W.R.P.; Boesten, L.S.M.; de Vries, M.R.; Zwinderman, A.H.; de Maat, M.P.M.; Doevendans, P.A.F.M.; de Winter, R.J.; et al. Tumor Necrosis Factor-Alpha Plays an Important Role in Restenosis Development. FASEB J. 2005, 19, 1998–2004. [Google Scholar] [CrossRef]
- Lee, M.Y.; Martin, A.S.; Mehta, P.K.; Dikalova, A.E.; Garrido, A.M.; Datla, S.R.; Lyons, E.; Krause, K.-H.; Banfi, B.; Lambeth, J.D.; et al. Mechanisms of Vascular Smooth Muscle NADPH Oxidase 1 (Nox1) Contribution to Injury-Induced Neointimal Formation. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 480–487. [Google Scholar] [CrossRef]
- Choi, H.; Dikalova, A.; Stark, R.J.; Lamb, F.S. C-Jun N-Terminal Kinase Attenuates TNFα Signaling by Reducing Nox1-Dependent Endosomal ROS Production in Vascular Smooth Muscle Cells. Free Radic. Biol. Med. 2015, 86, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.-I.; Lau, L.F. The Matricellular Protein CCN1 Induces Fibroblast Senescence and Restricts Fibrosis in Cutaneous Wound Healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Bai, T.; Chen, C.-C.; Lau, L.F. Matricellular Protein CCN1 Activates a Proinflammatory Genetic Program in Murine Macrophages. J. Immunol. 2010, 184, 3223–3232. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Dong, W.; Lin, Z.; Lu, J.; Wan, H.; Zhou, Z.; Liu, Z. CCN4 Regulates Vascular Smooth Muscle Cell Migration and Proliferation. Mol. Cells 2013, 36, 112–118. [Google Scholar] [CrossRef]
- Rodríguez, A.I.; Csányi, G.; Ranayhossaini, D.J.; Feck, D.M.; Blose, K.J.; Assatourian, L.; Vorp, D.A.; Pagano, P.J. MEF2B-Nox1 Signaling Is Critical for Stretch-Induced Phenotypic Modulation of Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Boosani, C.; Goswami, R. Epigenetics in Organ Specific Disorders; Translational Epigenetics; Academic Press: Cambridge, MA, USA, 2022; Volume 34. [Google Scholar]
- Jeong, K.; Murphy, J.M.; Kim, J.-H.; Campbell, P.M.; Park, H.; Rodriguez, Y.A.R.; Choi, C.-S.; Kim, J.-S.; Park, S.; Kim, H.J.; et al. FAK Activation Promotes SMC Dedifferentiation via Increased DNA Methylation in Contractile Genes. Circ. Res. 2021, 129, e215–e233. [Google Scholar] [CrossRef]
- Murphy, J.M.; Jeong, K.; Rodriguez, Y.A.R.; Kim, J.-H.; Ahn, E.-Y.E.; Lim, S.-T.S. FAK and Pyk2 Activity Promote TNF-α and IL-1β-Mediated pro-Inflammatory Gene Expression and Vascular Inflammation. Sci. Rep. 2019, 9, 7617. [Google Scholar] [CrossRef]
- Shu, Y.-N.; Dong, L.-H.; Li, H.; Pei, Q.-Q.; Miao, S.-B.; Zhang, F.; Zhang, D.-D.; Chen, R.; Yin, Y.-J.; Lin, Y.-L.; et al. CKII-SIRT1-SM22α Loop Evokes a Self-Limited Inflammatory Response in Vascular Smooth Muscle Cells. Cardiovasc. Res. 2017, 113, 1198–1207. [Google Scholar] [CrossRef]
- Lu, H.; Du, W.; Ren, L.; Hamblin, M.H.; Becker, R.C.; Chen, Y.E.; Fan, Y. Vascular Smooth Muscle Cells in Aortic Aneurysm: From Genetics to Mechanisms. J. Am. Heart Assoc. 2021, 10, e023601. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Z.; Wang, F.; Gao, P.; Pei, J.-F.; Liu, Y.; Xu, T.-T.; Tang, X.; Fu, W.-Y.; Lu, J.; Yan, Y.-F.; et al. Age-Associated Sirtuin 1 Reduction in Vascular Smooth Muscle Links Vascular Senescence and Inflammation to Abdominal Aortic Aneurysm. Circ. Res. 2016, 119, 1076–1088. [Google Scholar] [CrossRef]
- Salmon, M.; Gomez, D.; Greene, E.; Shankman, L.; Owens, G.K. Cooperative Binding of KLF4, pELK-1, and HDAC2 to a G/C Repressor Element in the SM22α Promoter Mediates Transcriptional Silencing During SMC Phenotypic Switching In Vivo. Circ. Res. 2012, 111, 685–696. [Google Scholar] [CrossRef]
