
Heat Shock Proteins, a Double-Edged Sword: Significance in Cancer Progression, Chemotherapy Resistance and Novel Therapeutic Perspectives

 ,
,  ,
,  ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
2. General Features of Hsps
2.1. Small Hsps: Classification, Structure–Function Relationship and Activities
2.2. An Overview of High-Molecular-Weight Hsps and Their Mechanism of Action
2.3. Hsps—Major Cancer Chaperones
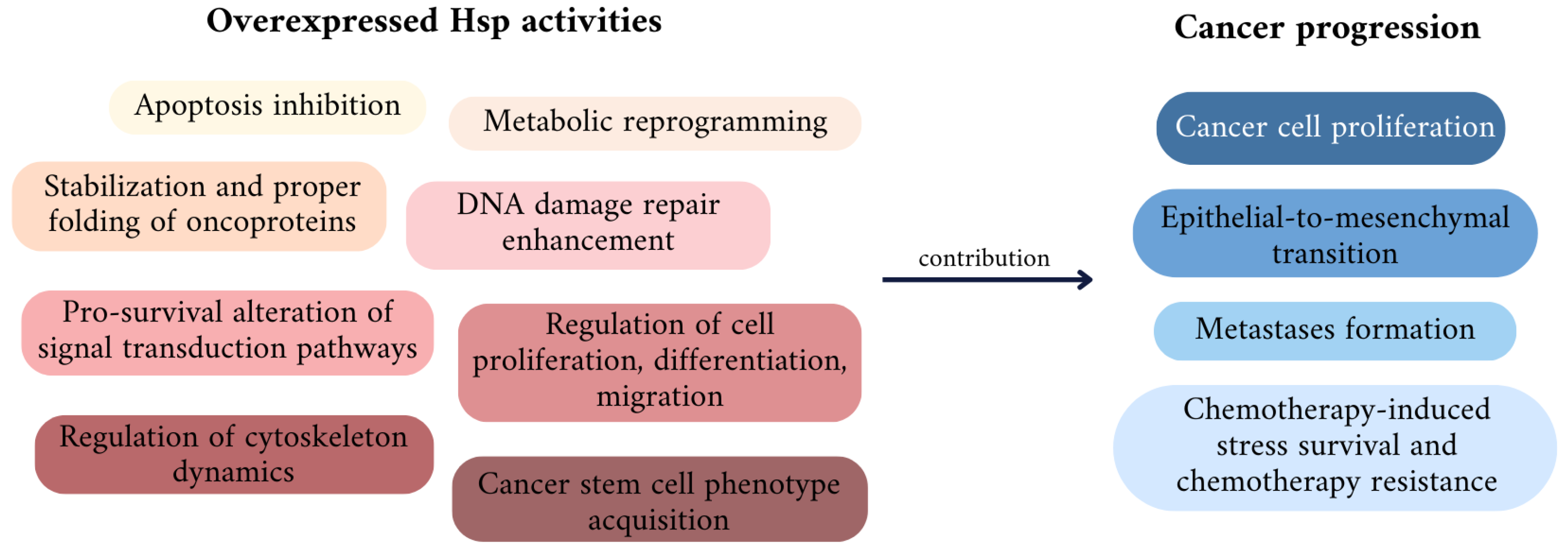
3. The Role of Particular Hsps in Cancer Development and Drug Resistance
3.1. Hsp27
3.1.1. Hsp27 as a Small Hsp Family Member
3.1.2. Involvement in Cancer Progression
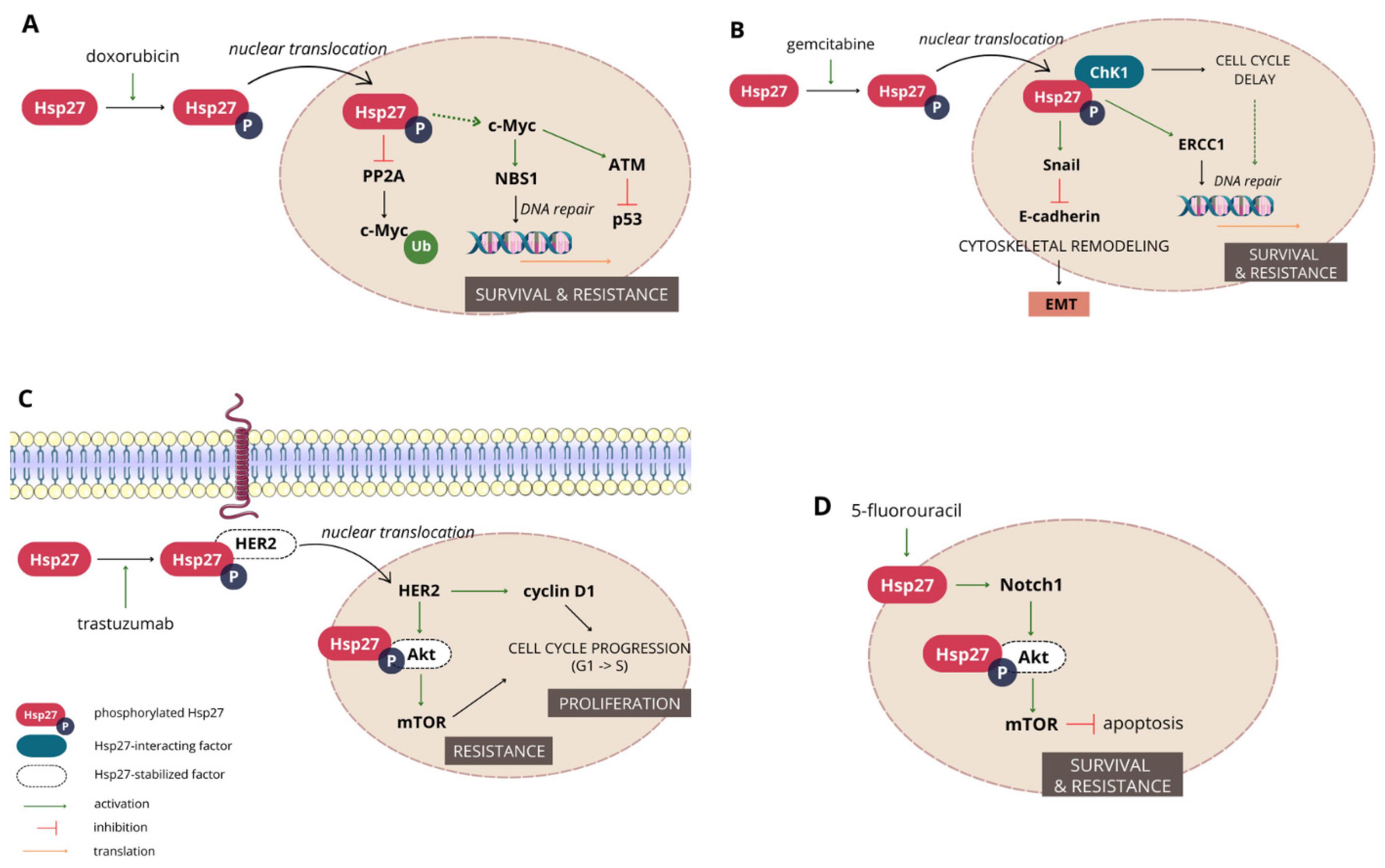
3.1.3. Hsp27 Mediates Resistance to a Wide Range of Chemotherapeutics
3.2. Hsp40
3.2.1. Hsp40s—A Diverse Co-Chaperone Family
3.2.2. Less-Known Carcinogenesis Contributors
3.2.3. Chemoresistance and Hsp40s
3.3. Hsp60
3.3.1. Hsp60—Mitochondrial Chaperonin
3.3.2. Hsp60 as a Player in Cancer Progression
3.3.3. Hsp60 and Drug Resistance
3.4. Hsp70
3.4.1. Hsp70 Protein Family
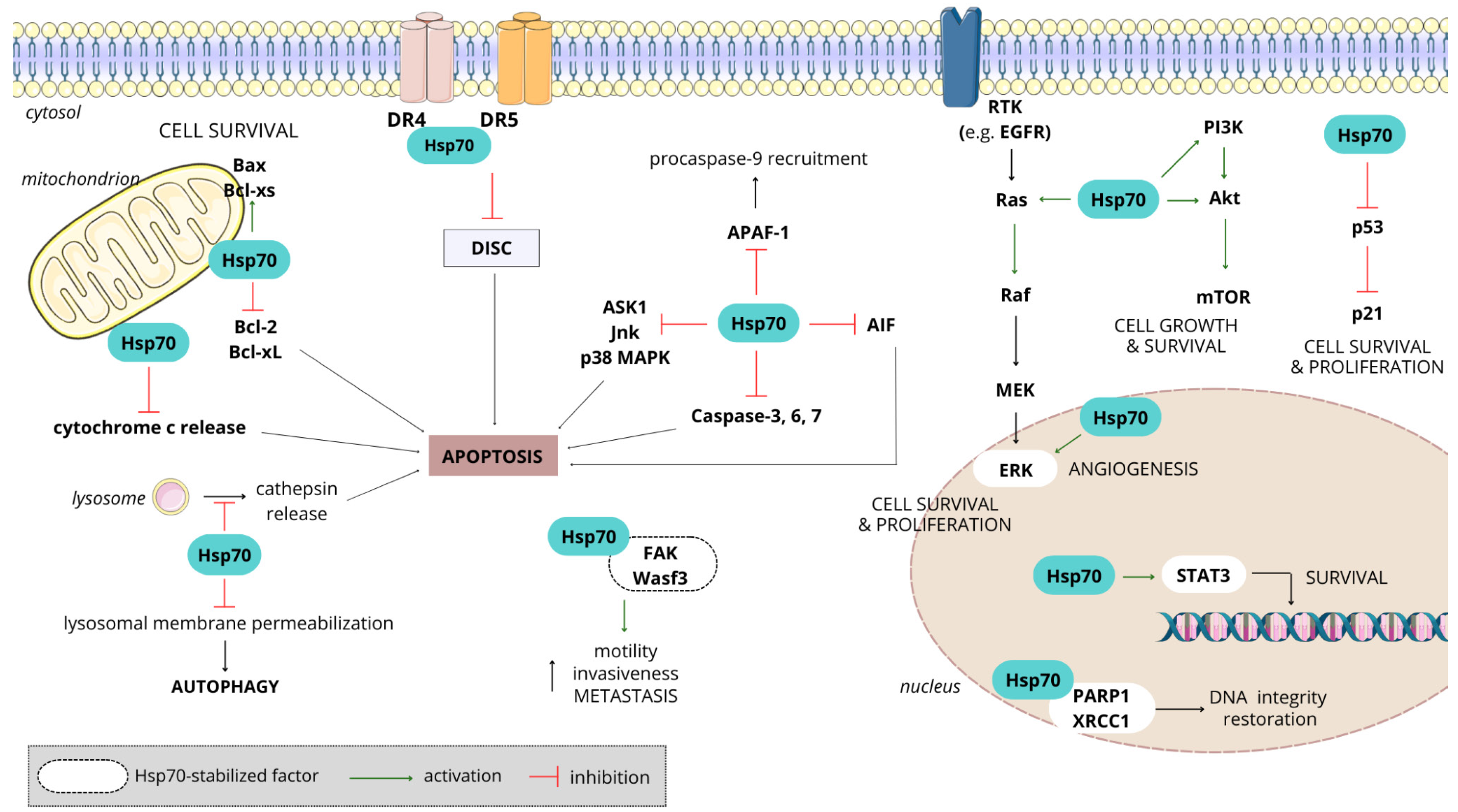
3.4.2. Hsp70 in Cancer
3.4.3. Significance of Hsp70 in Chemotherapy Resistance
3.5. Hsp90
3.5.1. Hsp90—Another ATP-Dependent Chaperone
3.5.2. Roles of Hsp90 in Carcinogenesis
3.5.3. Underexplored Issue of Hsp90 Contribution to Chemoresistance
3.6. Hsp110
3.6.1. Hsp110—Not Only a Co-Chaperone for Hsp70
3.6.2. Dual Role of Hsp110 in Cancer
3.6.3. Hsp110 as a Driver of Drug Resistance
4. Therapeutic Strategies
4.1. Inhibitors of Heat Shock Proteins in the Fight against Cancer
4.1.1. Hsp27 in Therapeutic Strategies
4.1.2. Hsp60-Aimed Therapies
4.1.3. Hsp70–Hsp40 and Hsp70–Hsp110 Axis
4.1.4. Hsp90 Strategies
4.1.5. Hsp110
4.2. Heat Shock Proteins in Cancer Immunotherapies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, B.; Feder, M.E.; Kang, L. Evolution of heat-shock protein expression underlying adaptive responses to environmental stress. Mol. Ecol. 2018, 27, 3040–3054. [Google Scholar] [CrossRef] [PubMed]
- Ikwegbue, P.C.; Masamba, P.; Oyinloye, B.E.; Kappo, A.P. Roles of Heat Shock Proteins in Apoptosis, Oxidative Stress, Human Inflammatory Diseases, and Cancer. Pharmaceuticals 2017, 11, 2. [Google Scholar] [CrossRef]
- Wu, Y.; Zhao, J.; Tian, Y.; Jin, H. Cellular functions of heat shock protein 20 (HSPB6) in cancer: A review. Cell Signal. 2023, 112, 110928. [Google Scholar] [CrossRef] [PubMed]
- Bozaykut, P.; Ozer, N.K.; Karademir, B. Regulation of protein turnover by heat shock proteins. Free Radic. Biol. Med. 2014, 77, 195–209. [Google Scholar] [CrossRef]
- Richter, K.; Haslbeck, M.; Buchner, J. The heat shock response: Life on the verge of death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Park, C.-J.; Seo, Y.-S. Heat Shock Proteins: A Review of the Molecular Chaperones for Plant Immunity. Plant Pathol. J. 2015, 31, 323–333. [Google Scholar] [CrossRef]
- Lanneau, D.; Wettstein, G.; Bonniaud, P.; Garrido, C. Heat shock proteins: Cell protection through protein triage. Sci. World J. 2010, 10, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Kim, H.J.; Lim, J.H.; Lee, S.H. Heat Shock Proteins: Agents of Cancer Development and Therapeutic Targets in Anti-Cancer Therapy. Cells 2019, 9, 60. [Google Scholar] [CrossRef]
- Potapov, O.; Sanchez Margallo, F.; Komorowski, A. The effectiveness of a live animal model in a laparoscopic partial nephrectomy for renal cancer training—A survey study. Nowotw. J. Oncol. 2022, 72, 155–160. [Google Scholar] [CrossRef]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef]
- Albakova, Z.; Mangasarova, Y.; Albakov, A.; Gorenkova, L. HSP70 and HSP90 in Cancer: Cytosolic, Endoplasmic Reticulum and Mitochondrial Chaperones of Tumorigenesis. Front. Oncol. 2022, 12, 829520. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.J.; Guerrero-Giménez, M.E.; Prince, T.L.; Ackerman, A.; Bonorino, C.; Calderwood, S.K. Heat Shock Proteins Are Essential Components in Transformation and Tumor Progression: Cancer Cell Intrinsic Pathways and Beyond. Int. J. Mol. Sci. 2019, 20, 4507. [Google Scholar] [CrossRef] [PubMed]
- Buttacavoli, M.; Di Cara, G.; D’Amico, C.; Geraci, F.; Pucci-Minafra, I.; Feo, S.; Cancemi, P. Prognostic and Functional Significant of Heat Shock Proteins (HSPs) in Breast Cancer Unveiled by Multi-Omics Approaches. Biology 2021, 10, 247. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Xiao, H.; Cao, L. Recent advances in heat shock proteins in cancer diagnosis, prognosis, metabolism and treatment. Biomed. Pharmacother. 2021, 142, 112074. [Google Scholar] [CrossRef] [PubMed]
- Jouzova, A.; Hruban, L.; Huptych, M.; Janku, P.; Polisenska, M. Maternal body mass index and external cephalic version success rate—Are they related? Ginekol. Pol. 2021, 92, 423–427. [Google Scholar] [CrossRef]
- Zou, T.; Liu, J.-Y.; She, L.; Yin, J.-Y.; Li, X.; Li, X.-P.; Zhou, H.-H.; Chen, J.; Liu, Z.-Q. The Association Between Heat-Shock Protein Polymorphisms and Prognosis in Lung Cancer Patients Treated With Platinum-Based Chemotherapy. Front. Pharmacol. 2020, 11, 1029. [Google Scholar] [CrossRef] [PubMed]
- Shevtsov, M.; Multhoff, G.; Mikhaylova, E.; Shibata, A.; Guzhova, I.; Margulis, B. Combination of Anti-Cancer Drugs with Molecular Chaperone Inhibitors. Int. J. Mol. Sci. 2019, 20, 5284. [Google Scholar] [CrossRef]
- Wang, X.; Chen, M.; Zhou, J.; Zhang, X. HSP27, 70 and 90, anti-apoptotic proteins, in clinical cancer therapy (Review). Int. J. Oncol. 2014, 45, 18–30. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Gong, J. Heat Shock Proteins Promote Cancer: It’s a Protection Racket. Trends Biochem. Sci. 2016, 41, 311–323. [Google Scholar] [CrossRef]
- Hu, C.; Yang, J.; Qi, Z.; Wu, H.; Wang, B.; Zou, F.; Mei, H.; Liu, J.; Wang, W.; Liu, Q. Heat shock proteins: Biological functions, pathological roles, and therapeutic opportunities. Med. Comm. 2022, 3, e161. [Google Scholar] [CrossRef]
- Sottile, M.L.; Nadin, S.B. Heat shock proteins and DNA repair mechanisms: An updated overview. Cell Stress Chaperones 2018, 23, 303–315. [Google Scholar] [CrossRef]
- Kanagasabai, R.; Krishnamurthy, K.; Druhan, L.J.; Ilangovan, G. Forced expression of heat shock protein 27 (Hsp27) reverses P-glycoprotein (ABCB1)-mediated drug efflux and MDR1 gene expression in Adriamycin-resistant human breast cancer cells. J. Biol. Chem. 2011, 286, 33289–33300. [Google Scholar] [CrossRef] [PubMed]
- Pokharel, D.; Roseblade, A.; Oenarto, V.; Lu, J.F.; Bebawy, M. Proteins regulating the intercellular transfer and function of P-glycoprotein in multidrug-resistant cancer. Ecancermedicalscience 2017, 11, 768. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Alam, Q.; Zubair Alam, M.; I. Azhar, E.; Hussain Wali Sait, K.; Anfinan, N.; Mushtaq, G.; Amjad Kamal, M.; Rasool, M. Current Understanding of HSP90 as a Novel Therapeutic Target: An Emerging Approach for the Treatment of Cancer. Curr. Pharm. Des. 2016, 22, 2947–2959. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Zhang, N.; Li, X.; Huang, H.; Peng, C.; Huang, W.; Foster, L.J.; He, G.; Han, B. Small-molecule dual inhibitors targeting heat shock protein 90 for cancer targeted therapy. Bioorg. Chem. 2023, 139, 106721. [Google Scholar] [CrossRef] [PubMed]
- Narayanankutty, V.; Narayanankutty, A.; Nair, A. Heat Shock Proteins (HSPs): A Novel Target for Cancer Metastasis Prevention. Curr. Drug Targets 2019, 20, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, T.; Taha, E.A.; Calderwood, S.K.; Ono, K. A Novel Model of Cancer Drug Resistance: Oncosomal Release of Cytotoxic and Antibody-Based Drugs. Biology 2020, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Parma, B.; Wurdak, H.; Ceppi, P. Harnessing mitochondrial metabolism and drug resistance in non-small cell lung cancer and beyond by blocking heat-shock proteins. Drug Resist. Updates 2022, 65, 100888. [Google Scholar] [CrossRef] [PubMed]
- Bakthisaran, R.; Tangirala, R.; Rao, C.M. Small heat shock proteins: Role in cellular functions and pathology. Biochim. Biophys. Acta 2015, 1854, 291–319. [Google Scholar] [CrossRef]
- Taylor, R.P.; Benjamin, I.J. Small heat shock proteins: A new classification scheme in mammals. J. Mol. Cell Cardiol. 2005, 38, 433–444. [Google Scholar] [CrossRef]
- Janowska, M.K.; Baughman, H.E.R.; Woods, C.N.; Klevit, R.E. Mechanisms of Small Heat Shock Proteins. Cold Spring Harb. Perspect. Biol. 2019, 11, a034025. [Google Scholar] [CrossRef] [PubMed]
- Mymrikov, E.V.; Seit-Nebi, A.S.; Gusev, N.B. Large potentials of small heat shock proteins. Physiol. Rev. 2011, 91, 1123–1159. [Google Scholar] [CrossRef]
