
Measurable (Minimal) Residual Disease in Myelodysplastic Neoplasms (MDS): Current State and Perspectives
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Molecular, Genomic-Based Detection of MRD in MDS
3. Cytogenomic-Based Detection of MRD in MDS
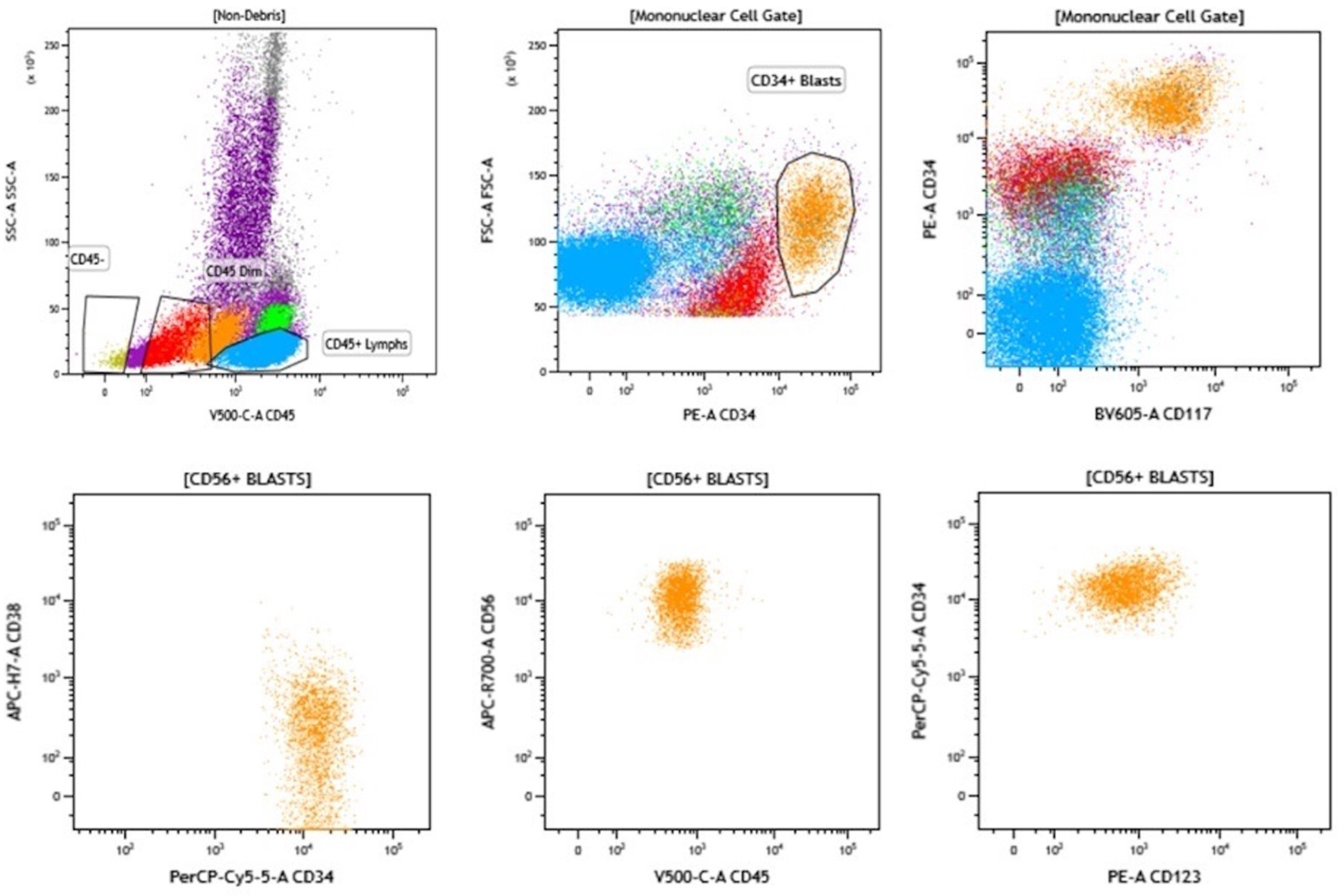
4. Multiparameter Flow Cytometry-Based Detection of MRD in MDS
5. MRD Monitoring Prior to alloHSCT or in the Non-alloHSCT Setting
6. Peri-Transplant MRD Monitoring in Patients with MDS Undergoing Allogeneic Hematopoietic Stem Cell Transplantation (alloHSCT)

{kind=link}
{kind=link}
| Study | Number of Patients Undergoing alloHSCT | Treatment(s) | Method of MRD Monitoring | Limit of Detection or “Cut-Off” for Positive Result | Setting of MRD Monitoring (Pre- and/or Post-Transplantation) | Conclusions |
|---|---|---|---|---|---|---|
| * Ma et al. [135] | 103 patients with MDS-EB with pre-transplantation MRD analysis | Patients received at least one cycle of chemotherapy prior to alloHSCT | Multiparameter flow cytometry | <0.05% to <0.01% throughout duration of study | Pre-transplantation | Worse overall survival (OS) and disease free survival (DFS) in MRD-positive patients; higher cumulative relapse rate in MRD-positive patients. |
| Sallman et al. [140] | Forty patients with TP53-mutated MDS were included in the study | APR-246 + Azacitidine | Next generation sequencing | VAF 5% | Pre-transplantation | TP53-mutated patients achieving CR/PR and NGS-negativity prior to alloHSCT had improved OS compared to those undergoing alloHSCT in CR/PR with NGS-positivity. |
| Dillon et al. [141] | 48 patients with MDS with NGS results prior to initiation of conditioning chemotherapy | MAC (myeloablative conditioning) vs. RIC (reduced-intensity conditioning) regimens according to BMT CTN 0901 protocol | DNA sequencing using a custom anchored multiplex polymerase chain reaction–based panel including 10 genes | Minimum allele frequency of 0.001% | Pre-transplantation | Those with detectable pre-transplant mutations had increased rates of relapse and decreased OS. In those with detectable mutations, RIC (compared to MAC) was associated with lower relapse free survival. |
| Sallman et al. [143] | 34 patients with higher-risk MDS | Magrolimab + Azacitidine; 5 patients treated with additional MDS-directed therapy prior to alloHSCT | Multiparameter flow cytometry | 0.02% | Pre-transplantation | 7 of 34 patients were MRD-negative (median OS not reached) prior to alloHSCT. Median survival was not reached in the MRD-negative cohort. |
| Bejar et al. [144] | 87 patients with MDS | 48% had <5% blasts | Deep, massively parallel sequencing examining 40 genes | Not reported | Pre-transplantation | After adjusting for clinical factors associated with poor outcomes, TP53, TET2, and DNMT3A mutations detected prior to alloHSCT were associated with worse survival. |
| Kharfan-Dabaja et al. [145] | 89 patients with MDS | 82% received Azacitidine prior to alloHSCT; 92% received MAC regimen | Next generation sequencing examining 26 genes | VAF 10% | Pre-transplantation | TP53 and IDH2 mutations were associated with inferior 3-year OS in multivariate analysis. |
| Hunter et al. [147] | 16 patients with TP53-mutated MDS proceeded to alloHSCT | All treated with HMA prior to alloHSCT | Next generation sequencing | VAF 5% | Pre-transplantation | 7 patients achieved TP53 mutation clearance prior to alloHSCT, although this did not result in a statistically significant survival advantage compared to 9 patients with TP53 mutation persistence (p = 0.1). |
| * Festuccia et al. [148] | 223 patients with MDS, 66 patients with secondary AML also included | 76% had <5% blasts at the time of alloHSCT | Multiparameter flow cytometry and cytogenetics | MFC ranged from 0.1% to 0.001% | Pre-transplantation | Patients with identifiable disease by cytogenetics and treated with RIC regimens had worse survival. |
| Craddock et al. [149] | 80 patients with MDS, 164 patients with AML were also included | Patients randomized to fludarabine-based RIC regimen or FLAMSA-Bu (fludarabine/amsacrine/cytarabine-busulfan) | Multiparameter flow cytometry | Approximately 0.02–0.05% | Pre-transplantation and post-transplantation | Detectable pre-transplantation MRD associated with increased 2-year cumulative incidence of relapse, although results were not stratified by disease (AML vs. MDS). |
| Yun et al. [153] | 37 patients with MDS, CMML, or secondary AML proceeded to alloHSCT | Patients treated with various pre-transplant regimens at a single institution | Next generation sequencing including 37 genes | VAF 5% | Pre-transplantation and post-transplantation | Post-transplantation NGS-negativity associated with significantly improved overall survival. |
| Bernal et al. [150] | 38 patients with MDS; patients with AML also included | Majority received RIC regimen | Multiparameter flow cytometry | 0.0001% | Pre-transplantation and day +100 post-transplantation | Day +100 MRD-positivity associated with increased relapse risk and worse overall survival, although results were not stratified by disease (AML vs. MDS), |
| Mo et al. [151] | 78 high-risk or very high-risk patients with MDS | Patients who were MRD-positive after alloHSCT received DLI, interferon-α, chemotherapy, or discontinued immunosuppression | Multiparameter flow cytometry and WT1 PCR | MFC 0.01%, WT1 considered positive if >0.60% | 1, 2, 3, 4.5, 6, 9, 12 months post-transplantation and at 6 month intervals thereafter | Two-year cumulative incidence of relapse significantly higher in MRD-positive by either MFC or WT1 PCR. |