- Ali, M.S.; Starke, R.M.; Jabbour, P.M.; Tjoumakaris, S.I.; Gonzalez, L.F.; Rosenwasser, R.H.; Owens, G.K.; Koch, W.J.; Greig, N.H.; Dumont, A.S. TNF-α Induces Phenotypic Modulation in Cerebral Vascular Smooth Muscle Cells: Implications for Cerebral Aneurysm Pathology. J. Cereb. Blood Flow. Metab. 2013, 33, 1564–1573. [Google Scholar] [CrossRef]
- Dostert, C.; Grusdat, M.; Letellier, E.; Brenner, D. The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol. Rev. 2019, 99, 115–160. [Google Scholar] [CrossRef]
- Micheau, O.; Rizzi, M.; Smulski, C.R. Editorial: TNFR Superfamily Oligomerization and Signaling. Front. Cell Dev. Biol. 2021, 9, 682472. [Google Scholar] [CrossRef] [PubMed]
- Simone, T.M.; Higgins, S.P.; Archambeault, J.; Higgins, C.E.; Ginnan, R.G.; Singer, H.; Higgins, P.J. A Small Molecule PAI-1 Functional Inhibitor Attenuates Neointimal Hyperplasia and Vascular Smooth Muscle Cell Survival by Promoting PAI-1 Cleavage. Cell. Signal. 2015, 27, 923–933. [Google Scholar] [CrossRef]
- Zhao, D.; Li, J.; Xue, C.; Feng, K.; Liu, L.; Zeng, P.; Wang, X.; Chen, Y.; Li, L.; Zhang, Z.; et al. TL1A Inhibits Atherosclerosis in apoE-Deficient Mice by Regulating the Phenotype of Vascular Smooth Muscle Cells. J. Biol. Chem. 2020, 295, 16314–16327. [Google Scholar] [CrossRef]
- Bamias, G.; Stamatelopoulos, K.; Zampeli, E.; Protogerou, A.; Sigala, F.; Papamichael, C.; Christopoulos, P.; Kitas, G.D.; Sfikakis, P.P. Circulating Levels of TNF-like Cytokine 1A Correlate with the Progression of Atheromatous Lesions in Patients with Rheumatoid Arthritis. Clin. Immunol. 2013, 147, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Weng, H.; Pei, Q.; Yang, P.; Fan, W.; Liu, R.; Yi, Q. The Relationship between TNF-like Protein 1A and Coronary Artery Aneurysms in Children with Kawasaki Disease. Clin. Exp. Med. 2022, 22, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Li, B.; Yang, P.; Chen, R.; Wang, C.; Wang, Z.; Shao, C.; Yuan, W.; Yan, J. CD137–CD137L Interaction Modulates Neointima Formation and the Phenotype Transformation of Vascular Smooth Muscle Cells via NFATc1 Signaling. Mol. Cell Biochem. 2018, 439, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Karpurapu, M.; Wang, D.; Singh, N.K.; Li, Q.; Rao, G.N. NFATc1 Targets Cyclin A in the Regulation of Vascular Smooth Muscle Cell Multiplication during Restenosis. J. Biol. Chem. 2008, 283, 26577–26590. [Google Scholar] [CrossRef] [PubMed]
- Farina, F.M.; Hall, I.F.; Serio, S.; Zani, S.; Climent, M.; Salvarani, N.; Carullo, P.; Civilini, E.; Condorelli, G.; Elia, L.; et al. miR-128-3p Is a Novel Regulator of Vascular Smooth Muscle Cell Phenotypic Switch and Vascular Diseases. Circ. Res. 2020, 126, e120–e135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, T.; Zhai, H.; Peng, W.; Zhou, Y.; Li, Q.; Yang, H. Inhibition of Lysine-Specific Demethylase 1A Suppresses Neointimal Hyperplasia by Targeting Bone Morphogenetic Protein 2 and Mediating Vascular Smooth Muscle Cell Phenotype. Cell Prolif. 2020, 53, e12711. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, L.; Zhan, Y.; Zhang, Z.; Chen, D.; Xiang, Y.; Xie, C. The Expression of SAH, IL-1β, Hcy, TNF-α and BDNF in Coronary Heart Disease and Its Relationship with the Severity of Coronary Stenosis. BMC Cardiovasc. Disord. 2022, 22, 101. [Google Scholar] [CrossRef]