- Boelens, W.C. Structural aspects of the human small heat shock proteins related to their functional activities. Cell Stress Chaperones 2020, 25, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Carra, S.; Alberti, S.; Arrigo, P.A.; Benesch, J.L.; Benjamin, I.J.; Boelens, W.; Bartelt-Kirbach, B.; Brundel, B.J.J.M.; Buchner, J.; Bukau, B.; et al. The growing world of small heat shock proteins: From structure to functions. Cell Stress Chaperones 2017, 22, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Delbecq, S.P.; Rosenbaum, J.C.; Klevit, R.E. A Mechanism of Subunit Recruitment in Human Small Heat Shock Protein Oligomers. Biochemistry 2015, 54, 4276–4284. [Google Scholar] [CrossRef] [PubMed]
- Carra, S.; Alberti, S.; Benesch, J.L.P.; Boelens, W.; Buchner, J.; Carver, J.A.; Cecconi, C.; Ecroyd, H.; Gusev, N.; Hightower, L.E.; et al. Small heat shock proteins: Multifaceted proteins with important implications for life. Cell Stress Chaperones 2019, 24, 295–308. [Google Scholar] [CrossRef]
- Mitra, R.; Wu, K.; Lee, C.; Bardwell, J.C.A. ATP-Independent Chaperones. Annu. Rev. Biophys. 2022, 51, 409–429. [Google Scholar] [CrossRef]
- Hall, D. On the nature of the optimal form of the holdase-type chaperone stress response. FEBS Lett. 2020, 594, 43–66. [Google Scholar] [CrossRef]
- Treweek, T.M.; Meehan, S.; Ecroyd, H.; Carver, J.A. Small heat-shock proteins: Important players in regulating cellular proteostasis. Cell Mol. Life Sci. 2015, 72, 429–451. [Google Scholar] [CrossRef]
- Jaya, N.; Garcia, V.; Vierling, E. Substrate binding site flexibility of the small heat shock protein molecular chaperones. Proc. Natl. Acad. Sci. USA 2009, 106, 15604–15609. [Google Scholar] [CrossRef]
- Mogk, A.; Bukau, B.; Kampinga, H.H. Cellular Handling of Protein Aggregates by Disaggregation Machines. Mol. Cell 2018, 69, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Haslbeck, M.; Vierling, E. A first line of stress defense: Small heat shock proteins and their function in protein homeostasis. J. Mol. Biol. 2015, 427, 1537–1548. [Google Scholar] [CrossRef] [PubMed]
- Arrigo, A.-P. Human small heat shock proteins: Protein interactomes of homo- and hetero-oligomeric complexes: An update. FEBS Lett. 2013, 587, 1959–1969. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Pirkkala, L.; Sistonen, L. Heat Shock Proteins (HSPs): Structure, Function and Genetics. In Encyclopedia of Life Sciences; John Wiley & Sons: Hoboken, NJ, USA, 2006; ISBN 9780470015902. [Google Scholar] [CrossRef]
- Rampelt, H.; Kirstein-Miles, J.; Nillegoda, N.B.; Chi, K.; Scholz, S.R.; Morimoto, R.I.; Bukau, B. Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J. 2012, 31, 4221–4235. [Google Scholar] [CrossRef] [PubMed]
- Lanneau, D.; Brunet, M.; Frisan, E.; Solary, E.; Fontenay, M.; Garrido, C. Heat shock proteins: Essential proteins for apoptosis regulation. J. Cell Mol. Med. 2008, 12, 743–761. [Google Scholar] [CrossRef] [PubMed]
- Zuo, D.; Subjeck, J.; Wang, X.Y. Unfolding the Role of Large Heat Shock Proteins: New Insights and Therapeutic Implications. Front. Immunol. 2016, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular chaperones and protein quality control. Cell 2006, 125, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Dai, S.; Cao, J. Proteotoxic stress of cancer: Implication of the heat-shock response in oncogenesis. J. Cell Physiol. 2012, 227, 2982–2987. [Google Scholar] [CrossRef]
- Vaklavas, C.; Blume, S.W.; Grizzle, W.E. Translational Dysregulation in Cancer: Molecular Insights and Potential Clinical Applications in Biomarker Development. Front. Oncol. 2017, 7, 158. [Google Scholar] [CrossRef]
- Dai, C.; Whitesell, L.; Rogers, A.B.; Lindquist, S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 2007, 130, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Dubrez, L.; Causse, S.; Borges Bonan, N.; Dumétier, B.; Garrido, C. Heat-shock proteins: Chaperoning DNA repair. Oncogene 2020, 39, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.K.; Sharma, B.; Tiwari, P.K. The small heat shock protein Hsp27: Present understanding and future prospects. J. Therm. Biol. 2017, 69, 149–154. [Google Scholar] [CrossRef]
- McDonald, E.T.; Bortolus, M.; Koteiche, H.A.; Mchaourab, H.S. Sequence, structure, and dynamic determinants of Hsp27 (HspB1) equilibrium dissociation are encoded by the N-terminal domain. Biochemistry 2012, 51, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Keith Calderwood, S.; Multhoff, G.; Garrido, C.; O’Brien, E.R.; Venu, P.V.; Batulan, Z.; Krishna Pulakazhi Venu, V.; Li, Y.; Koumbadinga, G.; Gisela Alvarez-Olmedo, D.; et al. Extracellular Release and Signaling by Heat Shock Protein 27: Role in Modifying vascular inflammation. Front. Immunol. 2016, 7, 188545. [Google Scholar]
- Shin, K.D.; Lee, M.-Y.; Shin, D.-S.; Lee, S.; Son, K.-H.; Koh, S.; Paik, Y.-K.; Kwon, B.-M.; Han, D.C. Blocking tumor cell migration and invasion with biphenyl isoxazole derivative KRIBB3, a synthetic molecule that inhibits Hsp27 phosphorylation. J. Biol. Chem. 2005, 280, 41439–41448. [Google Scholar] [CrossRef] [PubMed]
- Lampros, M.; Vlachos, N.; Voulgaris, S.; Alexiou, G.A. The Role of Hsp27 in Chemotherapy Resistance. Biomedicines 2022, 10, 897. [Google Scholar] [CrossRef] [PubMed]
- Vahid, S.; Thaper, D.; Gibson, K.F.; Bishop, J.L.; Zoubeidi, A. Molecular chaperone Hsp27 regulates the Hippo tumor suppressor pathway in cancer. Sci. Rep. 2016, 6, 31842. [Google Scholar] [CrossRef]
- Tan, C.Y.; Ban, H.; Kim, Y.-H.; Lee, S.-K. The heat shock protein 27 (Hsp27) operates predominantly by blocking the mitochondrial-independent/extrinsic pathway of cellular apoptosis. Mol. Cells 2009, 27, 533–538. [Google Scholar] [CrossRef]
- Grotegut, P.; Hoerdemann, P.J.; Reinehr, S.; Gupta, N.; Dick, H.B.; Joachim, S.C. Heat Shock Protein 27 Injection Leads to Caspase Activation in the Visual Pathway and Retinal T-Cell Response. Int. J. Mol. Sci. 2021, 22, 513. [Google Scholar] [CrossRef]
- Schmitt, E.; Gehrmann, M.; Brunet, M.; Multhoff, G.; Garrido, C. Intracellular and extracellular functions of heat shock proteins: Repercussions in cancer therapy. J. Leukoc. Biol. 2007, 81, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Havasi, A.; Li, Z.; Wang, Z.; Martin, J.L.; Botla, V.; Ruchalski, K.; Schwartz, J.H.; Borkan, S.C. Hsp27 inhibits Bax activation and apoptosis via a phosphatidylinositol 3-kinase-dependent mechanism. J. Biol. Chem. 2008, 283, 12305–12313. [Google Scholar] [CrossRef] [PubMed]
- Shan, R.; Liu, N.; Yan, Y.; Liu, B. Apoptosis, autophagy and atherosclerosis: Relationships and the role of Hsp27. Pharmacol. Res. 2021, 166, 105169. [Google Scholar] [CrossRef] [PubMed]
- Charette, S.J.; Landry, J. The interaction of HSP27 with Daxx identifies a potential regulatory role of HSP27 in Fas-induced apoptosis. Ann. N. Y. Acad. Sci. 2000, 926, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Maugeri-Saccà, M.; De Maria, R. The Hippo pathway in normal development and cancer. Pharmacol. Ther. 2018, 186, 60–72. [Google Scholar] [CrossRef]
- Vahid, S.; Thaper, D.; Zoubeidi, A. Abstract 4956: Hsp27 negatively affects Hippo tumor suppressor pathway to regulate cell survival in cancer. Cancer Res. 2015, 75, 4956. [Google Scholar] [CrossRef]
- Noubissi Nzeteu, G.A.; Geismann, C.; Arlt, A.; Hoogwater, F.J.H.; Nijkamp, M.W.; Meyer, N.H.; Bockhorn, M. Role of Epithelial-to-Mesenchymal Transition for the Generation of Circulating Tumors Cells and Cancer Cell Dissemination. Cancers 2022, 14, 5483. [Google Scholar] [CrossRef]
- Shiota, M.; Bishop, J.L.; Nip, K.M.; Zardan, A.; Takeuchi, A.; Cordonnier, T.; Beraldi, E.; Bazov, J.; Fazli, L.; Chi, K.; et al. Hsp27 regulates epithelial mesenchymal transition, metastasis, and circulating tumor cells in prostate cancer. Cancer Res. 2013, 73, 3109–3119. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; He, L.; Gan, Y.; Liu, J.; Tang, J.; Long, Z.; Tan, J. HMGN5 promotes IL-6-induced epithelial-mesenchymal transition of bladder cancer by interacting with Hsp27. Aging 2020, 12, 7282–7298. [Google Scholar] [CrossRef]
- Cordonnier, T.; Bishop, J.L.; Shiota, M.; Nip, K.M.; Thaper, D.; Vahid, S.; Heroux, D.; Gleave, M.; Zoubeidi, A. Hsp27 regulates EGF/β-catenin mediated epithelial to mesenchymal transition in prostate cancer. Int. J. Cancer 2015, 136, E496–E507. [Google Scholar] [CrossRef]
- Gui, T.; Sun, Y.; Shimokado, A.; Muragaki, Y. The Roles of Mitogen-Activated Protein Kinase Pathways in TGF-β-Induced Epithelial-Mesenchymal Transition. J. Signal Transduct. 2012, 2012, 289243. [Google Scholar] [CrossRef]