| Duncavage et al. [152] | 65 patients with MDS, 21 patients with secondary AML also included | 58% received MAC regimen | Enhanced exome sequencing | Considered positive if >0.5% | Day +30 and day +100 post-transplantation | Increased risk of disease progression if detectable mutation at day +30 or +100 even after adjusting for conditioning regimen. |
| Nakamura et al. [154] | 14 patients with MDS, 39 patients with AML also included | Received MAC regimen | Digital droplet PCR (ddPCR) and circulating tumor DNA (ctDNA) | 0.04% for ddPCR | 1- and 3 months post-transplantation | Results were not stratified by disease (AML vs. MDS), but both ctDNA and ddPCR-positivity at 1- and 3 months post-transplantation were associated with increased risk of relapse and death. |
| * Tobiasson et al. [37] | 177 patients with MDS, 89 additional patients with MDS/MPN or AML with dysplastic features and 20–29% blasts included | Conditioning intensity and previous therapies not reported | ddPCR | Cut-off 0.1% | Bone marrow samples at 1- and 3 months post-transplantation, then every 3 months until month 24 or relapse/death; Peripheral blood samples obtained monthly | Bone marrow MRD-positivity preceded clinical relapse in 42/44 patients by a median of 71 days. |
| Hou et al. [103] | 115 patients with MDS | Received MAC regimen | Multiparameter flow cytometry (MFC) and Next generation sequencing | MFC “high” defined as ≥0.1% | Day +30 post-transplantation | Progression free survival significantly worse for MFC “high” and mutation positive patients. |
| Loke et al. [155] | 66 patients with MDS, 121 patients with AML also included | Received RIC regimen according to FIGARO protocol | Multiparameter flow cytometry | 0.05% | Day +42 up to month 12 post-transplantation | MRD-positivity was associated with inferior overall survival and relapse free survival, although this analysis was not stratified according to underlying hematologic malignancy. Full donor T cell chimerism associated with lower rate of MRD-positivity. |
7. Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Li, H.; Hu, F.; Gale, R.P.; Sekeres, M.A.; Liang, Y. Myelodysplastic Syndromes. Nat. Rev. Dis. Primers 2022, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Cogle, C.R. Incidence and Burden of the Myelodysplastic Syndromes. Curr. Hematol. Malig. Rep. 2015, 10, 272–281. [Google Scholar] [CrossRef]
- Maggioni, G.; Della Porta, M.G. Molecular Landscape of Myelodysplastic Neoplasms in Disease Classification and Prognostication. Curr. Opin. Hematol. 2023, 30, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Carraway, H.E.; Saygin, C. Therapy for Lower-Risk MDS. Hematol. Am. Soc. Hematol. Educ. Program. 2020, 2020, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Giagounidis, A. Current Treatment Algorithm for the Management of Lower-Risk MDS. Hematology 2017, 2017, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; Stahl, M.; DeVeaux, M.; Giri, S.; Huntington, S.; Podoltsev, N.; Wang, R.; Ma, X.; Davidoff, A.J.; Gore, S.D. Counseling Patients with Higher-Risk MDS Regarding Survival with Azacitidine Therapy: Are We Using Realistic Estimates? Blood Cancer J. 2018, 8, 55. [Google Scholar] [CrossRef]
- Garcia, J.S.; Swords, R.T.; Roboz, G.J.; Jacoby, M.A.; Garcia-Manero, G.; Hong, W.-J.; Yang, X.; Zhou, Y.; Platzbecker, U.; Steensma, D.P.; et al. A Systematic Review of Higher-Risk Myelodysplastic Syndromes Clinical Trials to Determine the Benchmark of Azacitidine and Explore Alternative Endpoints for Overall Survival. Leuk. Res. 2021, 104, 106555. [Google Scholar] [CrossRef]
- Stahl, M.; Bewersdorf, J.P.; Xie, Z.; Della Porta, M.G.; Komrokji, R.; Xu, M.L.; Abdel-Wahab, O.; Taylor, J.; Steensma, D.P.; Starczynowski, D.T.; et al. Classification, Risk Stratification and Response Assessment in Myelodysplastic Syndromes/Neoplasms (MDS): A State-of-the-Art Report on Behalf of the International Consortium for MDS (IcMDS). Blood Rev. 2023, 62, 101128. [Google Scholar] [CrossRef]
- Cheson, B.D.; Bennett, J.M.; Kantarjian, H.; Pinto, A.; Schiffer, C.A.; Nimer, S.D.; Löwenberg, B.; Beran, M.; de Witte, T.M.; Stone, R.M.; et al. Report of an International Working Group to Standardize Response Criteria for Myelodysplastic Syndromes. Blood 2000, 96, 3671–3674. [Google Scholar]
- Cheson, B.D. Clinical Application and Proposal for Modification of the International Working Group (IWG) Response Criteria in Myelodysplasia. Blood 2006, 108, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; Platzbecker, U.; Bewersdorf, J.P.; Stahl, M.; Adès, L.; Borate, U.; Bowen, D.T.; Buckstein, R.J.; Brunner, A.M.; Carraway, H.E.; et al. Consensus Proposal for Revised International Working Group Response Criteria for Higher Risk Myelodysplastic Syndromes. Blood 2023, 141, 2047–2061. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent Pathway Mutations of Splicing Machinery in Myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M.; Della Porta, M.G.; Malcovati, L. The Genetic Basis of Myelodysplasia and Its Clinical Relevance. Blood 2013, 122, 4021–4034. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of Genetic Lesions in 944 Patients with Myelodysplastic Syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and Biological Implications of Driver Mutations in Myelodysplastic Syndromes. Blood 2013, 122, 3616–3627, quiz 3699. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, S.M.; Cerami, E.; Baras, A.; Pugh, T.J.; Schultz, N.; Stricker, T.; Lindsay, J.; Del Vecchio Fitz, C.; Kumari, P.; Micheel, C.; et al. AACR Project Genie: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- de Bruijn, I.; Kundra, R.; Mastrogiacomo, B.; Tran, T.N.; Sikina, L.; Mazor, T.; Li, X.; Ochoa, A.; Zhao, G.; Lai, B.; et al. Analysis and Visualization of Longitudinal Genomic and Clinical Data from the AACR Project GENIE Biopharma Collaborative in CBioPortal. Cancer Res. 2023, 83, 3861–3867. [Google Scholar] [CrossRef]