- Gonzalez Rodriguez, A.; Schroeder, M.E.; Grim, J.C.; Walker, C.J.; Speckl, K.F.; Weiss, R.M.; Anseth, K.S. Tumor Necrosis Factor-α Promotes and Exacerbates Calcification in Heart Valve Myofibroblast Populations. FASEB J. 2021, 35, e21382. [Google Scholar] [CrossRef]
- Cao, G.; Xuan, X.; Hu, J.; Zhang, R.; Jin, H.; Dong, H. How Vascular Smooth Muscle Cell Phenotype Switching Contributes to Vascular Disease. Cell Commun. Signal. 2022, 20, 180. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.C.; Feldman, T.E.; Hirshfeld, J.W.; Jacobs, A.K.; Kern, M.J.; King, S.B.; Morrison, D.A.; O’Neill, W.W.; Schaff, H.V.; Whitlow, P.L.; et al. ACC/AHA/SCAI 2005 Guideline Update for Percutaneous Coronary Intervention: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/SCAI Writing Committee to Update the 2001 Guidelines for Percutaneous Coronary Intervention). J. Am. Coll. Cardiol. 2006, 47, e1–e121. [Google Scholar] [CrossRef] [PubMed]
- Bonta, P.I.; Pols, T.W.H.; van Tiel, C.M.; Vos, M.; Arkenbout, E.K.; Rohlena, J.; Koch, K.T.; de Maat, M.P.M.; Tanck, M.W.T.; de Winter, R.J.; et al. Nuclear Receptor Nurr1 Is Expressed in and Is Associated with Human Restenosis and Inhibits Vascular Lesion Formation In Mice Involving Inhibition of Smooth Muscle Cell Proliferation and Inflammation. Circulation 2010, 121, 2023–2032. [Google Scholar] [CrossRef]
- Matsumae, H.; Yoshida, Y.; Ono, K.; Togi, K.; Inoue, K.; Furukawa, Y.; Nakashima, Y.; Kojima, Y.; Nobuyoshi, M.; Kita, T.; et al. CCN1 Knockdown Suppresses Neointimal Hyperplasia in a Rat Artery Balloon Injury Model. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1077–1083. [Google Scholar] [CrossRef]
- Rao, V.H.; Rai, V.; Stoupa, S.; Subramanian, S.; Agrawal, D.K. Tumor Necrosis Factor-α Regulates Triggering Receptor Expressed on Myeloid Cells-1-Dependent Matrix Metalloproteinases in the Carotid Plaques of Symptomatic Patients with Carotid Stenosis. Atherosclerosis 2016, 248, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.H.; Rai, V.; Stoupa, S.; Subramanian, S.; Agrawal, D.K. Data on TREM-1 Activation Destabilizing Carotid Plaques. Data Brief. 2016, 8, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Herring, B.P.; Hoggatt, A.M.; Griffith, S.L.; McClintick, J.N.; Gallagher, P.J. Inflammation and Vascular Smooth Muscle Cell Dedifferentiation Following Carotid Artery Ligation. Physiol. Genom. 2017, 49, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Peppel, K.; Brian, L.; Chien, L.; Freedman, N.J. Vein Graft Neointimal Hyperplasia Is Exacerbated by Tumor Necrosis Factor Receptor-1 Signaling in Graft-Intrinsic Cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2277–2283. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Shukla, A.; Miller, B.L.; Espino, D.R.; Tao, M.; Berceli, S.A.; Ozaki, C.K. Tumor Necrosis Factor-α and the Early Vein Graft. J. Vasc. Surg. 2007, 45, 169–176. [Google Scholar] [CrossRef][Green Version]
- Zhang, L.; Sivashanmugam, P.; Wu, J.-H.; Brian, L.; Exum, S.T.; Freedman, N.J.; Peppel, K. Tumor Necrosis Factor Receptor-2 Signaling Attenuates Vein Graft Neointima Formation by Promoting Endothelial Recovery. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Steger, C.M.; Hartmann, A.; Rieker, R.J. Molecular Differences between Arterial and Venous Grafts in the First Year after Coronary Artery Bypass Grafting. Histochem. Cell Biol. 2020, 154, 405–419. [Google Scholar] [CrossRef] [PubMed]