- Liu, Y.; Qian, J.; Li, X.; Chen, W.; Xu, A.; Zhao, K.; Hua, Y.; Huang, Z.; Zhang, J.; Liang, C.; et al. Long noncoding RNA BX357664 regulates cell proliferation and epithelial-to-mesenchymal transition via inhibition of TGF-β1/p38/HSP27 signaling in renal cell carcinoma. Oncotarget 2016, 7, 81410–81422. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, H.; Okano, T.; Minegishi, Y.; Matsuda, K.; Sudoh, J.; Kitamura, K.; Noro, R.; Soeno, C.; Yoshimura, A.; Seike, M.; et al. HSP27 modulates epithelial to mesenchymal transition of lung cancer cells in a Smad-independent manner. Oncol. Lett. 2010, 1, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Wettstein, G.; Bellaye, P.-S.; Kolb, M.; Hammann, A.; Crestani, B.; Soler, P.; Marchal-Somme, J.; Hazoume, A.; Gauldie, J.; Gunther, A.; et al. Inhibition of HSP27 blocks fibrosis development and EMT features by promoting Snail degradation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 1549–1560. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Chou, K.-T.; Hsu, J.-W.; Lin, J.-H.; Hsu, T.-W.; Yen, D.H.-T.; Hung, S.-C.; Hsu, H.-S. High metabolic rate and stem cell characteristics of esophageal cancer stem-like cells depend on the Hsp27-AKT-HK2 pathway. Int. J. Cancer 2019, 145, 2144–2156. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Liu, T.-T.; Wang, H.-H.; Hong, H.-M.; Yu, A.L.; Feng, H.-P.; Chang, W.-W. Hsp27 participates in the maintenance of breast cancer stem cells through regulation of epithelial-mesenchymal transition and nuclear factor-κB. Breast Cancer Res. 2011, 13, R101. [Google Scholar] [CrossRef] [PubMed]
- Thuringer, D.; Jego, G.; Wettstein, G.; Terrier, O.; Cronier, L.; Yousfi, N.; Hébrard, S.; Bouchot, A.; Hazoumé, A.; Joly, A.-L.; et al. Extracellular HSP27 mediates angiogenesis through Toll-like receptor 3. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 4169–4183. [Google Scholar] [CrossRef]
- Ramani, S.; Park, S. HSP27 role in cardioprotection by modulating chemotherapeutic doxorubicin-induced cell death. J. Mol. Med. 2021, 99, 771–784. [Google Scholar] [CrossRef]
- Xu, Y.; Diao, Y.; Qi, S.; Pan, X.; Wang, Q.; Xin, Y.; Cao, X.; Ruan, J.; Zhao, Z.; Luo, L.; et al. Phosphorylated Hsp27 activates ATM-dependent p53 signaling and mediates the resistance of MCF-7 cells to doxorubicin-induced apoptosis. Cell Signal. 2013, 25, 1176–1185. [Google Scholar] [CrossRef]
- Bi, X.; Zhang, M.; Zhou, J.; Yan, X.; Cheng, L.; Luo, L.; Huang, C.; Yin, Z. Phosphorylated Hsp27 promotes adriamycin resistance in breast cancer cells through regulating dual phosphorylation of c-Myc. Cell Signal. 2023, 112, 110913. [Google Scholar] [CrossRef]
- Maadi, H.; Soheilifar, M.H.; Choi, W.-S.; Moshtaghian, A.; Wang, Z. Trastuzumab Mechanism of Action; 20 Years of Research to Unravel a Dilemma. Cancers 2021, 13, 3540. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Kang, K.W.; Kim, K.-H.; Kwon, B.; Kim, S.-K.; Lee, H.-Y.; Kong, S.-Y.; Lee, E.S.; Jang, S.-G.; Yoo, B.C. Upregulated HSP27 in human breast cancer cells reduces Herceptin susceptibility by increasing Her2 protein stability. BMC Cancer 2008, 8, 286. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.-Y.; Choi, S.-K.; Seo, S.H.; Jo, H.; Shin, J.-H.; Na, Y.; Lee, Y.-S.; Kwon, Y. Specific Roles of HSP27 S15 Phosphorylation Augmenting the Nuclear Function of HER2 to Promote Trastuzumab Resistance. Cancers 2020, 12, 1540. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, X.; Huang, S.; Chen, M.; Shen, S.; Ding, X.; Lv, Y.; Zou, X. The Effects of HSP27 on Gemcitabine-Resistant Pancreatic Cancer Cell Line Through Snail. Pancreas 2015, 44, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, A.; Jalili-Nik, M.; Avan, A.; Ferns, G.A.; Khazaei, M.; Hassanian, S.M. The role of HSP27 in the development of drug resistance of gastrointestinal malignancies: Current status and perspectives. J. Cell Physiol. 2019, 234, 8241–8248. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Li, Z.; Tang, T.; Du, J.; Zhou, X.; Wu, H.; Li, X.; Qin, G. ERp29 downregulation enhances lung adenocarcinoma cell chemosensitivity to gemcitabine by upregulating HSP27 phosphorylation. Exp. Ther. Med. 2019, 17, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Choi, H.J.; Kang, S.; Kim, S.Y.; Hwang, Y.-S.; Je, S.; Han, Z.; Kim, J.-H.; Song, J.J. Ratio of phosphorylated HSP27 to nonphosphorylated HSP27 biphasically acts as a determinant of cellular fate in gemcitabine-resistant pancreatic cancer cells. Cell Signal. 2015, 27, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Neizer-Ashun, F.; Bhattacharya, R. Reality CHEK: Understanding the biology and clinical potential of CHK1. Cancer Lett. 2021, 497, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, M.; Adachi, S.; Yasuda, I.; Yamauchi, T.; Kawaguchi, J.; Itani, M.; Yoshioka, T.; Matsushima-Nishiwaki, R.; Hirose, Y.; Kozawa, O.; et al. Phosphorylation status of heat shock protein 27 plays a key role in gemcitabine-induced apoptosis of pancreatic cancer cells. Cancer Lett. 2011, 313, 218–225. [Google Scholar] [CrossRef]
- Zhang, N.; Yin, Y.; Xu, S.-J.; Chen, W.-S. 5-Fluorouracil: Mechanisms of resistance and reversal strategies. Molecules 2008, 13, 1551–1569. [Google Scholar] [CrossRef]
- Tausif, Y.M.; Thekkekkara, D.; Sai, T.E.; Jahagirdar, V.; Arjun, H.R.; Meheronnisha, S.K.; Babu, A.; Banerjee, A. Heat shock protein paradigms in cancer progression: Future therapeutic perspectives. 3 Biotech 2024, 14, 96. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liu, Y.; Long, Y.; Liu, B.; Wang, X. Role of HSP27 in the multidrug sensitivity and resistance of colon cancer cells. Oncol. Lett. 2020, 19, 2021–2027. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bao, Y.; Yang, G.-K.; Wan, J.; Du, L.-J.; Ma, Z.-H. MiR-214 sensitizes human colon cancer cells to 5-FU by targeting Hsp27. Cell. Mol. Biol. Lett. 2019, 24, 22. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Upadhyay, A.K.; Bhat, M.K. Inhibition of Hsp27 and Hsp40 potentiates 5-fluorouracil and carboplatin mediated cell killing in hepatoma cells. Cancer Biol. Ther. 2009, 8, 2106–2113. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liang, C.; Zhou, L. Structural and functional analysis of the Hsp70/Hsp40 chaperone system. Protein Sci. 2020, 29, 378–390. [Google Scholar] [CrossRef]
- Sterrenberg, J.N.; Blatch, G.L.; Edkins, A.L. Human DNAJ in cancer and stem cells. Cancer Lett. 2011, 312, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.J.; Hageman, J.; Carra, S.; Kampinga, H.H. Structural and functional diversities between members of the human HSPB, HSPH, HSPA, and DNAJ chaperone families. Biochemistry 2008, 47, 7001–7011. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-Y.; Hong, S. Multi-Faceted Roles of DNAJB Protein in Cancer Metastasis and Clinical Implications. Int. J. Mol. Sci. 2022, 23, 14970. [Google Scholar] [CrossRef]
- Hageman, J.; Rujano, M.A.; van Waarde, M.A.W.H.; Kakkar, V.; Dirks, R.P.; Govorukhina, N.; Oosterveld-Hut, H.M.J.; Lubsen, N.H.; Kampinga, H.H. A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol. Cell 2010, 37, 355–369. [Google Scholar] [CrossRef]
- Qiu, X.B.; Shao, Y.M.; Miao, S.; Wang, L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol. Life Sci. 2006, 63, 2560–2570. [Google Scholar] [CrossRef] [PubMed]
- Faust, O.; Rosenzweig, R. Structural and Biochemical Properties of Hsp40/Hsp70 Chaperone System. Adv. Exp. Med. Biol. 2020, 1243, 3–20. [Google Scholar] [PubMed]
- Lu, B.; Garrido, N.; Spelbrink, J.N.; Suzuki, C.K. Tid1 isoforms are mitochondrial DnaJ-like chaperones with unique carboxyl termini that determine cytosolic fate. J. Biol. Chem. 2006, 281, 13150–13158. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, T.; Rios, Z.; Mei, Q.; Lin, X.; Cao, S. Heat Shock Proteins and Cancer. Trends Pharmacol. Sci. 2017, 38, 226–256. [Google Scholar] [CrossRef] [PubMed]
- Javid, H.; Hashemian, P.; Yazdani, S.; Sharbaf Mashhad, A.; Karimi-Shahri, M. The role of heat shock proteins in metastatic colorectal cancer: A review. J. Cell Biochem. 2022, 123, 1704–1735. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Craig, E.A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Choi, H.-K.; Choi, Y.-S.; Park, S.-Y.; Sung, G.-J.; Lee, Y.-H.; Lee, J.; Jun, W.J.; Kim, K.; Choi, K.-C.; et al. DNAJB1 destabilizes PDCD5 to suppress p53-mediated apoptosis. Cancer Lett. 2015, 357, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Li, X.-N.; Li, X.-G.; Li, M.; Gao, P.-Z. DNAJC6 promotes hepatocellular carcinoma progression through induction of epithelial-mesenchymal transition. Biochem. Biophys. Res. Commun. 2014, 455, 298–304. [Google Scholar] [CrossRef]
- Menezes, M.E.; Mitra, A.; Shevde, L.A.; Samant, R.S. DNAJB6 governs a novel regulatory loop determining Wnt/β-catenin signalling activity. Biochem. J. 2012, 444, 573–580. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Hsueh, C.-M.; Yu, S.-L.; Su, C.-C.; Shum, W.-Y.; Yeh, K.-C.; Chang, G.-C.; Chen, J.J.W. HLJ1 is a novel caspase-3 substrate and its expression enhances UV-induced apoptosis in non-small cell lung carcinoma. Nucleic Acids Res. 2010, 38, 6148–6158. [Google Scholar] [CrossRef]
- Liu, T.; Jiang, W.; Han, D.; Yu, L. DNAJC25 is downregulated in hepatocellular carcinoma and is a novel tumor suppressor gene. Oncol. Lett. 2012, 4, 1274–1280. [Google Scholar] [CrossRef] [PubMed]
- Zanini, C.; Giribaldi, G.; Mandili, G.; Carta, F.; Crescenzio, N.; Bisaro, B.; Doria, A.; Foglia, L.; di Montezemolo, L.C.; Timeus, F.; et al. Inhibition of heat shock proteins (HSP) expression by quercetin and differential doxorubicin sensitization in neuroblastoma and Ewing’s sarcoma cell lines. J. Neurochem. 2007, 103, 1344–1354. [Google Scholar] [CrossRef]