- Gundry, M.; Vijg, J. Direct Mutation Analysis by High-Throughput Sequencing: From Germline to Low-Abundant, Somatic Variants. Mutat. Res. 2012, 729, 1–15. [Google Scholar] [CrossRef]
- Schmitt, M.W.; Kennedy, S.R.; Salk, J.J.; Fox, E.J.; Hiatt, J.B.; Loeb, L.A. Detection of Ultra-Rare Mutations by next-Generation Sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 14508–14513. [Google Scholar] [CrossRef]
- Au, C.H.; Wa, A.; Ho, D.N.; Chan, T.L.; Ma, E.S.K. Clinical Evaluation of Panel Testing by Next-Generation Sequencing (NGS) for Gene Mutations in Myeloid Neoplasms. Diagn. Pathol. 2016, 11, 11. [Google Scholar] [CrossRef]
- Levis, M.J.; Perl, A.E.; Altman, J.K.; Gocke, C.D.; Bahceci, E.; Hill, J.; Liu, C.; Xie, Z.; Carson, A.R.; McClain, V.; et al. A Next-Generation Sequencing-Based Assay for Minimal Residual Disease Assessment in AML Patients with FLT3-ITD Mutations. Blood Adv. 2018, 2, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Craven, K.E.; Fischer, C.G.; Jiang, L.Q.; Pallavajjala, A.; Lin, M.T.; Eshleman, J.R. Optimizing Insertion and Deletion Detection Using Next-Generation Sequencing in the Clinical Laboratory. J. Mol. Diagn. 2022, 24, 1217–1231. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.B. Perspective on Measurable Residual Disease Testing in Acute Myeloid Leukemia. Leukemia 2024, 38, 10–13. [Google Scholar] [CrossRef]
- Harankhedkar, S.; Patkar, N. Molecular MRD Assessment in Acute Myeloid Leukemias. Indian J. Med. Paediatr. Oncol. 2023, 44, 566–577. [Google Scholar] [CrossRef]
- Hiatt, J.B.; Pritchard, C.C.; Salipante, S.J.; O’Roak, B.J.; Shendure, J. Single Molecule Molecular Inversion Probes for Targeted, High-Accuracy Detection of Low-Frequency Variation. Genome Res. 2013, 23, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Heuser, M.; Freeman, S.D.; Ossenkoppele, G.J.; Buccisano, F.; Hourigan, C.S.; Ngai, L.L.; Tettero, J.M.; Bachas, C.; Baer, C.; Béné, M.-C.; et al. 2021 Update on MRD in Acute Myeloid Leukemia: A Consensus Document from the European LeukemiaNet MRD Working Party. Blood 2021, 138, 2753–2767. [Google Scholar] [CrossRef]
- Dillon, L.W.; Higgins, J.; Nasif, H.; Othus, M.; Beppu, L.; Smith, T.H.; Schmidt, E.; Valentine III, C.C.; Salk, J.J.; Wood, B.L.; et al. Quantification of Measurable Residual Disease Using Duplex Sequencing in Adults with Acute Myeloid Leukemia. Haematologica 2023, 109, 401–410. [Google Scholar] [CrossRef]
- Stengel, A.; Baer, C.; Walter, W.; Meggendorfer, M.; Kern, W.; Haferlach, T.; Haferlach, C. Mutational Patterns and Their Correlation to CHIP-Related Mutations and Age in Hematological Malignancies. Blood Adv. 2021, 5, 4426–4434. [Google Scholar] [CrossRef] [PubMed]
- Gaulin, C.; Kelemen, K.; Arana Yi, C. Molecular Pathways in Clonal Hematopoiesis: From the Acquisition of Somatic Mutations to Transformation into Hematologic Neoplasm. Life 2022, 12, 1135. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.J.; Shen, D.; Shao, J.; Ding, L.; White, B.S.; Kandoth, C.; Miller, C.A.; Niu, B.; McLellan, M.D.; Dees, N.D.; et al. Clonal Diversity of Recurrently Mutated Genes in Myelodysplastic Syndromes. Leukemia 2013, 27, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Mossner, M.; Jann, J.-C.; Wittig, J.; Nolte, F.; Fey, S.; Nowak, V.; Obländer, J.; Pressler, J.; Palme, I.; Xanthopoulos, C.; et al. Mutational Hierarchies in Myelodysplastic Syndromes Dynamically Adapt and Evolve upon Therapy Response and Failure. Blood 2016, 128, 1246–1259. [Google Scholar] [CrossRef] [PubMed]
- Makishima, H. Founder and Subclonal Mutations in Myelodysplastic Syndromes and Related Myeloid Neoplasms. Best. Pract. Res. Clin. Haematol. 2020, 33, 101189. [Google Scholar] [CrossRef] [PubMed]
- El Hussein, S.; Loghavi, S. The Impact of Clonal Hierarchy and Heterogeneity on Phenotypic Manifestations of Myelodysplastic Neoplasms. Cancers 2022, 14, 5690. [Google Scholar] [CrossRef]
- Tobiasson, M.; Pandzic, T.; Illman, J.; Nilsson, L.; Weström, S.; Ejerblad, E.; Olesen, G.; Björklund, A.; Olsnes Kittang, A.; Werlenius, O.; et al. Patient-Specific Measurable Residual Disease Markers Predict Outcome in Patients with Myelodysplastic Syndrome and Related Diseases after Hematopoietic Stem-Cell Transplantation. J. Clin. Oncol. 2024; online ahead of print. [Google Scholar] [CrossRef]