- Prasongsukarn, K.; Chaisri, U.; Chartburus, P.; Wetchabut, K.; Benjathummarak, S.; Khachansaksumet, V.; Maneerat, Y. Phenotypic Alterations in Human Saphenous Vein Culture Induced by Tumor Necrosis Factor-Alpha and Lipoproteins: A Preliminary Development of an Initial Atherosclerotic Plaque Model. Lipids Health Dis. 2013, 12, 132. [Google Scholar] [CrossRef] [PubMed]
- Brahmbhatt, A.; Remuzzi, A.; Franzoni, M.; Misra, S. The Molecular Mechanisms of Hemodialysis Vascular Access Failure. Kidney Int. 2016, 89, 303–316. [Google Scholar] [CrossRef]
- Wasse, H.; Huang, R.; Naqvi, N.; Smith, E.; Wang, D.; Husain, A. Inflammation, Oxidation and Venous Neointimal Hyperplasia Precede Vascular Injury from AVF Creation in CKD Patients. J. Vasc. Access 2012, 13, 168–174. [Google Scholar] [CrossRef]
- Strik, A.S.; Löwenberg, M.; Buskens, C.J.; Gecse, K.B.; Ponsioen, C.I.; Bemelman, W.A.; D’Haens, G.R. Higher Anti-TNF Serum Levels Are Associated with Perianal Fistula Closure in Crohn’s Disease Patients. Scand. J. Gastroenterol. 2019, 54, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Yarur, A.J.; Kanagala, V.; Stein, D.J.; Czul, F.; Quintero, M.A.; Agrawal, D.; Patel, A.; Best, K.; Fox, C.; Idstein, K.; et al. Higher Infliximab Trough Levels Are Associated with Perianal Fistula Healing in Patients with Crohn’s Disease. Aliment. Pharmacol. Ther. 2017, 45, 933–940. [Google Scholar] [CrossRef]
- Davidov, Y.; Ungar, B.; Bar-Yoseph, H.; Carter, D.; Haj-Natour, O.; Yavzori, M.; Chowers, Y.; Eliakim, R.; Ben-Horin, S.; Kopylov, U. Association of Induction Infliximab Levels with Clinical Response in Perianal Crohn’s Disease. J. Crohn’s Colitis 2017, 11, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Frischknecht, K.; Greutert, H.; Weisshaupt, C.; Kaspar, M.; Yang, Z.; Lüscher, T.F.; Carrel, T.P.; Tanner, F.C. Different Vascular Smooth Muscle Cell Apoptosis in the Human Internal Mammary Artery and the Saphenous Vein: Implications for Bypass Graft Disease. J. Vasc. Res. 2006, 43, 338–346. [Google Scholar] [CrossRef]
- Bakken, A.M.; Protack, C.D.; Roztocil, E.; Nicholl, S.M.; Davies, M.G. Cell Migration in Response to the Amino-Terminal Fragment of Urokinase Requires Epidermal Growth Factor Receptor Activation through an ADAM-Mediated Mechanism. J. Vasc. Surg. 2009, 49, 1296–1303. [Google Scholar] [CrossRef]
- Kwon, H.J.; Cot, T.R.; Cuffe, M.S.; Kramer, J.M.; Braun, M.M. Case Reports of Heart Failure after Therapy with a Tumor Necrosis Factor Antagonist. Ann. Intern. Med. 2003, 138, 807–811. [Google Scholar] [CrossRef]
- Sinagra, E.; Perricone, G.; Romano, C.; Cottone, M. Heart Failure and Anti Tumor Necrosis Factor-Alpha in Systemic Chronic Inflammatory Diseases. Eur. J. Intern. Med. 2013, 24, 385–392. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sinh, P.; Cross, R. Cardiovascular Risk Assessment and Impact of Medications on Cardiovascular Disease in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2021, 27, 1107–1115. [Google Scholar] [CrossRef]
- Page, R.L.; O’Bryant, C.L.; Cheng, D.; Dow, T.J.; Ky, B.; Stein, C.M.; Spencer, A.P.; Trupp, R.J.; Lindenfeld, J. Drugs That May Cause or Exacerbate Heart Failure: A Scientific Statement From the American Heart Association. Circulation 2016, 134, e32–e69. [Google Scholar] [CrossRef]