- Yamashita, M.; Hirohashi, Y.; Torigoe, T.; Kusumoto, H.; Murai, A.; Imagawa, T.; Sato, N. Dnajb8, a Member of the Heat Shock Protein 40 Family Has a Role in the Tumor Initiation and Resistance to Docetaxel but Is Dispensable for Stress Response. PLoS ONE 2016, 11, e0146501. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, P.; Rashid, N. HSP60 as Modulators of Apoptosis. Heat Shock. Protein 60 Hum. Dis. Disord. 2019, 18, 41–55. [Google Scholar]
- Sadat, A.; Tiwari, S.; Sunidhi, S.; Chaphalkar, A.; Kochar, M.; Ali, M.; Zaidi, Z.; Sharma, A.; Verma, K.; Narayana Rao, K.B.; et al. Conserved and divergent chaperoning effects of Hsp60/10 chaperonins on protein folding landscapes. Proc. Natl. Acad. Sci. USA 2022, 119, e2118465119. [Google Scholar] [CrossRef] [PubMed]
- Ishida, R.; Okamoto, T.; Motojima, F.; Kubota, H.; Takahashi, H.; Tanabe, M.; Oka, T.; Kitamura, A.; Kinjo, M.; Yoshida, M.; et al. Physicochemical Properties of the Mammalian Molecular Chaperone HSP60. Int. J. Mol. Sci. 2018, 19, 489. [Google Scholar] [CrossRef]
- Okamoto, T.; Yamamoto, H.; Kudo, I.; Matsumoto, K.; Odaka, M.; Grave, E.; Itoh, H. HSP60 possesses a GTPase activity and mediates protein folding with HSP10. Sci. Rep. 2017, 7, 16931. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Ishida, R.; Yamamoto, H.; Tanabe-Ishida, M.; Haga, A.; Takahashi, H.; Takahashi, K.; Goto, D.; Grave, E.; Itoh, H. Functional structure and physiological functions of mammalian wild-type HSP60. Arch. Biochem. Biophys. 2015, 586, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Bie, A.S.; Cömert, C.; Körner, R.; Corydon, T.J.; Palmfeldt, J.; Hipp, M.S.; Hartl, F.U.; Bross, P. An inventory of interactors of the human HSP60/HSP10 chaperonin in the mitochondrial matrix space. Cell Stress Chaperones 2020, 25, 407–416. [Google Scholar] [CrossRef]
- Vilasi, S.; Bulone, D.; Bavisotto, C.C.; Campanella, C.; Gammazza, A.M.; San Biagio, P.L.; Cappello, F.; de Macario, E.C.; Macario, A.J.L. Chaperonin of Group I: Oligomeric spectrum and biochemical and biological implications. Front. Mol. Biosci. 2018, 4, 327889. [Google Scholar] [CrossRef]
- Cappello, F.; De Macario, E.C.; Marasà, L.; Zummo, G.; Macario, A.J.L. Hsp60 expression, new locations, functions, and perspectives for cancer diagnosis and therapy. Cancer Biol. Ther. 2008, 7, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Campanella, C.; Bucchieri, F.; Merendino, A.M.; Fucarino, A.; Burgio, G.; Corona, D.F.V.; Barbieri, G.; David, S.; Farina, F.; Zummo, G.; et al. The odyssey of Hsp60 from tumor cells to other destinations includes plasma membrane-associated stages and Golgi and exosomal protein-trafficking modalities. PLoS ONE 2012, 7, e42008. [Google Scholar] [CrossRef]
- Tutar, L.; Tutar, Y. Heat shock proteins; an overview. Curr. Pharm. Biotechnol. 2010, 11, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Cohen, I.R. The HSP60 immune system network. Trends Immunol. 2011, 32, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhou, Y.; Fan, S.; Wen, Q. The multiple roles and therapeutic potential of HSP60 in cancer. Biochem. Pharmacol. 2022, 201, 115096. [Google Scholar] [CrossRef] [PubMed]
- Chandra, D.; Choy, G.; Tang, D.G. Cytosolic accumulation of HSP60 during apoptosis with or without apparent mitochondrial release: Evidence that its pro-apoptotic or pro-survival functions involve differential interactions with caspase-3. J. Biol. Chem. 2007, 282, 31289–31301. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.N.; Choi, B.; Lee, K.W.; Lee, D.J.; Kang, D.H.; Lee, J.Y.; Song, I.S.; Kim, H.I.; Lee, S.-H.; Kim, H.S.; et al. Cytosolic Hsp60 is involved in the NF-kappaB-dependent survival of cancer cells via IKK regulation. PLoS ONE 2010, 5, e9422. [Google Scholar] [CrossRef]
- Shan, Y.-X.; Liu, T.-J.; Su, H.-F.; Samsamshariat, A.; Mestril, R.; Wang, P.H. Hsp10 and Hsp60 modulate Bcl-2 family and mitochondria apoptosis signaling induced by doxorubicin in cardiac muscle cells. J. Mol. Cell. Cardiol. 2003, 35, 1135–1143. [Google Scholar] [CrossRef]
- Dubińska-Magiera, M.; Jabłońska, J.; Saczko, J.; Kulbacka, J.; Jagla, T.; Daczewska, M. Contribution of small heat shock proteins to muscle development and function. FEBS Lett. 2014, 588, 517–530. [Google Scholar] [CrossRef]
- Kumar, S.; O’Malley, J.; Chaudhary, A.K.; Inigo, J.R.; Yadav, N.; Kumar, R.; Chandra, D. Hsp60 and IL-8 axis promotes apoptosis resistance in cancer. Br. J. Cancer 2019, 121, 934–943. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Yeh, C.-T. Functional Compartmentalization of HSP60-Survivin Interaction between Mitochondria and Cytosol in Cancer Cells. Cells 2020, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-H.; Lin, K.-H.; Yu, J.-S.; Wu, T.-J.; Lee, W.-C.; Chao, C.C.-K.; Pan, T.-L.; Yeh, C.-T. Targeting HSP60 by subcutaneous injections of jetPEI/HSP60-shRNA destabilizes cytoplasmic survivin and inhibits hepatocellular carcinoma growth. Mol. Carcinog. 2018, 57, 1087–1101. [Google Scholar] [CrossRef] [PubMed]
- Chaiwatanasirikul, K.-A.; Sala, A. The tumour-suppressive function of CLU is explained by its localisation and interaction with HSP60. Cell Death Dis. 2011, 2, e219. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.-P.; Yang, M.-H.; Huang, C.-H.; Chang, S.-Y.; Chen, P.-M.; Liu, C.-J.; Teng, S.-C.; Wu, K.-J. Interaction between HSP60 and beta-catenin promotes metastasis. Carcinogenesis 2009, 30, 1049–1057. [Google Scholar] [CrossRef]
- Tang, H.; Li, J.; Liu, X.; Wang, G.; Luo, M.; Deng, H. Down-regulation of HSP60 Suppresses the Proliferation of Glioblastoma Cells via the ROS/AMPK/mTOR Pathway. Sci. Rep. 2016, 6, 28388. [Google Scholar] [CrossRef] [PubMed]
- Abu-Hadid, M.; Wilkes, J.D.; Elakawi, Z.; Pendyala, L.; Perez, R.P. Relationship between heat shock protein 60 (HSP60) mRNA expression and resistance to platinum analogues in human ovarian and bladder carcinoma cell lines. Cancer Lett. 1997, 119, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Nakata, B.; Barton, R.; Robbins, K.; Howell, S.; Los, G. Association between hsp60 messenger-RNA levels and Cisplatin resistance in human head and neck-cancer cell-lines. Int. J. Oncol. 1994, 5, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.W.; Pouliot, L.M.; Hall, M.D.; Gottesman, M.M. Cisplatin resistance: A cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol. Rev. 2012, 64, 706–721. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.W.; Akiyama, S.; Schoenlein, P.; Pastan, I.; Gottesman, M.M. Characterisation of high-level cisplatin-resistant cell lines established from a human hepatoma cell line and human KB adenocarcinoma cells: Cross-resistance and protein changes. Br. J. Cancer 1995, 71, 676–683. [Google Scholar] [CrossRef]
- Zhang, K.; Jiang, K.; Hong, R.; Xu, F.; Xia, W.; Qin, G.; Lee, K.; Zheng, Q.; Lu, Q.; Zhai, Q.; et al. Identification and characterization of critical genes associated with tamoxifen resistance in breast cancer. PeerJ 2020, 8, e10468. [Google Scholar] [CrossRef]
- Harper, A.K.; Fletcher, N.M.; Fan, R.; Morris, R.T.; Saed, G.M. Heat Shock Protein 60 (HSP60) Serves as a Potential Target for the Sensitization of Chemoresistant Ovarian Cancer Cells. Reprod. Sci. 2020, 27, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.S.C.; Wai-Ki Wong, V.; Ming-Lok Chan, C.; Buig-Yue Ma, B.; Pun Hui, E.; Chi-Keung Wong, M.; Yan-Yee Lam, M.; Chi-Chuen, T.; Chan, W.H.; Cheuk, W.; et al. Identification of 5-fluorouracil response proteins in colorectal carcinoma cell line SW480 by two-dimensional electrophoresis and MALDI-TOF mass spectrometry. Oncol. Rep. 2008, 20, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Vostakolaei, M.A.; Hatami-Baroogh, L.; Babaei, G.; Molavi, O.; Kordi, S.; Abdolalizadeh, J. Hsp70 in cancer: A double agent in the battle between survival and death. J. Cell Physiol. 2021, 236, 3420–3444. [Google Scholar] [CrossRef] [PubMed]
- Albakova, Z.; Armeev, G.A.; Kanevskiy, L.M.; Kovalenko, E.I.; Sapozhnikov, A.M. HSP70 Multi-Functionality in Cancer. Cells 2020, 9, 587. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Fernández, M.R.; Valpuesta, J.M. Hsp70 chaperone: A master player in protein homeostasis. F1000Research 2018, 7, 1497. [Google Scholar] [CrossRef] [PubMed]
- Shevtsov, M.; Huile, G.; Multhoff, G. Membrane heat shock protein 70: A theranostic target for cancer therapy. Philos. Trans. R. Soc. Ser. B Biol. Sci. 2016, 373, 20160526. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stokes, J., 3rd; Singh, U.P.; Scissum Gunn, K.; Acharya, A.; Manne, U.; Mishra, M. Targeting Hsp70: A possible therapy for cancer. Cancer Lett. 2016, 374, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Gastpar, R.; Gehrmann, M.; Bausero, M.A.; Asea, A.; Gross, C.; Schroeder, J.A.; Multhoff, G. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res. 2005, 65, 5238–5247. [Google Scholar] [CrossRef] [PubMed]