- Makishima, H.; Yoshizato, T.; Yoshida, K.; Sekeres, M.A.; Radivoyevitch, T.; Suzuki, H.; Przychodzen, B.; Nagata, Y.; Meggendorfer, M.; Sanada, M.; et al. Dynamics of Clonal Evolution in Myelodysplastic Syndromes. Nat. Genet. 2017, 49, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; Stahl, M.; Hu, X.; Wang, R.; Huntington, S.F.; Podoltsev, N.A.; Gore, S.D.; Ma, X.; Davidoff, A.J. Long-Term Survival of Older Patients with MDS Treated with HMA Therapy without Subsequent Stem Cell Transplantation. Blood 2018, 131, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Mohamedali, A.M.; Gäken, J.; Ahmed, M.; Malik, F.; Smith, A.E.; Best, S.; Mian, S.; Gaymes, T.; Ireland, R.; Kulasekararaj, A.G.; et al. High Concordance of Genomic and Cytogenetic Aberrations between Peripheral Blood and Bone Marrow in Myelodysplastic Syndrome (MDS). Leukemia 2015, 29, 1928–1938. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Fenaux, P.; Han, X.; James, D.A.; Malek, K.; Marques Ramos, P.; Miyazaki, Y.; Platzbecker, U. Molecular Measurable Residual Disease (MRD) Clearance (≤1%) Is Associated with Improved Clinical Outcomes in Patients with Higher-Risk Myelodysplastic Neoplasms (HR-MDS): An Exploratory Analysis of Stimulus-MDS1 in Patients Receiving Sabatolimab or Placebo + Hypomethylating Agent (HMA). Blood 2023, 142, 3236. [Google Scholar] [CrossRef]
- Osman, A.E.G.; Mencia-Trinchant, N.; Saygin, C.; Moma, L.; Kim, A.; Housman, G.; Pozsgai, M.; Sinha, E.; Chandra, P.; Hassane, D.C.; et al. Paired Bone Marrow and Peripheral Blood Samples Demonstrate Lack of Widespread Dissemination of Some CH Clones. Blood Adv. 2023, 7, 1910–1914. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Lang, W.; Mei, C.; Ren, Y.; Ma, L.; Jiang, L.; Ye, L.; Xu, G.; Luo, Y.; Liu, L.; et al. Serial Monitoring of Circulating Tumour DNA on Clinical Outcome in Myelodysplastic Syndromes and Acute Myeloid Leukaemia. Clin. Transl. Med. 2023, 13, e1349. [Google Scholar] [CrossRef] [PubMed]
- Iriyama, C.; Tomita, A.; Hoshino, H.; Adachi-Shirahata, M.; Furukawa-Hibi, Y.; Yamada, K.; Kiyoi, H.; Naoe, T. Using Peripheral Blood Circulating DNAs to Detect CpG Global Methylation Status and Genetic Mutations in Patients with Myelodysplastic Syndrome. Biochem. Biophys. Res. Commun. 2012, 419, 662–669. [Google Scholar] [CrossRef]
- Suzuki, Y.; Tomita, A.; Nakamura, F.; Iriyama, C.; Shirahata-Adachi, M.; Shimada, K.; Akashi, A.; Ishikawa, Y.; Kaneda, N.; Kiyoi, H. Peripheral Blood Cell-Free DNA Is an Alternative Tumor DNA Source Reflecting Disease Status in Myelodysplastic Syndromes. Cancer Sci. 2016, 107, 1329–1337. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gisbert, N.; Garcia-Ávila, S.; Merchán, B.; Salido, M.; Fernández-Rodríguez, C.; Gibert, J.; Fernández-Ibarrondo, L.; Camacho, L.; Lafuente, M.; Longarón, R.; et al. Molecular and Cytogenetic Characterization of Myelodysplastic Syndromes in Cell-Free DNA. Blood Adv. 2022, 6, 3178–3188. [Google Scholar] [CrossRef]
- Dietz, A.C.; DeFor, T.E.; Brunstein, C.G.; Wagner, J.E. Donor-Derived Myelodysplastic Syndrome and Acute Leukaemia after Allogeneic Haematopoietic Stem Cell Transplantation: Incidence, Natural History and Treatment Response. Br. J. Haematol. 2014, 166, 209–212. [Google Scholar] [CrossRef]
- Lee, J.-M.; Kim, Y.-J.; Park, S.-S.; Han, E.; Kim, M.; Kim, Y. Simultaneous Monitoring of Mutation and Chimerism Using Next-Generation Sequencing in Myelodysplastic Syndrome. J. Clin. Med. 2019, 8, 2077. [Google Scholar] [CrossRef]
- Hecker, J.S.; Hartmann, L.; Rivière, J.; Buck, M.C.; van der Garde, M.; Rothenberg-Thurley, M.; Fischer, L.; Winter, S.; Ksienzyk, B.; Ziemann, F.; et al. CHIP and Hips: Clonal Hematopoiesis Is Common in Patients Undergoing Hip Arthroplasty and Is Associated with Autoimmune Disease. Blood 2021, 138, 1727–1732. [Google Scholar] [CrossRef] [PubMed]
- Risques, R.A.; Kennedy, S.R. Aging and the Rise of Somatic Cancer-Associated Mutations in Normal Tissues. PLoS Genet. 2018, 14, e1007108. [Google Scholar] [CrossRef]
- Malcovati, L.; Gallì, A.; Travaglino, E.; Ambaglio, I.; Rizzo, E.; Molteni, E.; Elena, C.; Ferretti, V.V.; Catricalà, S.; Bono, E.; et al. Clinical Significance of Somatic Mutation in Unexplained Blood Cytopenia. Blood 2017, 129, 3371–3378. [Google Scholar] [CrossRef]
- Nannya, Y.; Tobiasson, M.; Sato, S.; Bernard, E.; Ohtake, S.; Takeda, J.; Creignou, M.; Zhao, L.; Kusakabe, M.; Shibata, Y.; et al. Postazacitidine Clone Size Predicts Long-Term Outcome of Patients with Myelodysplastic Syndromes and Related Myeloid Neoplasms. Blood Adv. 2023, 7, 3624–3636. [Google Scholar] [CrossRef] [PubMed]
- Cook, M.R.; Karp, J.E.; Lai, C. The Spectrum of Genetic Mutations in Myelodysplastic Syndrome: Should We Update Prognostication? EJHaem 2022, 3, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Arango Ossa, J.E.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L.; et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evid. 2022, 1, EVIDoa2200008. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating Morphologic, Clinical, and Genomic Data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Grimes, H.L. Why Single-Cell Sequencing Has Promise in MDS. Front. Oncol. 2021, 11, 769753. [Google Scholar] [CrossRef] [PubMed]
- Nangalia, J.; Mitchell, E.; Green, A.R. Clonal Approaches to Understanding the Impact of Mutations on Hematologic Disease Development. Blood 2019, 133, 1436–1445. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Chin, V.; Fairfax, K.; Moutinho, C.; Suan, D.; Ji, H.; Powell, J.E. Transitioning Single-Cell Genomics into the Clinic. Nat. Rev. Genet. 2023, 24, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.; Lin, Y.; Navin, N. Advancing Cancer Research and Medicine with Single-Cell Genomics. Cancer Cell 2020, 37, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Brierley, C.K.; Mead, A.J. Single-Cell Sequencing in Hematology. Curr. Opin. Oncol. 2020, 32, 139–145. [Google Scholar] [CrossRef]