- Sack, M.N. Tumor Necrosis Factor-α in Cardiovascular Biology and the Potential Role for Anti-Tumor Necrosis Factor-α Therapy in Heart Disease. Pharmacol. Ther. 2002, 94, 123–135. [Google Scholar] [CrossRef]
- Robertson, I.G.C.; JernstrÖm, B. The Enzymatic Conjugation of Glutathione with Bay-Region Diol-Epoxides of Benzo[a]Pyrene, Benz[a]Anthracene and Chrysene. Carcinogenesis 1986, 7, 1633–1636. [Google Scholar] [CrossRef] [PubMed]
- Bausinger, J.; Schütz, P.; Piberger, A.L.; Speit, G. Further Characterization of Benzo[a]Pyrene Diol-Epoxide (BPDE)-Induced Comet Assay Effects. Mutagenesis 2016, 31, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Iyer, P.C.; Yagi, H.; Sayer, J.M.; Jerina, D.M. 3′-H-Phosphonate Synthesis of Chiral Benzo[a]Pyrene Diol Epoxide Adducts at N(2) of Deoxyguanosine in Oligonucleotides. Chem. Res. Toxicol. 2007, 20, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Richards, P.J.; Williams, A.S.; Goodfellow, R.M.; Williams, B.D. Liposomal Clodronate Eliminates Synovial Macrophages, Reduces Inflammation and Ameliorates Joint Destruction in Antigen-Induced Arthritis. Rheumatology 1999, 38, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-J.; Zhan, Y.; Li, C.; Sun, J.; Yang, C. CPI-1189 Protects Neuronal Cells from Oxygen Glucose Deprivation/Re-Oxygenation-Induced Oxidative Injury and Cell Death. Aging 2021, 13, 6712–6723. [Google Scholar] [CrossRef]
- Bjugstad, K.B.; Flitter, W.D.; Garland, W.A.; Philpot, R.M.; Kirstein, C.L.; Arendash, G.W. CPI-1189 Prevents Apoptosis and Reduces Glial Fibrillary Acidic Protein Immunostaining in a TNF-Alpha Infusion Model for AIDS Dementia Complex. J. Neurovirol. 2000, 6, 478–491. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Hu, W.; Wu, J.; Zhou, H.; Zhou, H.; Li, X. Quinoxalinone as a Privileged Platform in Drug Development. Mini Rev. Med. Chem. 2018, 18, 392–413. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Wang, J.; Huo, C. Copper Catalyzed Aerobic Oxidative Amination of 3,4-Dihydroquinoxalin-2(1H)-Ones. Tetrahedron Lett. 2021, 78, 153271. [Google Scholar] [CrossRef]
- Yang, Q.; Yang, Z.; Tan, Y.; Zhao, J.; Sun, Q.; Zhang, H.-Y.; Zhang, Y. Direct C(Sp2)−H Amination to Synthesize Primary 3-Aminoquinoxalin-2(1H)-Ones under Simple and Mild Conditions. Adv. Synth. Catal. 2019, 361, 1662–1667. [Google Scholar] [CrossRef]
- Ma, L.; Gong, H.; Zhu, H.; Ji, Q.; Su, P.; Liu, P.; Cao, S.; Yao, J.; Jiang, L.; Han, M.; et al. A Novel Small-Molecule Tumor Necrosis Factor α Inhibitor Attenuates Inflammation in a Hepatitis Mouse Model. J. Biol. Chem. 2014, 289, 12457–12466. [Google Scholar] [CrossRef]
- Montana, J.G.; Dyke, H.J. Update on the Therapeutic Potential of PDE4 Inhibitors. Expert. Opin. Investig. Drugs 2002, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Wei, K.; Zhao, J.; Jin, Y.; Chang, C.; Zhang, R.; Xu, L.; Xu, L.; Shi, Y.; Guo, S.; et al. Trends of Treatment Development in Rheumatoid Arthritis: Promise, Progress, and Challenges. Preprints 2022, 2022030071. [Google Scholar] [CrossRef]
- Vieujean, S.; D’Amico, F.; Netter, P.; Danese, S.; Peyrin-Biroulet, L. Landscape of New Drugs and Targets in Inflammatory Bowel Disease. United Eur. Gastroenterol. J. 2022, 10, 1129–1166. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-D.; Maeng, H.; Park, Y.-H.; Shin, K.-J.; Heo, T.-H. THU0047 IA-14069, a Novel Small-Molecule Inhibitor Direct-Targeting Tumor Necrosis Factor-α, Attenuates Collagen Induced Arthritis. BMJ 2019, 78, 291. [Google Scholar] [CrossRef]