- Rothammer, A.; Sage, E.K.; Werner, C.; Combs, S.E.; Multhoff, G. Increased heat shock protein 70 (Hsp70) serum levels and low NK cell counts after radiotherapy—Potential markers for predicting breast cancer recurrence? Radiat. Oncol. 2019, 14, 78. [Google Scholar] [CrossRef]
- Linder, M.; Pogge von Strandmann, E. The Role of Extracellular HSP70 in the Function of Tumor-Associated Immune Cells. Cancers 2021, 13, 4721. [Google Scholar] [CrossRef]
- Pocaly, M.; Lagarde, V.; Etienne, G.; Ribeil, J.-A.; Claverol, S.; Bonneu, M.; Moreau-Gaudry, F.; Guyonnet-Duperat, V.; Hermine, O.; Melo, J.V.; et al. Overexpression of the heat-shock protein 70 is associated to imatinib resistance in chronic myeloid leukemia. Leukemia 2007, 21, 93–101. [Google Scholar] [CrossRef]
- Pocaly, M.; Lagarde, V.; Etienne, G.; Dupouy, M.; Lapaillerie, D.; Claverol, S.; Vilain, S.; Bonneu, M.; Turcq, B.; Mahon, F.-X.; et al. Proteomic analysis of an imatinib-resistant K562 cell line highlights opposing roles of heat shock cognate 70 and heat shock 70 proteins in resistance. Proteomics 2008, 8, 2394–2406. [Google Scholar] [CrossRef]
- Frezzato, F.; Visentin, A.; Severin, F.; Pizzo, S.; Ruggeri, E.; Mouawad, N.; Martinello, L.; Pagnin, E.; Trimarco, V.; Tonini, A.; et al. Targeting of HSP70/HSF1 Axis Abrogates In Vitro Ibrutinib-Resistance in Chronic Lymphocytic Leukemia. Cancers 2021, 13, 5453. [Google Scholar] [CrossRef]
- Wang, L.; Jia, Z.; Xie, D.; Zhao, T.; Tan, Z.; Zhang, S.; Kong, F.; Wei, D.; Xie, K. Methylation of HSP70 Orchestrates Its Binding to and Stabilization of BCL2 mRNA and Renders Pancreatic Cancer Cells Resistant to Therapeutics. Cancer Res. 2020, 80, 4500–4513. [Google Scholar] [CrossRef] [PubMed]
- García-Aranda, M.; Pérez-Ruiz, E.; Redondo, M. Bcl-2 Inhibition to Overcome Resistance to Chemo- and Immunotherapy. Int. J. Mol. Sci. 2018, 19, 3950. [Google Scholar] [CrossRef]
- Sliutz, G.; Karlseder, J.; Tempfer, C.; Orel, L.; Holzer, G.; Simon, M.M. Drug resistance against gemcitabine and topotecan mediated by constitutive hsp70 overexpression in vitro: Implication of quercetin as sensitiser in chemotherapy. Br. J. Cancer 1996, 74, 172–177. [Google Scholar] [CrossRef]
- Grivicich, I.; Regner, A.; Zanoni, C.; Correa, L.P.; Jotz, G.P.; Henriques, J.A.P.; Schwartsmann, G.; da Rocha, A.B. Hsp70 response to 5-fluorouracil treatment in human colon cancer cell lines. Int. J. Colorectal Dis. 2007, 22, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Guo, Z.; Chen, X.; Liu, K.; Li, H.; Jia, W.; Wang, C.; Luo, F.; Ji, X.; Zhang, T.; et al. Excessive HSP70/TLR2 activation leads to remodeling of the tumor immune microenvironment to resist chemotherapy sensitivity of mFOLFOX in colorectal cancer. Clin. Immunol. 2022, 245, 109157. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Lu, Y.; Kaufmann, S.H.; Gustafson, W.C.; Karp, J.E.; Boldogh, I.; Fields, A.P.; Brasier, A.R. Genomic mechanisms of p210BCR-ABL signaling: Induction of heat shock protein 70 through the GATA response element confers resistance to paclitaxel-induced apoptosis. J. Biol. Chem. 2004, 279, 35604–35615. [Google Scholar] [CrossRef]
- Liu, T.; Singh, R.; Rios, Z.; Bhushan, A.; Li, M.; Sheridan, P.P.; Bearden, S.E.; Lai, J.C.K.; Agbenowu, S.; Cao, S.; et al. Tyrosine phosphorylation of HSC70 and its interaction with RFC mediates methotrexate resistance in murine L1210 leukemia cells. Cancer Lett. 2015, 357, 231–241. [Google Scholar] [CrossRef]
- Vargas-Roig, L.M.; Gago, F.E.; Tello, O.; Aznar, J.C.; Ciocca, D.R. Heat shock protein expression and drug resistance in breast cancer patients treated with induction chemotherapy. Int. J. Cancer 1998, 79, 468–475. [Google Scholar] [CrossRef]
- Yang, X.; Wang, J.; Zhou, Y.; Wang, Y.; Wang, S.; Zhang, W. Hsp70 promotes chemoresistance by blocking Bax mitochondrial translocation in ovarian cancer cells. Cancer Lett. 2012, 321, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Sheng, L.; Tang, T.; Liu, Y.; Ma, Y.; Wang, Z.; Tao, H.; Zhang, Y.; Qi, Z. Inducible HSP70 antagonizes cisplatin-induced cell apoptosis through inhibition of the MAPK signaling pathway in HGC-27 cells. Int. J. Mol. Med. 2018, 42, 2089–2097. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Yuan, Q.; Li, H.; Wang, W.; Xie, G.; Zhu, K.; Li, D. miR-223/Hsp70/JNK/JUN/miR-223 feedback loop modulates the chemoresistance of osteosarcoma to cisplatin. Biochem. Biophys. Res. Commun. 2018, 497, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, E.; Maingret, L.; Puig, P.E.; Rerole, A.L.; Ghiringhelli, F.; Hammann, A.; Solary, E.; Kroemer, G.; Garrido, C. Heat shock protein 70 neutralization exerts potent antitumor effects in animal models of colon cancer and melanoma. Cancer Res. 2006, 66, 4191–4197. [Google Scholar] [CrossRef] [PubMed]
- Cordonnier, M.; Chanteloup, G.; Isambert, N.; Seigneuric, R.; Fumoleau, P.; Garrido, C.; Gobbo, J. Exosomes in cancer theranostic: Diamonds in the rough. Cell Adh. Migr. 2017, 11, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Xu, Z.; Zhu, S.; Sun, W.; Wang, X.; Tan, C.; Zhang, Y.; Zhang, G.; Xu, Y.; Tang, J. Small extracellular vesicle-mediated Hsp70 intercellular delivery enhances breast cancer adriamycin resistance. Free Radic. Biol. Med. 2021, 164, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Alberti, G.; Vergilio, G.; Paladino, L.; Barone, R.; Cappello, F.; Conway de Macario, E.; Macario, A.J.L.; Bucchieri, F.; Rappa, F. The Chaperone System in Breast Cancer: Roles and Therapeutic Prospects of the Molecular Chaperones Hsp27, Hsp60, Hsp70, and Hsp90. Int. J. Mol. Sci. 2022, 23, 7792. [Google Scholar] [CrossRef]
- Barrott, J.J.; Haystead, T.A.J. Hsp90, an unlikely ally in the war on cancer. FEBS J. 2013, 280, 1381–1396. [Google Scholar] [CrossRef]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef]
- Poggio, P.; Sorge, M.; Seclì, L.; Brancaccio, M. Extracellular HSP90 Machineries Build Tumor Microenvironment and Boost Cancer Progression. Front. Cell Dev. Biol. 2021, 9, 735529. [Google Scholar] [CrossRef] [PubMed]
- Chern, Y.-J.; Tai, I.T. Adaptive response of resistant cancer cells to chemotherapy. Cancer Biol. Med. 2020, 17, 842–863. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.S.; Jay, D.G. Emerging Roles of Extracellular Hsp90 in Cancer. Adv. Cancer Res. 2016, 129, 141–163. [Google Scholar] [PubMed]
- Sidera, K.; Patsavoudi, E. HSP90 Inhibitors: Current Development and Potential in Cancer Therapy. Recent Pat. Anticancer Drug Discov. 2013, 9, 1–20. [Google Scholar] [CrossRef]
- Barzegar, S.; Pirouzpanah, S. Zinc finger proteins and ATP-binding cassette transporter-dependent multidrug resistance. Eur. J. Clin. Investig. 2024, 54, e14120. [Google Scholar] [CrossRef] [PubMed]
- Jafari, A.; Rezaei-Tavirani, M.; Farhadihosseinabadi, B.; Taranejoo, S.; Zali, H. HSP90 and Co-chaperones: Impact on Tumor Progression and Prospects for Molecular-Targeted Cancer Therapy. Cancer Investig. 2020, 38, 310–328. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Abrams, S.L.; Steelman, L.S.; Shelton, J.G.; Wong, E.W.T.; Chappell, W.H.; Bäsecke, J.; Stivala, F.; Donia, M.; Nicoletti, F.; Libra, M.; et al. The Raf/MEK/ERK pathway can govern drug resistance, apoptosis and sensitivity to targeted therapy. Cell Cycle 2010, 9, 1781–1791. [Google Scholar] [CrossRef]
- Yin, L.; Yang, Y.; Zhu, W.; Xian, Y.; Han, Z.; Huang, H.; Peng, L.; Zhang, K.; Zhao, Y. Heat Shock Protein 90 Triggers Multi-Drug Resistance of Ovarian Cancer via AKT/GSK3β/β-Catenin Signaling. Front. Oncol. 2021, 11, 620907. [Google Scholar] [CrossRef]
- Kumar, P.; Devaki, B.; Jonnala, U.K.; Amere Subbarao, S. Hsp90 facilitates acquired drug resistance of tumor cells through cholesterol modulation however independent of tumor progression. Biochim. Biophys. Acta. Mol. Cell Res. 2020, 1867, 118728. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, G.P.; Zakka, K.M.; Landry, J.C.; Shaib, W.L.; Lesinski, G.B.; El-Rayes, B.F. Inhibition of HSP90 overcomes resistance to chemotherapy and radiotherapy in pancreatic cancer. Int. J. Cancer 2019, 145, 1529–1537. [Google Scholar] [CrossRef]
- Tabata, M.; Tsubaki, M.; Takeda, T.; Tateishi, K.; Maekawa, S.; Tsurushima, K.; Imano, M.; Satou, T.; Ishizaka, T.; Nishida, S. Inhibition of HSP90 overcomes melphalan resistance through downregulation of Src in multiple myeloma cells. Clin. Exp. Med. 2020, 20, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Ogata, K.; Altan, B.; Yokobori, T.; Ide, M.; Mochiki, E.; Toyomasu, Y.; Kogure, N.; Yanoma, T.; Suzuki, M.; et al. Nuclear heat shock protein 110 expression is associated with poor prognosis and chemotherapy resistance in gastric cancer. Oncotarget 2016, 7, 18415–18423. [Google Scholar] [CrossRef] [PubMed]