- Ediriwickrema, A.; Aleshin, A.; Reiter, J.G.; Corces, M.R.; Kohnke, T.; Stafford, M.; Liedtke, M.; Medeiros, B.C.; Majeti, R. Single-Cell Mutational Profiling Enhances the Clinical Evaluation of AML MRD. Blood Adv. 2020, 4, 943–952. [Google Scholar] [CrossRef]
- Robinson, T.M.; Bowman, R.L.; Persaud, S.; Liu, Y.; Neigenfind, R.; Gao, Q.; Zhang, J.; Sun, X.; Miles, L.A.; Cai, S.F.; et al. Single-Cell Genotypic and Phenotypic Analysis of Measurable Residual Disease in Acute Myeloid Leukemia. Sci. Adv. 2023, 9, eadg0488. [Google Scholar] [CrossRef] [PubMed]
- Dillon, L.W.; Ghannam, J.; Nosiri, C.; Gui, G.; Goswami, M.; Calvo, K.R.; Lindblad, K.E.; Oetjen, K.A.; Wilkerson, M.D.; Soltis, A.R.; et al. Personalized Single-Cell Proteogenomics to Distinguish Acute Myeloid Leukemia from Nonmalignant Clonal Hematopoiesis. Blood Cancer Discov. 2021, 2, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Grimes, H.L. Improved Detection of Measurable Residual Disease in Acute Myeloid Leukemia. Sci. Adv. 2023, 9, eadk2533. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.; Aplan, P.D.; Freeman, S.D.; Pavletic, S.Z. Moving toward a Conceptualization of Measurable Residual Disease in Myelodysplastic Syndromes. Blood Adv. 2023, 7, 4381–4394. [Google Scholar] [CrossRef] [PubMed]
- Acha, P.; Palomo, L.; Fuster-Tormo, F.; Xicoy, B.; Mallo, M.; Manzanares, A.; Grau, J.; Marcé, S.; Granada, I.; Rodríguez-Luaces, M.; et al. Analysis of Intratumoral Heterogeneity in Myelodysplastic Syndromes with Isolated Del(5q) Using a Single Cell Approach. Cancers 2021, 13, 841. [Google Scholar] [CrossRef]
- Liu, Y.; Niu, H.; Song, N.; Zhang, W.; Li, L.; Wang, H.; Fu, R.; Shao, Z. Single-Cell RNA Sequencing Identifies the Properties of Myelodysplastic Syndrome Stem Cells. J. Transl. Med. 2022, 20, 499. [Google Scholar] [CrossRef]
- Schanz, J.; Tüchler, H.; Solé, F.; Mallo, M.; Luño, E.; Cervera, J.; Granada, I.; Hildebrandt, B.; Slovak, M.L.; Ohyashiki, K.; et al. New Comprehensive Cytogenetic Scoring System for Primary Myelodysplastic Syndromes (MDS) and Oligoblastic Acute Myeloid Leukemia after MDS Derived from an International Database Merge. J. Clin. Oncol. 2012, 30, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Haase, D.; Germing, U.; Schanz, J.; Pfeilstöcker, M.; Nösslinger, T.; Hildebrandt, B.; Kundgen, A.; Lübbert, M.; Kunzmann, R.; Giagounidis, A.A.N.; et al. New Insights into the Prognostic Impact of the Karyotype in MDS and Correlation with Subtypes: Evidence from a Core Dataset of 2124 Patients. Blood 2007, 110, 4385–4395. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Rigolin, G.; Bigoni, R.; Milani, R.; Cavazzini, F.; Roberti, M.; Bardi, A.; Agostini, P.; Della Porta, M.; Tieghi, A.; Piva, N.; et al. Clinical Importance of Interphase Cytogenetics Detecting Occult Chromosome Lesions in Myelodysplastic Syndromes with Normal Karyotype. Leukemia 2001, 15, 1841–1847. [Google Scholar] [CrossRef]
- Bernasconi, P.; Cavigliano, P.M.; Boni, M.; Calatroni, S.; Klersy, C.; Giardini, I.; Rocca, B.; Crosetto, N.; Caresana, M.; Lazzarino, M.; et al. Is FISH a Relevant Prognostic Tool in Myelodysplastic Syndromes with a Normal Chromosome Pattern on Conventional Cytogenetics? A Study on 57 Patients. Leukemia 2003, 17, 2107–2112. [Google Scholar] [CrossRef] [PubMed]
- Cherry, A.M.; Brockman, S.R.; Paternoster, S.F.; Hicks, G.A.; Neuberg, D.; Higgins, R.R.; Bennett, J.M.; Greenberg, P.L.; Miller, K.; Tallman, M.S.; et al. Comparison of Interphase FISH and Metaphase Cytogenetics to Study Myelodysplastic Syndrome: An Eastern Cooperative Oncology Group (ECOG) Study. Leuk. Res. 2003, 27, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Mohamedali, A.; Gäken, J.; Twine, N.A.; Ingram, W.; Westwood, N.; Lea, N.C.; Hayden, J.; Donaldson, N.; Aul, C.; Gattermann, N.; et al. Prevalence and Prognostic Significance of Allelic Imbalance by Single-Nucleotide Polymorphism Analysis in Low-Risk Myelodysplastic Syndromes. Blood 2007, 110, 3365–3373. [Google Scholar] [CrossRef] [PubMed]
- Maciejewski, J.P.; Mufti, G.J. Whole Genome Scanning as a Cytogenetic Tool in Hematologic Malignancies. Blood 2008, 112, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Tiu, R.V.; Gondek, L.P.; O’keefe, C.L.; Elson, P.; Huh, J.; Mohamedali, A.; Kulasekararaj, A.; Advani, A.S.; Paquette, R.; List, A.F.; et al. Prognostic Impact of SNP Array Karyotyping in Myelodysplastic Syndromes and Related Myeloid Malignancies. Blood 2011, 117, 4552–4560. [Google Scholar] [CrossRef] [PubMed]
- Kanagal-Shamanna, R.; Hodge, J.C.; Tucker, T.; Shetty, S.; Yenamandra, A.; Dixon-McIver, A.; Bryke, C.; Huxley, E.; Lennon, P.A.; Raca, G.; et al. Assessing Copy Number Aberrations and Copy Neutral Loss of Heterozygosity across the Genome as Best Practice: An Evidence Based Review of Clinical Utility from the Cancer Genomics Consortium (CGC) Working Group for Myelodysplastic Syndrome, Myelodysplastic/Myeloproliferative and Myeloproliferative Neoplasms. Cancer Genet. 2018, 228–229, 197–217. [Google Scholar] [CrossRef]
- Gondek, L.P.; Dunbar, A.J.; Szpurka, H.; McDevitt, M.A.; Maciejewski, J.P. SNP Array Karyotyping Allows for the Detection of Uniparental Disomy and Cryptic Chromosomal Abnormalities in MDS/MPD-U and MPD. PLoS ONE 2007, 2, e1225. [Google Scholar] [CrossRef]