- Cho, O.; Jeong, Y.-J.; Heo, T.-H. TNF Targeting Small Molecule Attenuates Colonic Inflammation in Acute DSS-Induced Colitis Mice. J. Immunol. 2019, 202 (Suppl. S1), 52.11. [Google Scholar] [CrossRef]
- Liang, T.-H.; Lee, C.-S.; Lee, S.-S.; Wu, C.-S.; Chen, K.-H.; Hsu, P.-N.; Lin, H.-Y. Efficacy and Safety of Opinercept Tumor Necrosis Factor Inhibitor Therapy for Drug-Refractory Rheumatoid Arthritis: A Randomized Clinical Trial. Arch. Rheumatol. 2020, 35, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Fujii, K.; Takagi, H.; Shibuya, I.; Oka, D.; Yamaya, N.; Hagita, H.; Matsumoto, M.; Inagaki, K. Antibacterial and Immunosuppressive Effects of OPS-2071, a Candidate Therapy for Inflammatory Bowel Disease. Dig. Dis. Sci. 2022, 67, 3993–4007. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Sato, M.; Kazuyuki, F.; Shibuya, I.; Oka, D.; Yamaya, N.; Inagaki, K. P031 Therapeutic Effect of OPS-2071 in a Murine Model of Crohn’s Disease and in In Vitro Anti-Inflammatory Assays. J. Crohn’s Colitis 2020, 14 (Suppl. S1), S147. [Google Scholar] [CrossRef]
- Chanan-Khan, A.A.; Swaika, A.; Paulus, A.; Kumar, S.K.; Mikhael, J.R.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q. Pomalidomide: The New Immunomodulatory Agent for the Treatment of Multiple Myeloma. Blood Cancer J. 2013, 3, e143. [Google Scholar] [CrossRef] [PubMed]
- Vukanovic, J.; Isaacs, J.T. Linomide Inhibits Angiogenesis, Growth, Metastasis, and Macrophage Infiltration within Rat Prostatic Cancers1. Cancer Res. 1995, 55, 1499–1504. [Google Scholar]
- Dong, Y.; Wang, S.; Cong, L.; Zhang, T.; Cheng, J.; Yang, N.; Qu, X.; Li, D.; Zhou, X.; Wang, H.; et al. TNF-α Inhibitor Tanfanercept (HBM9036) Improves Signs and Symptoms of Dry Eye in a Phase 2 Trial in the Controlled Adverse Environment in China. Int. Ophthalmol. 2022, 42, 2459–2472. [Google Scholar] [CrossRef]
- Sultana, S.; Bishayi, B. Potential Anti-Arthritic and Anti-Inflammatory Effects of TNF-α Processing Inhibitor-1 (TAPI-1): A New Approach to the Treatment of S. Aureus Arthritis. Immunobiology 2020, 225, 151887. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Chen, B.; Huang, W.-H.; Lee, A.-R.; Media, J.; Valeriote, F.A. The Small-Molecule TNF-Alpha Modulator, UTL-5g, Reduces Side Effects Induced by Cisplatin and Enhances the Therapeutic Effect of Cisplatin In Vivo. J. Exp. Ther. Oncol. 2011, 9, 129–137. [Google Scholar] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boosani, C.S.; Burela, L. The Exacerbating Effects of the Tumor Necrosis Factor in Cardiovascular Stenosis: Intimal Hyperplasia. Cancers 2024, 16, 1435. https://doi.org/10.3390/cancers16071435
Boosani CS, Burela L. The Exacerbating Effects of the Tumor Necrosis Factor in Cardiovascular Stenosis: Intimal Hyperplasia. Cancers. 2024; 16(7):1435. https://doi.org/10.3390/cancers16071435
Chicago/Turabian StyleBoosani, Chandra Shekhar, and Laxminarayana Burela. 2024. "The Exacerbating Effects of the Tumor Necrosis Factor in Cardiovascular Stenosis: Intimal Hyperplasia" Cancers 16, no. 7: 1435. https://doi.org/10.3390/cancers16071435
APA StyleBoosani, C. S., & Burela, L. (2024). The Exacerbating Effects of the Tumor Necrosis Factor in Cardiovascular Stenosis: Intimal Hyperplasia. Cancers, 16(7), 1435. https://doi.org/10.3390/cancers16071435