- Teshima, H.; Watanabe, H.; Yasutake, R.; Ikeda, Y.; Yonezu, Y.; Okamoto, N.; Kakihana, A.; Yuki, R.; Nakayama, Y.; Saito, Y. Functional differences between Hsp105/110 family proteins in cell proliferation, cell division, and drug sensitivity. J. Cell Biochem. 2021, 122, 1958–1967. [Google Scholar] [CrossRef] [PubMed]
- Andréasson, C.; Fiaux, J.; Rampelt, H.; Mayer, M.P.; Bukau, B. Hsp110 is a nucleotide-activated exchange factor for Hsp70. J. Biol. Chem. 2008, 283, 8877–8884. [Google Scholar] [CrossRef]
- Morano, K.A. New tricks for an old dog: The evolving world of Hsp70. Ann. N. Y. Acad. Sci. 2007, 1113, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Finka, A.; Sharma, S.K.; Goloubinoff, P. Multi-layered molecular mechanisms of polypeptide holding, unfolding and disaggregation by HSP70/HSP110 chaperones. Front. Mol. Biosci. 2015, 2, 29. [Google Scholar] [CrossRef] [PubMed]
- Velasco, L.; Dublang, L.; Moro, F.; Muga, A. The Complex Phosphorylation Patterns that Regulate the Activity of Hsp70 and Its Cochaperones. Int. J. Mol. Sci. 2019, 20, 4122. [Google Scholar] [CrossRef]
- Jego, G.; Hermetet, F.; Girodon, F.; Garrido, C. Chaperoning STAT3/5 by Heat Shock Proteins: Interest of Their Targeting in Cancer Therapy. Cancers 2020, 12, 21. [Google Scholar] [CrossRef]
- Binder, R.J. Functions of heat shock proteins in pathways of the innate and adaptive immune system. J. Immunol. 2014, 193, 5765–5771. [Google Scholar] [CrossRef] [PubMed]
- Chakafana, G.; Shonhai, A. The Role of Non-Canonical Hsp70s (Hsp110/Grp170) in Cancer. Cells 2021, 10, 254. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, Y.; Guo, K.; Wang, N.; Jin, H.; Liu, Y.; Qin, W. Heat shock proteins in hepatocellular carcinoma: Molecular mechanism and therapeutic potential. Int. J. Cancer 2016, 138, 1824–1834. [Google Scholar] [CrossRef] [PubMed]
- Gozzi, G.J.; Gonzalez, D.; Boudesco, C.; Dias, A.M.M.; Gotthard, G.; Uyanik, B.; Dondaine, L.; Marcion, G.; Hermetet, F.; Denis, C.; et al. Selecting the first chemical molecule inhibitor of HSP110 for colorectal cancer therapy. Cell Death Differ. 2019, 27, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Berthenet, K.; Bokhari, A.; Lagrange, A.; Marcion, G.; Boudesco, C.; Causse, S.; De Thonel, A.; Svrcek, M.; Goloudina, A.R.; Dumont, S.; et al. HSP110 promotes colorectal cancer growth through STAT3 activation. Oncogene 2016, 36, 2328–2336. [Google Scholar] [CrossRef] [PubMed]
- Causse, S.Z.; Marcion, G.; Chanteloup, G.; Uyanik, B.; Boudesco, C.; Grigorash, B.B.; Douhard, R.; Dias, A.M.M.; Dumetier, B.; Dondaine, L.; et al. HSP110 translocates to the nucleus upon genotoxic chemotherapy and promotes DNA repair in colorectal cancer cells. Oncogene 2018, 38, 2767–2777. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, M.; Abbaszadeh, S.; Rashidi, M.; Amini, N.; Talebi Anaraki, K.; Motahhary, M.; Khalilipouya, E.; Harif Nashtifani, A.; Shafiei, S.; Ramezani Farani, M.; et al. STAT3 as a newly emerging target in colorectal cancer therapy: Tumorigenesis, therapy response, and pharmacological/nanoplatform strategies. Environ. Res. 2023, 233, 116458. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ming, T.; Tang, S.; Ren, S.; Yang, H.; Liu, M.; Tao, Q.; Xu, H. Wnt signaling in colorectal cancer: Pathogenic role and therapeutic target. Mol. Cancer 2022, 21, 144. [Google Scholar] [CrossRef]
- Groenewald, W.; Lund, A.H.; Gay, D.M. The Role of WNT Pathway Mutations in Cancer Development and an Overview of Therapeutic Options. Cells 2023, 12, 990. [Google Scholar] [CrossRef]
- Hrudka, J.; Jelínková, K.; Fišerová, H.; Matěj, R.; Mandys, V.; Waldauf, P. Heat Shock Proteins 27, 70, and 110: Expression and Prognostic Significance in Colorectal Cancer. Cancers 2021, 13, 4407. [Google Scholar] [CrossRef]
- Chen, S.; Guttridge, D.C.; You, Z.; Zhang, Z.; Fribley, A.; Mayo, M.W.; Kitajewski, J.; Wang, C.Y. Wnt-1 signaling inhibits apoptosis by activating beta-catenin/T cell factor-mediated transcription. J. Cell Biol. 2001, 152, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Liu, H.; Liu, L.; Wang, Y.D.; Chen, W.D. Emerging Roles of Wnt Ligands in Human Colorectal Cancer. Front. Oncol. 2020, 10, 537296. [Google Scholar] [CrossRef] [PubMed]
- Yamane, T.; Saito, Y.; Teshima, H.; Hagino, M.; Kakihana, A.; Sato, S.; Shimada, M.; Kato, Y.; Kuga, T.; Yamagishi, N.; et al. Hsp105α suppresses Adriamycin-induced cell death via nuclear localization signal-dependent nuclear accumulation. J. Cell Biochem. 2019, 120, 17951–17962. [Google Scholar] [CrossRef] [PubMed]
- Das, J.K.; Xiong, X.; Ren, X.; Yang, J.-M.; Song, J. Heat Shock Proteins in Cancer Immunotherapy. J. Oncol. 2019, 2019, 3267207. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, J.C.; Donakonda, S.; Haupt, V.J.; Lennig, P.; Zhang, Y.; Schroeder, M. New HSP27 inhibitors efficiently suppress drug resistance development in cancer cells. Oncotarget 2016, 7, 68156–68169. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jung, Y.J.; Choi, B.; Lee, N.L.; Lee, H.J.; Kwak, S.Y.; Kwon, Y.; Na, Y.; Lee, Y.-S.; Kim, J.H.; et al. Overcoming HSP27-mediated resistance by altered dimerization of HSP27 using small molecules. Oncotarget 2016, 7, 53178–53190. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, J.-C.; Tuukkanen, A.; Schroeder, M.; Fahrig, T.; Fahrig, R. RP101 (brivudine) binds to heat shock protein HSP27 (HSPB1) and enhances survival in animals and pancreatic cancer patients. J. Cancer Res. Clin. Oncol. 2011, 137, 1349–1361. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-F.; Nieh, S.; Jao, S.-W.; Liu, C.-L.; Wu, C.-H.; Chang, Y.-C.; Yang, C.-Y.; Lin, Y.-S. Quercetin suppresses drug-resistant spheres via the p38 MAPK-Hsp27 apoptotic pathway in oral cancer cells. PLoS ONE 2012, 7, e49275. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.K.; Oesterreich, S.; Lemieux, P.; Sarge, K.D.; Fuqua, S.A. Quercetin inhibits heat shock protein induction but not heat shock factor DNA-binding in human breast carcinoma cells. Biochem. Biophys. Res. Commun. 1997, 239, 851–856. [Google Scholar] [CrossRef]
- Borgo, C.; Vilardell, J.; Bosello-Travain, V.; Pinna, L.A.; Venerando, A.; Salvi, M. Dependence of HSP27 cellular level on protein kinase CK2 discloses novel therapeutic strategies. Biochim. Biophys. Acta. Gen. Subj. 2018, 1862, 2902–2910. [Google Scholar] [CrossRef]
- Reyes-Farias, M.; Carrasco-Pozo, C. The Anti-Cancer Effect of Quercetin: Molecular Implications in Cancer Metabolism. Int. J. Mol. Sci. 2019, 20, 3177. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-K.; Kam, H.; Kim, K.-Y.; Park, S.I.; Lee, Y.-S. Targeting Heat Shock Protein 27 in Cancer: A Druggable Target for Cancer Treatment? Cancers 2019, 11, 1195. [Google Scholar] [CrossRef]
- Chi, K.N.; Yu, E.Y.; Jacobs, C.; Bazov, J.; Kollmannsberger, C.; Higano, C.S.; Mukherjee, S.D.; Gleave, M.E.; Stewart, P.S.; Hotte, S.J. A phase I dose-escalation study of apatorsen (OGX-427), an antisense inhibitor targeting heat shock protein 27 (Hsp27), in patients with castration-resistant prostate cancer and other advanced cancers. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 1116–1122. [Google Scholar] [CrossRef]
- Chi, K.N.; Hotte, S.J.; Ellard, S.; Gingerich, J.R.; Joshua, A.M.; Kollmannsberger, C.K.; Yu, E.Y.; Gleave, M.E. A randomized phase II study of OGX-427 plus prednisone versus prednisone alone in patients with chemotherapy-naive metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2012, 30, 121. [Google Scholar] [CrossRef]
- Spigel, D.R.; Shipley, D.L.; Waterhouse, D.M.; Jones, S.F.; Ward, P.J.; Shih, K.C.; Hemphill, B.; McCleod, M.; Whorf, R.C.; Page, R.D.; et al. A Randomized, Double-Blinded, Phase II Trial of Carboplatin and Pemetrexed with or without Apatorsen (OGX-427) in Patients with Previously Untreated Stage IV Non-Squamous-Non-Small-Cell Lung Cancer: The SPRUCE Trial. Oncologist 2019, 24, e1409–e1416. [Google Scholar] [CrossRef]
- Gibert, B.; Hadchity, E.; Czekalla, A.; Aloy, M.-T.; Colas, P.; Rodriguez-Lafrasse, C.; Arrigo, A.-P.; Diaz-Latoud, C. Inhibition of heat shock protein 27 (HspB1) tumorigenic functions by peptide aptamers. Oncogene 2011, 30, 3672–3681. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Li, Y.; Tan, X.; Fu, L. Small Heat Shock Proteins in Cancers: Functions and Therapeutic Potential for Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 6611. [Google Scholar] [CrossRef]
- Fahrig, R.; Quietzsch, D.; Heinrich, J.-C.; Heinemann, V.; Boeck, S.; Schmid, R.M.; Praha, C.; Liebert, A.; Sonntag, D.; Krupitza, G.; et al. RP101 improves the efficacy of chemotherapy in pancreas carcinoma cell lines and pancreatic cancer patients. Anticancer Drugs 2006, 17, 1045–1056. [Google Scholar] [CrossRef]
- Kamgar-Dayhoff, P.; Brelidze, T.I.; Kamgar-Dayhoff, P.; Brelidze, T.I. Multifaceted effect of chlorpromazine in cancer: Implications for cancer treatment. Oncotarget 2021, 12, 1406–1426. [Google Scholar] [CrossRef]