- Stengel, A.; Kern, W.; Haferlach, T.; Meggendorfer, M.; Haferlach, C. The 5q Deletion Size in Myeloid Malignancies Is Correlated to Additional Chromosomal Aberrations and to TP53 Mutations. Genes Chromosomes Cancer 2016, 55, 777–785. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Sekeres, M.A.; Makishima, H.; Traina, F.; Visconte, V.; Jankowska, A.; Jerez, A.; Szpurka, H.; O’Keefe, C.L.; Guinta, K.; et al. Cytogenetic and Molecular Predictors of Response in Patients with Myeloid Malignancies without Del[5q] Treated with Lenalidomide. J. Hematol. Oncol. 2012, 5, 4. [Google Scholar] [CrossRef]
- Malcovati, L.; Hellström-Lindberg, E.; Bowen, D.; Adès, L.; Cermak, J.; del Cañizo, C.; Della Porta, M.G.; Fenaux, P.; Gattermann, N.; Germing, U.; et al. Diagnosis and Treatment of Primary Myelodysplastic Syndromes in Adults: Recommendations from the European LeukemiaNet. Blood 2013, 122, 2943–2964. [Google Scholar] [CrossRef]
- Yang, H.; Garcia-Manero, G.; Rush, D.; Montalban-Bravo, G.; Mallampati, S.; Medeiros, L.J.; Levy, B.; Luthra, R.; Kanagal-Shamanna, R. Application of Optical Genome Mapping For Comprehensive Assessment of Chromosomal Structural Variants for Clinical Evaluation of Myelodysplastic Syndromes. medRxiv 2021. [Google Scholar] [CrossRef]
- Yang, H.; Garcia-Manero, G.; Sasaki, K.; Montalban-Bravo, G.; Tang, Z.; Wei, Y.; Kadia, T.; Chien, K.; Rush, D.; Nguyen, H.; et al. High-Resolution Structural Variant Profiling of Myelodysplastic Syndromes by Optical Genome Mapping Uncovers Cryptic Aberrations of Prognostic and Therapeutic Significance. Leukemia 2022, 36, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Pei, Z.; Lei, C.; Zhu, S.; Deng, K.; Zhou, J.; Yang, J.; Lu, D.; Sun, X.; Xu, C.; et al. Detection of Cryptic Balanced Chromosomal Rearrangements Using High-Resolution Optical Genome Mapping. J. Med. Genet. 2023, 60, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Gerding, W.M.; Tembrink, M.; Nilius-Eliliwi, V.; Mika, T.; Dimopoulos, F.; Ladigan-Badura, S.; Eckhardt, M.; Pohl, M.; Wünnenberg, M.; Farshi, P.; et al. Optical Genome Mapping Reveals Additional Prognostic Information Compared to Conventional Cytogenetics in AML/MDS Patients. Int. J. Cancer 2022, 150, 1998–2011. [Google Scholar] [CrossRef] [PubMed]
- Vangala, D.B.; Nilius-Eliliwi, V.; Gerding, W.M.; Schroers, R.; Nguyen, H.P. Optical Genome Mapping in MDS and AML as Tool for Structural Variant Profiling-Comment and Data Update on Yang et al. “High-Resolution Structural Variant Profiling of Myelodysplastic Syndromes by Optical Genome Mapping Uncovers Cryptic Aberrations of Prognostic and Therapeutic Significance”. Leukemia 2023, 37, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.J.; Garcia-Manero, G.; Strati, P.; Mishra, A.; Al Ali, N.H.; Padron, E.; Lancet, J.; Kadia, T.; Daver, N.; O’Brien, S.; et al. Outcome of Patients with Low-Risk and Intermediate-1-Risk Myelodysplastic Syndrome after Hypomethylating Agent Failure: A Report on Behalf of the MDS Clinical Research Consortium. Cancer 2015, 121, 876–882. [Google Scholar] [CrossRef]
- da Frota França, I.G.; de Melo, M.M.L.; Teixeira, M.S.C.; Cordeiro, J.V.A.; de Paula Borges, D.; de Oliveira, R.T.G.; Furtado, S.R.; Magalhães, S.M.M.; Pinheiro, R.F. Role of Conventional Cytogenetics in Sequential Karyotype Analysis of Myelodysplastic Syndrome: A Patient with Der(1;7)(Q10;P10). Hematol. Transfus. Cell Ther. 2019, 41, 91–94. [Google Scholar] [CrossRef]
- Neukirchen, J.; Lauseker, M.; Hildebrandt, B.; Nolting, A.-C.; Kaivers, J.; Kobbe, G.; Gattermann, N.; Haas, R.; Germing, U. Cytogenetic Clonal Evolution in Myelodysplastic Syndromes Is Associated with Inferior Prognosis. Cancer 2017, 123, 4608–4616. [Google Scholar] [CrossRef]
- Stasik, S.; Burkhard-Meier, C.; Kramer, M.; Middeke, J.M.; Oelschlaegel, U.; Sockel, K.; Ehninger, G.; Serve, H.; Müller-Tidow, C.; Baldus, C.D.; et al. Deep Sequencing in CD34+ Cells from Peripheral Blood Enables Sensitive Detection of Measurable Residual Disease in AML. Blood Adv. 2022, 6, 3294–3303. [Google Scholar] [CrossRef]
- Barbet, R. Purification of CD34+ Stem Cells from Myelodysplastic Syndrome with Ring Sideroblasts Using Immunomagnetic Strategy. Biomed. J. Sci. Tech. Res. 2021, 34, 27120–27125. [Google Scholar] [CrossRef]
- Shin, S.Y.; Jang, S.; Park, C.-J.; Chi, H.-S.; Lee, J.-H.; Lee, J.H.; Lee, K.H.; Suh, C.; Lim, S.E.; Seo, E.-J. Application of an Immune-Magnetic Cell Sorting Method for CD138-Positive Plasma Cells in FISH Analysis of Multiple Myeloma. Int. J. Lab. Hematol. 2012, 34, 541–546. [Google Scholar] [CrossRef]
- Tehranchi, R.; Woll, P.S.; Anderson, K.; Buza-Vidas, N.; Mizukami, T.; Mead, A.J.; Astrand-Grundström, I.; Strömbeck, B.; Horvat, A.; Ferry, H.; et al. Persistent Malignant Stem Cells in Del(5q) Myelodysplasia in Remission. N. Engl. J. Med. 2010, 363, 1025–1037. [Google Scholar] [CrossRef] [PubMed]
- Orvain, C.; Wilson, J.A.; Fang, M.; Sandmaier, B.M.; Rodríguez-Arbolí, E.; Wood, B.L.; Othus, M.; Appelbaum, F.R.; Walter, R.B. Relative Impact of Residual Cytogenetic Abnormalities and Flow Cytometric Measurable Residual Disease on Outcome after Allogeneic Hematopoietic Cell Transplantation in Adult Acute Myeloid Leukemia. Haematologica 2023, 108, 420–432. [Google Scholar] [CrossRef]
- Chen, Y.; Cortes, J.; Estrov, Z.; Faderl, S.; Qiao, W.; Abruzzo, L.; Garcia-Manero, G.; Pierce, S.; Huang, X.; Kebriaei, P.; et al. Persistence of Cytogenetic Abnormalities at Complete Remission after Induction in Patients with Acute Myeloid Leukemia: Prognostic Significance and the Potential Role of Allogeneic Stem-Cell Transplantation. J. Clin. Oncol. 2011, 29, 2507–2513. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, P.; Labopin, M.; Viguié, F.; Perot, C.; Isnard, F.; Mamez, A.-C.; Bilhou-Nabera, C.; Marzac, C.; Delhommeau, F.; Lapusan, S.; et al. Interest of Cytogenetic and FISH Evaluation for Prognosis Evaluation in 198 Patients with Acute Myeloid Leukemia in First Complete Remission in a Single Institution. Leuk. Res. 2014, 38, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, G.; Mrózek, K.; Ruppert, A.S.; Archer, K.J.; Pettenati, M.J.; Heerema, N.A.; Carroll, A.J.; Koduru, P.R.K.; Kolitz, J.E.; Sterling, L.J.; et al. Abnormal Cytogenetics at Date of Morphologic Complete Remission Predicts Short Overall and Disease-Free Survival, and Higher Relapse Rate in Adult Acute Myeloid Leukemia: Results from Cancer and Leukemia Group B Study 8461. J. Clin. Oncol. 2004, 22, 2410–2418. [Google Scholar] [CrossRef] [PubMed]
- Balleisen, S.; Kuendgen, A.; Hildebrandt, B.; Haas, R.; Germing, U. Prognostic Relevance of Achieving Cytogenetic Remission in Patients with Acute Myelogenous Leukemia or High-Risk Myelodysplastic Syndrome Following Induction Chemotherapy. Leuk. Res. 2009, 33, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Saini, L.; Brandwein, J.; Szkotak, A.; Ghosh, S.; Sandhu, I. Persistent Cytogenetic Abnormalities in Patients Undergoing Intensive Chemotherapy for Acute Myeloid Leukemia. Leuk. Lymphoma 2018, 59, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Storer, B.; Wood, B.; Gyurkocza, B.; Sandmaier, B.M.; Appelbaum, F.R. Prognostic Impact of Discordant Results from Cytogenetics and Flow Cytometry in Patients with Acute Myeloid Leukemia Undergoing Hematopoietic Cell Transplantation. Cancer 2012, 118, 2411–2419. [Google Scholar] [CrossRef]
- Norkin, M.; Katragadda, L.; Zou, F.; Xiong, S.; Chang, M.; Dai, Y.; Hsu, J.W.; Moreb, J.S.; Leather, H.; Murthy, H.S.; et al. Minimal Residual Disease by Either Flow Cytometry or Cytogenetics Prior to an Allogeneic Hematopoietic Stem Cell Transplant Is Associated with Poor Outcome in Acute Myeloid Leukemia. Blood Cancer J. 2017, 7, 634. [Google Scholar] [CrossRef]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Béne, M.C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S.; et al. Minimal/Measurable Residual Disease in AML: A Consensus Document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Zhou, L.; Yang, M.; Jiang, S.; Shen, H.; Zhu, M.; Chen, J.; Miao, M.; Xu, Y.; Wu, D. The Prognostic Value of Early Detection of Minimal Residual Disease as Defined by Flow Cytometry and Gene Mutation Clearance for Myelodysplastic Syndrome Patients After Myeloablative Allogeneic Hematopoietic Stem-Cell Transplantation. Front. Oncol. 2021, 11, 700234. [Google Scholar] [CrossRef] [PubMed]
- Kriegsmann, K.; Löffler, H.; Eckstein, V.; Schulz, R.; Kräker, S.; Braun, U.; Luft, T.; Hegenbart, U.; Schönland, S.; Dreger, P.; et al. CD7 Is Expressed on a Subset of Normal CD34-Positive Myeloid Precursors. Eur. J. Haematol. 2018, 101, 318–325. [Google Scholar] [CrossRef]
- Barreau, S.; Green, A.S.; Dussiau, C.; Alary, A.S.; Raimbault, A.; Mathis, S.; Willems, L.; Bouscary, D.; Kosmider, O.; Bardet, V.; et al. Phenotypic Landscape of Granulocytes and Monocytes by Multiparametric Flow Cytometry: A Prospective Study of a 1-Tube Panel Strategy for Diagnosis and Prognosis of Patients with MDS. Cytom. B Clin. Cytom. 2020, 98, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Talati, C.; Zhang, L.; Shaheen, G.; Kuykendall, A.; Ball, M.; Zhang, Q.; Lancet, J.E.; Zuckerman, K.S.; List, A.F.; Komrokji, R.; et al. Monocyte Subset Analysis Accurately Distinguishes CMML from MDS and Is Associated with a Favorable MDS Prognosis. Blood 2017, 129, 1881–1883. [Google Scholar] [CrossRef] [PubMed]
- van de Loosdrecht, A.A.; Kern, W.; Porwit, A.; Valent, P.; Kordasti, S.; Cremers, E.; Alhan, C.; Duetz, C.; Dunlop, A.; Hobo, W.; et al. Clinical Application of Flow Cytometry in Patients with Unexplained Cytopenia and Suspected Myelodysplastic Syndrome: A Report of the European LeukemiaNet International MDS-Flow Cytometry Working Group. Cytom. B Clin. Cytom. 2023, 104, 77–86. [Google Scholar] [CrossRef]
- Maynadié, M.; Picard, F.; Husson, B.; Chatelain, B.; Cornet, Y.; Le Roux, G.L.; Campos, L.; Dromelet, A.; Lepelley, P.; Jouault, H.; et al. Immunophenotypic Clustering of Myelodysplastic Syndromes. Blood 2002, 100, 2349–2356. [Google Scholar] [CrossRef]
- Wangen, J.R.; Eidenschink Brodersen, L.; Stolk, T.T.; Wells, D.A.; Loken, M.R. Assessment of Normal Erythropoiesis by Flow Cytometry: Important Considerations for Specimen Preparation. Int. J. Lab. Hematol. 2014, 36, 184–196. [Google Scholar] [CrossRef]
- Porwit, A.; Béné, M.C.; Duetz, C.; Matarraz, S.; Oelschlaegel, U.; Westers, T.M.; Wagner-Ballon, O.; Kordasti, S.; Valent, P.; Preijers, F.; et al. Multiparameter Flow Cytometry in the Evaluation of Myelodysplasia: Analytical Issues: Recommendations from the European LeukemiaNet/International Myelodysplastic Syndrome Flow Cytometry Working Group. Cytom. B Clin. Cytom. 2023, 104, 27–50. [Google Scholar] [CrossRef]