- Lelj-Garolla, B.; Kumano, M.; Beraldi, E.; Nappi, L.; Rocchi, P.; Ionescu, D.N.; Fazli, L.; Zoubeidi, A.; Gleave, M.E. Hsp27 inhibition with OGX-427 sensitizes Non-small cell lung cancer cells to erlotinib and chemotherapy. Mol. Cancer Ther. 2015, 14, 1107–1116. [Google Scholar] [CrossRef]
- Díaz-Chávez, J.; Fonseca-Sánchez, M.A.; Arechaga-Ocampo, E.; Flores-Pérez, A.; Palacios-Rodríguez, Y.; Domínguez-Gómez, G.; Marchat, L.A.; Fuentes-Mera, L.; Mendoza-Hernández, G.; Gariglio, P.; et al. Proteomic profiling reveals that resveratrol inhibits HSP27 expression and sensitizes breast cancer cells to doxorubicin therapy. PLoS ONE 2013, 8, e64378. [Google Scholar] [CrossRef]
- Umar, H.I.; Ajayi, A.T.; Mukerjee, N.; Aborode, A.T.; Hasan, M.M.; Maitra, S.; Bello, R.O.; Alabere, H.O.; Sanusi, A.A.; Awolaja, O.O.; et al. Discovery of Novel HSP27 Inhibitors as Prospective Anti-Cancer Agents Utilizing Computer-Assisted Therapeutic Discovery Approaches. Cells 2022, 11, 2412. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.; Abdeen, S.; Salim, N.; Ray, A.-M.; Washburn, A.; Chitre, S.; Sivinski, J.; Park, Y.; Hoang, Q.Q.; Chapman, E.; et al. HSP60/10 chaperonin systems are inhibited by a variety of approved drugs, natural products, and known bioactive molecules. Bioorg. Med. Chem. Lett. 2019, 29, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Cappello, F.; Marino Gammazza, A.; Palumbo Piccionello, A.; Campanella, C.; Pace, A.; Conway de Macario, E.; Macario, A.J.L. Hsp60 chaperonopathies and chaperonotherapy: Targets and agents. Expert Opin. Ther. Targets 2014, 18, 185–208. [Google Scholar] [CrossRef] [PubMed]
- Palumbo Piccionello, A.; Marzullo, P.; Buscemi, S.; Pace, A. Hsp60 Inhibitors and Modulators. Heat Shock. Protein 60 Hum. Dis. Disord. 2019, 18, 27–39. [Google Scholar]
- Tanabe, M.; Ishida, R.; Izuhara, F.; Komatsuda, A.; Wakui, H.; Sawada, K.; Otaka, M.; Nakamura, N.; Itoh, H.; Tanabe, M.; et al. The ATPase activity of molecular chaperone HSP60 is inhibited by immunosuppressant mizoribine. Am. J. Mol. Biol. 2012, 2, 93–102. [Google Scholar] [CrossRef]
- Wiechmann, K.; Müller, H.; König, S.; Wielsch, N.; Svatoš, A.; Jauch, J.; Werz, O. Mitochondrial Chaperonin HSP60 Is the Apoptosis-Related Target for Myrtucommulone. Cell Chem. Biol. 2017, 24, 614–623.e6. [Google Scholar] [CrossRef]
- Meng, Q.; Li, B.X.; Xiao, X. Toward Developing Chemical Modulators of Hsp60 as Potential Therapeutics. Front. Mol. Biosci. 2018, 5, 35. [Google Scholar] [CrossRef]
- Spinello, A.; Barone, G.; Cappello, F.; Pace, A.; Buscemi, S.; Palumbo Piccionello, A. The Binding Mechanism of Epolactaene to Hsp60 Unveiled by in Silico Modelling. ChemistrySelect 2016, 1, 759–765. [Google Scholar] [CrossRef]
- Caruso Bavisotto, C.; Marino Gammazza, A.; Lo Cascio, F.; Mocciaro, E.; Vitale, A.M.; Vergilio, G.; Pace, A.; Cappello, F.; Campanella, C.; Palumbo Piccionello, A. Curcumin Affects HSP60 Folding Activity and Levels in Neuroblastoma Cells. Int. J. Mol. Sci. 2020, 21, 661. [Google Scholar] [CrossRef]
- Marino Gammazza, A.; Campanella, C.; Barone, R.; Caruso Bavisotto, C.; Gorska, M.; Wozniak, M.; Carini, F.; Cappello, F.; D’Anneo, A.; Lauricella, M.; et al. Doxorubicin anti-tumor mechanisms include Hsp60 post-translational modifications leading to the Hsp60/p53 complex dissociation and instauration of replicative senescence. Cancer Lett. 2017, 385, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Gorska, M.; Marino Gammazza, A.; Zmijewski, M.A.; Campanella, C.; Cappello, F.; Wasiewicz, T.; Kuban-Jankowska, A.; Daca, A.; Sielicka, A.; Popowska, U.; et al. Geldanamycin-induced osteosarcoma cell death is associated with hyperacetylation and loss of mitochondrial pool of heat shock protein 60 (hsp60). PLoS ONE 2013, 8, e71135. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Li, G.; Yu, Q.; Liu, D.; Tang, X. HSP60 in cancer: A promising biomarker for diagnosis and a potentially useful target for treatment. J. Drug Target. 2022, 30, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, A.J.; Chapman, E. Function, Therapeutic Potential, and Inhibition of Hsp70 Chaperones. J. Med. Chem. 2021, 64, 7060–7082. [Google Scholar] [CrossRef] [PubMed]
- Goloudina, A.R.; Demidov, O.N.; Garrido, C. Inhibition of HSP70: A challenging anti-cancer strategy. Cancer Lett. 2012, 325, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.J.; Williamson, D.S.; Browne, H.; Murray, J.B.; Dokurno, P.; Shaw, T.; Macias, A.T.; Daniels, Z.; Geoffroy, S.; Dopson, M.; et al. A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother. Pharmacol. 2010, 66, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Jinwal, U.K.; Miyata, Y.; Koren, J., 3rd; Jones, J.R.; Trotter, J.H.; Chang, L.; O’Leary, J.; Morgan, D.; Lee, D.C.; Shults, C.L.; et al. Chemical manipulation of hsp70 ATPase activity regulates tau stability. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 12079–12088. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Zhou, G.; Liu, Y.; Zhang, J.; Chen, Y.; Liu, L.; Zhang, G. HSP70 Family in Cancer: Signaling Mechanisms and Therapeutic Advances. Biomolecules 2023, 13, 601. [Google Scholar] [CrossRef]
- Sha, G.; Jiang, Z.; Zhang, W.; Jiang, C.; Wang, D.; Tang, D. The multifunction of HSP70 in cancer: Guardian or traitor to the survival of tumor cells and the next potential therapeutic target. Int. Immunopharmacol. 2023, 122, 110492. [Google Scholar] [CrossRef]
- McKeon, A.M.; Egan, A.; Chandanshive, J.; McMahon, H.; Griffith, D.M. Novel Improved Synthesis of HSP70 Inhibitor, Pifithrin-μ. In Vitro Synergy Quantification of Pifithrin-μ Combined with Pt Drugs in Prostate and Colorectal Cancer Cells. Molecules 2016, 21, 949. [Google Scholar] [CrossRef]
- Zhou, Y.; Ma, J.; Zhang, J.; He, L.; Gong, J.; Long, C. Pifithrin-μ is efficacious against non-small cell lung cancer via inhibition of heat shock protein 70. Oncol. Rep. 2017, 37, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.; Kühnl, A.; Reins, J.; Fischer, S.; Ortiz-Tanchez, J.; Schlee, C.; Mochmann, L.H.; Heesch, S.; Benlasfer, O.; Hofmann, W.K.; et al. Antileukemic activity of the HSP70 inhibitor pifithrin-μ in acute leukemia. Blood Cancer J. 2011, 1, e28. [Google Scholar] [CrossRef] [PubMed]
- Nitzsche, B.; Höpfner, M.; Biersack, B. Synthetic Small Molecule Modulators of Hsp70 and Hsp40 Chaperones as Promising Anticancer Agents. Int. J. Mol. Sci. 2023, 24, 4083. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Ren, X.; Liang, Y.; Yan, Y.; Zhou, Y.; Hu, J.; Wang, Z.; Song, F.; Wang, F.; Liao, W.; et al. KNK437 restricts the growth and metastasis of colorectal cancer via targeting DNAJA1/CDC45 axis. Oncogene 2020, 39, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Tong, X.; Sun, L.; Li, H.; Jones, R.D.; Liao, J.; Yang, G.Y. Inhibition of mutant Kras and p53-driven pancreatic carcinogenesis by atorvastatin: Mainly via targeting of the farnesylated DNAJA1 in chaperoning mutant p53. Mol. Carcinog. 2019, 58, 2052–2064. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Choi, H.-K.; Seo, J.S.; Yoo, J.-Y.; Jeong, J.-W.; Choi, Y.; Choi, K.-C.; Yoon, H.-G. DNAJB1 negatively regulates MIG6 to promote epidermal growth factor receptor signaling. Biochim. Biophys. Acta 2015, 1853, 2722–2730. [Google Scholar] [CrossRef] [PubMed]
- Tatokoro, M.; Koga, F.; Yoshida, S.; Kihara, K. Heat shock protein 90 targeting therapy: State of the art and future perspective. EXCLI J. 2015, 14, 48–58. [Google Scholar] [PubMed]
- Lu, X.; Xiao, L.; Wang, L.; Ruden, D.M. Hsp90 inhibitors and drug resistance in cancer: The potential benefits of combination therapies of Hsp90 inhibitors and other anti-cancer drugs. Biochem. Pharmacol. 2012, 83, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.H. Organelle-specific Hsp90 inhibitors. Arch. Pharm. Res. 2015, 38, 1582–1590. [Google Scholar] [CrossRef]
- Dutta Gupta, S.; Bommaka, M.K.; Banerjee, A. Inhibiting protein-protein interactions of Hsp90 as a novel approach for targeting cancer. Eur. J. Med. Chem. 2019, 178, 48–63. [Google Scholar] [CrossRef]
- Youssef, M.E.; Cavalu, S.; Hasan, A.M.; Yahya, G.; Abd-Eldayem, M.A.; Saber, S. Role of Ganetespib, an HSP90 Inhibitor, in Cancer Therapy: From Molecular Mechanisms to Clinical Practice. Int. J. Mol. Sci. 2023, 24, 5014. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.E.; Wang, S.L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 2019, 10, 131. [Google Scholar] [CrossRef] [PubMed]
- Hyun, S.Y.; Le, H.T.; Nguyen, C.-T.; Yong, Y.-S.; Boo, H.-J.; Lee, H.J.; Lee, J.-S.; Min, H.-Y.; Ann, J.; Chen, J.; et al. Development of a novel Hsp90 inhibitor NCT-50 as a potential anticancer agent for the treatment of non-small cell lung cancer. Sci. Rep. 2018, 8, 13924. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.-Z.; Wang, X.-P. Immunogenic Cell Death-Based Cancer Vaccines. Front. Immunol. 2021, 12, 697964. [Google Scholar] [CrossRef] [PubMed]
- Dublang, L.; Underhaug, J.; Flydal, M.I.; Velasco-Carneros, L.; Maréchal, J.-D.; Moro, F.; Boyano, M.D.; Martinez, A.; Muga, A. Inhibition of the Human Hsc70 System by Small Ligands as a Potential Anticancer Approach. Cancers 2021, 13, 2936. [Google Scholar] [CrossRef] [PubMed]
- Wach, M.M.; Subjeck, J.R.; Wang, X.-Y.; Repasky, E.; Matsuzaki, J.; Yu, H.; Wang, C.; Fisher, D.; Skitzki, J.J.; Kane, J.M. 3rd Recombinant human Hsp110-gp100 chaperone complex vaccine is nontoxic and induces response in advanced stage melanoma patients. Melanoma Res. 2022, 32, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Guzhova, I.V.; Margulis, B.A. HSP70-based anti-cancer immunotherapy. Hum. Vaccin. Immunother. 2016, 12, 2529–2535. [Google Scholar] [CrossRef] [PubMed]
- Taha, E.A.; Ono, K.; Eguchi, T. Roles of Extracellular HSPs as Biomarkers in Immune Surveillance and Immune Evasion. Int. J. Mol. Sci. 2019, 20, 4588. [Google Scholar] [CrossRef] [PubMed]
- Murshid, A.; Borges, T.J.; Bonorino, C.; Lang, B.J.; Calderwood, S.K. Immunological Outcomes Mediated Upon Binding of Heat Shock Proteins to Scavenger Receptors SCARF1 and LOX-1, and Endocytosis by Mononuclear Phagocytes. Front. Immunol. 2020, 10, 510781. [Google Scholar] [CrossRef]
- Zininga, T.; Ramatsui, L.; Shonhai, A. Heat Shock Proteins as Immunomodulants. Molecules 2018, 23, 2846. [Google Scholar] [CrossRef]
- Wang, X.-Y.; Subjeck, J.R. High molecular weight stress proteins: Identification, cloning and utilisation in cancer immunotherapy. Int. J. Hyperth. 2013, 29, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Shevtsov, M.; Multhoff, G. Heat shock protein-Peptide and HSP-based immunotherapies for the treatment of cancer. Front. Immunol. 2016, 7, 180087. [Google Scholar] [CrossRef]
- Li, D.-Y.; Liang, S.; Wen, J.-H.; Tang, J.-X.; Deng, S.-L.; Liu, Y.-X. Extracellular HSPs: The Potential Target for Human Disease Therapy. Molecules 2022, 27, 2361. [Google Scholar] [CrossRef] [PubMed]
- Panayi, G.S.; Corrigall, V.C. Chaperonins and the regulation of immunity. Arthritis Res. Ther. 2005, 7, S2. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Gong, J.; Murshid, A. Extracellular HSPs: The Complicated Roles of Extracellular HSPs in Immunity. Front. Immunol. 2016, 7, 159. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, S.-Y.; Yuan, M.; Tang, Y.; Guo, Q.-Y.; Cui, X.-M.; Sui, X.; Peng, J. Prophylactic Antitumor Effect of Mixed Heat Shock Proteins/Peptides in Mouse Sarcoma. Chin. Med. J. 2015, 128, 2234–2241. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.; Koelch, W.; DeMaio, A.; Arispe, N.; Multhoff, G. Cell surface-bound heat shock protein 70 (Hsp70) mediates perforin-independent apoptosis by specific binding and uptake of granzyme B. J. Biol. Chem. 2003, 278, 41173–41181. [Google Scholar] [CrossRef]
- Manjili, M.H.; Henderson, R.; Wang, X.-Y.; Chen, X.; Li, Y.; Repasky, E.; Kazim, L.; Subjeck, J.R. Development of a recombinant HSP110-HER-2/neu vaccine using the chaperoning properties of HSP110. Cancer Res. 2002, 62, 1737–1742. [Google Scholar] [PubMed]
- Guo, C.; Subjeck, J.R.; Wang, X.-Y. Creation of Recombinant Chaperone Vaccine Using Large Heat Shock Protein for Antigen-Targeted Cancer Immunotherapy. Methods Mol. Biol. 2018, 1709, 345–357. [Google Scholar]
- Manjili, M.H.; Wang, X.-Y.; Chen, X.; Martin, T.; Repasky, E.A.; Henderson, R.; Subjeck, J.R. HSP110-HER2/neu chaperone complex vaccine induces protective immunity against spontaneous mammary tumors in HER-2/neu transgenic mice. J. Immunol. 2003, 171, 4054–4061. [Google Scholar] [CrossRef]
- Yu, X.; Subjeck, J.R.; Wang, X.-Y. Integrating a “danger” signal into molecular chaperoning to improve vaccination against cancer. Expert Rev. Vaccines 2013, 12, 581–583. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kakimi, K.; Karasaki, T.; Matsushita, H.; Sugie, T. Advances in personalized cancer immunotherapy. Breast Cancer 2017, 24, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, Y.; Bharti, A.; Khaleque, A.A.; Song, B.; Liu, C.; Apostolopoulos, V.; Xing, P.; Calderwood, S.K.; Gong, J. Enhanced immunogenicity of heat shock protein 70 peptide complexes from dendritic cell-tumor fusion cells. J. Immunol. 2006, 177, 5946–5955. [Google Scholar] [CrossRef] [PubMed]
- Weng, D.; Calderwood, S.K.; Gong, J. Preparation of a heat-shock protein 70-based vaccine from DC-tumor fusion cells. Methods Mol. Biol. 2011, 787, 255–265. [Google Scholar] [PubMed]
- Akhbariyoon, H.; Azizpour, Y.; Esfahani, M.F.; Firoozabad, M.S.M.; Rad, M.R.; Esfahani, K.S.; Khoshavi, N.; Karimi, N.; Shirinisaz, A.; Abedi, F.; et al. Immune checkpoint inhibition for the treatment of cancers: An update and critical review of ongoing clinical trials. Clin. Immunol. 2021, 232, 108873. [Google Scholar] [CrossRef] [PubMed]
- Zavareh, R.B.; Spangenberg, S.H.; Woods, A.; Martínez-Peña, F.; Lairson, L.L. HSP90 Inhibition Enhances Cancer Immunotherapy by Modulating the Surface Expression of Multiple Immune Checkpoint Proteins. Cell Chem. Biol. 2021, 28, 158–168.e5. [Google Scholar] [CrossRef] [PubMed]
- Marzec, M.; Zhang, Q.; Goradia, A.; Raghunath, P.N.; Liu, X.; Paessler, M.; Wang, H.Y.; Wysocka, M.; Cheng, M.; Ruggeri, B.A.; et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl. Acad. Sci. USA 2008, 105, 20852–20857. [Google Scholar] [CrossRef] [PubMed]
- Albakova, Z.; Mangasarova, Y. The HSP Immune Network in Cancer. Front. Immunol. 2021, 12, 796493. [Google Scholar] [CrossRef] [PubMed]
- Proia, D.A.; Kaufmann, G.F. Targeting Heat-Shock Protein 90 (HSP90) as a Complementary Strategy to Immune Checkpoint Blockade for Cancer Therapy. Cancer Immunol. Res. 2015, 3, 583–589. [Google Scholar] [CrossRef]
- Bae, J.; Munshi, A.; Li, C.; Samur, M.; Prabhala, R.; Mitsiades, C.; Anderson, K.C.; Munshi, N.C. Heat shock protein 90 is critical for regulation of phenotype and functional activity of human T lymphocytes and NK cells. J. Immunol. 2013, 190, 1360–1371. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Chemotherapeutic | Cancer Type (Cell Line Studied) | Ref. | |
|---|---|---|---|
| Hsp27 | doxorubicin | breast cancer (MCF7, MDA-MB-231, MDA-MB-435) colon cancer (CaCo2, HT-29), prostate cancer (LNCaP) | [58] |
| trastuzumab | breast cancer (SK-BR-3) | [83] | |
| gemcitabine | pancreatic cancer (SW1990) | [85] | |
| 5-fluorouracil | colon cancer (HT-29) | [93] | |
| hepatocellular carcinoma (Hep3B, HepG2) | [94] | ||
| Hsp40 | docetaxel | renal cell carcinoma (RenCa) | [114] |
| 5-fluorouracil | hepatocellular carcinoma (Hep3B, HepG2) | [94] | |
| Hsp60 | cisplatin, oxaliplatin | ovarian cancer (A2780), bladder cancer (UCRU-BL13) | [137] |
| cisplatin | head and neck cancer (UMSCC5, UMSCC10b) | [138] | |
| cervical cancer (HeLa) | [140] | ||
| tamoxifen | breast cancer (MCF-7) | [141] | |
| 5-fluorouracil | colorectal cancer (SW480) | [143] | |
| Hsp70 | imatinib | chronic myeloid leukemia (K562) | [152] |
| ibrutinib | chronic lymphocytic leukemia (patient-derived samples) | [154] | |
| gemcitabine | pancreatic ductal adenocarcinoma (human or mouse tissue samples) | [155] | |
| gemcitabine, topotecan | fibrosarcoma (WEHI) | [157] | |
| 5-fluorouracil | colon cancer (HT-29, SNU-C4) | [158] | |
| paclitaxel | chronic myeloid leukemia (K562), acute promyelocytic leukemia (HL-60), patients’ bone marrow aspirates | [160] | |
| cisplatin | gastric cancer (HGC-27) | [164] | |
| ovarian cancer (OV2008, A2780) | [163] | ||
| Hsp90 | paclitaxel, cisplatin | ovarian cancer (A2780) | [181] |
| 5-fluorouracil, gemcitabine | pancreatic ductal adenocarcinoma (HPAC, PANC-1) | [183] | |
| melphalan | multiple myeloma (RPMI8226) | [184] | |
| Hsp110 | doxorubicin | cervical cancer (HeLa) | [204] |
| oxaliplatin | colorectal cancer (SW480) | [197] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kunachowicz, D.; Król-Kulikowska, M.; Raczycka, W.; Sleziak, J.; Błażejewska, M.; Kulbacka, J. Heat Shock Proteins, a Double-Edged Sword: Significance in Cancer Progression, Chemotherapy Resistance and Novel Therapeutic Perspectives. Cancers 2024, 16, 1500. https://doi.org/10.3390/cancers16081500
Kunachowicz D, Król-Kulikowska M, Raczycka W, Sleziak J, Błażejewska M, Kulbacka J. Heat Shock Proteins, a Double-Edged Sword: Significance in Cancer Progression, Chemotherapy Resistance and Novel Therapeutic Perspectives. Cancers. 2024; 16(8):1500. https://doi.org/10.3390/cancers16081500
Chicago/Turabian StyleKunachowicz, Dominika, Magdalena Król-Kulikowska, Wiktoria Raczycka, Jakub Sleziak, Marta Błażejewska, and Julita Kulbacka. 2024. "Heat Shock Proteins, a Double-Edged Sword: Significance in Cancer Progression, Chemotherapy Resistance and Novel Therapeutic Perspectives" Cancers 16, no. 8: 1500. https://doi.org/10.3390/cancers16081500
APA StyleKunachowicz, D., Król-Kulikowska, M., Raczycka, W., Sleziak, J., Błażejewska, M., & Kulbacka, J. (2024). Heat Shock Proteins, a Double-Edged Sword: Significance in Cancer Progression, Chemotherapy Resistance and Novel Therapeutic Perspectives. Cancers, 16(8), 1500. https://doi.org/10.3390/cancers16081500