- Bradbury, C.; Houlton, A.E.; Akiki, S.; Gregg, R.; Rindl, M.; Khan, J.; Ward, J.; Khan, N.; Griffiths, M.; Nagra, S.; et al. Prognostic Value of Monitoring a Candidate Immunophenotypic Leukaemic Stem/Progenitor Cell Population in Patients Allografted for Acute Myeloid Leukaemia. Leukemia 2015, 29, 988–991. [Google Scholar] [CrossRef]
- Li, S.Q.; Xu, L.P.; Wang, Y.; Zhang, X.H.; Chen, H.; Chen, Y.H.; Wang, F.R.; Han, W.; Sun, Y.Q.; Yan, C.H.; et al. An LSC-Based MRD Assay to Complement the Traditional MFC Method for Prediction of AML Relapse: A Prospective Study. Blood 2022, 140, 516–520. [Google Scholar] [CrossRef]
- Gorczyca, W.; Sun, Z.-Y.; Cronin, W.; Li, X.; Mau, S.; Tugulea, S. Immunophenotypic Pattern of Myeloid Populations by Flow Cytometry Analysis. Methods Cell Biol. 2011, 103, 221–266. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Escribano, L.; Födinger, M.; Hartmann, K.; Brockow, K.; Castells, M.; Sperr, W.R.; Kluin-Nelemans, H.C.; Hamdy, N.A.T.; et al. Standards and Standardization in Mastocytosis: Consensus Statements on Diagnostics, Treatment Recommendations and Response Criteria. Eur. J. Clin. Investig. 2007, 37, 435–453. [Google Scholar] [CrossRef] [PubMed]
- Rose, G.S.; Liu, H. Dysplastic Agranular Basophils Identified by Flow Cytometry. Cytom. B Clin. Cytom. 2021, 100, 606–608. [Google Scholar] [CrossRef] [PubMed]
- Loken, M.R. Residual Disease in AML, a Target That Can Move in More than One Direction. Cytom. B Clin. Cytom. 2014, 86, 15–17. [Google Scholar] [CrossRef]
- Grimwade, D.; Freeman, S.D. Defining Minimal Residual Disease in Acute Myeloid Leukemia: Which Platforms Are Ready for “Prime Time”? Blood 2014, 124, 3345–3355. [Google Scholar] [CrossRef] [PubMed]
- Ko, B.S.; Wang, Y.F.; Li, J.L.; Li, C.C.; Weng, P.F.; Hsu, S.C.; Hou, H.A.; Huang, H.H.; Yao, M.; Lin, C.T.; et al. Clinically Validated Machine Learning Algorithm for Detecting Residual Diseases with Multicolor Flow Cytometry Analysis in Acute Myeloid Leukemia and Myelodysplastic Syndrome. EBioMedicine 2018, 37, 91–100. [Google Scholar] [CrossRef]
- van Rhenen, A.; Moshaver, B.; Kelder, A.; Feller, N.; Nieuwint, A.W.M.; Zweegman, S.; Ossenkoppele, G.J.; Schuurhuis, G.J. Aberrant Marker Expression Patterns on the CD34+CD38- Stem Cell Compartment in Acute Myeloid Leukemia Allows to Distinguish the Malignant from the Normal Stem Cell Compartment Both at Diagnosis and in Remission. Leukemia 2007, 21, 1700–1707. [Google Scholar] [CrossRef] [PubMed]
- Terwijn, M.; Zeijlemaker, W.; Kelder, A.; Rutten, A.P.; Snel, A.N.; Scholten, W.J.; Pabst, T.; Verhoef, G.; Löwenberg, B.; Zweegman, S.; et al. Leukemic Stem Cell Frequency: A Strong Biomarker for Clinical Outcome in Acute Myeloid Leukemia. PLoS ONE 2014, 9, e107587. [Google Scholar] [CrossRef]
- Hanekamp, D.; Denys, B.; Kaspers, G.J.L.; te Marvelde, J.G.; Schuurhuis, G.J.; De Haas, V.; De Moerloose, B.; de Bont, E.S.; Zwaan, C.M.; de Jong, A.; et al. Leukaemic Stem Cell Load at Diagnosis Predicts the Development of Relapse in Young Acute Myeloid Leukaemia Patients. Br. J. Haematol. 2018, 183, 512–516. [Google Scholar] [CrossRef]
- Zeijlemaker, W.; Grob, T.; Meijer, R.; Hanekamp, D.; Kelder, A.; Carbaat-Ham, J.C.; Oussoren-Brockhoff, Y.J.M.; Snel, A.N.; Veldhuizen, D.; Scholten, W.J.; et al. CD34+CD38− Leukemic Stem Cell Frequency to Predict Outcome in Acute Myeloid Leukemia. Leukemia 2019, 33, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Orfao, A.; Matarraz, S.; Pérez-Andrés, M.; Almeida, J.; Teodosio, C.; Berkowska, M.A.; van Dongen, J.J.M. EuroFlow Immunophenotypic Dissection of Normal Hematopoiesis. J. Immunol. Methods 2019, 475, 112684. [Google Scholar] [CrossRef]
- Van Dongen, J.J.M.; Lhermitte, L.; Böttcher, S.; Almeida, J.; Van Der Velden, V.H.J.; Flores-Montero, J.; Rawstron, A.; Asnafi, V.; Lécrevisse, Q.; Lucio, P.; et al. EuroFlow Antibody Panels for Standardized N-Dimensional Flow Cytometric Immunophenotyping of Normal, Reactive and Malignant Leukocytes. Leukemia 2012, 26, 1908–1975. [Google Scholar] [CrossRef] [PubMed]
- Aanei, C.-M.; Veyrat-Masson, R.; Rigollet, L.; Stagnara, J.; Tavernier Tardy, E.; Daguenet, E.; Guyotat, D.; Campos Catafal, L. Advanced Flow Cytometry Analysis Algorithms for Optimizing the Detection of “Different From Normal” Immunophenotypes in Acute Myeloid Blasts. Front. Cell Dev. Biol. 2021, 9, 735518. [Google Scholar] [CrossRef] [PubMed]
- van der Velden, V.H.J.; Preijers, F.; Johansson, U.; Westers, T.M.; Dunlop, A.; Porwit, A.; Béné, M.C.; Valent, P.; te Marvelde, J.; Wagner-Ballon, O.; et al. Flow Cytometric Analysis of Myelodysplasia: Pre-Analytical and Technical Issues—Recommendations from the European LeukemiaNet. Cytom. B Clin. Cytom. 2023, 104, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T.R.; Naim, I.; Rebhahn, J.; Datta, S.; Cavenaugh, J.S.; Weaver, J.M.; Sharma, G. SWIFT-Scalable Clustering for Automated Identification of Rare Cell Populations in Large, High-Dimensional Flow Cytometry Datasets, Part 2: Biological Evaluation. Cytom. Part. A 2014, 85, 422–433. [Google Scholar] [CrossRef]
- Naim, I.; Datta, S.; Rebhahn, J.; Cavenaugh, J.S.; Mosmann, T.R.; Sharma, G. SWIFT-Scalable Clustering for Automated Identification of Rare Cell Populations in Large, High-Dimensional Flow Cytometry Datasets, Part 1: Algorithm Design. Cytom. Part. A 2014, 85, 408–421. [Google Scholar] [CrossRef]
- Mair, F.; Hartmann, F.J.; Mrdjen, D.; Tosevski, V.; Krieg, C.; Becher, B. The End of Gating? An Introduction to Automated Analysis of High Dimensional Cytometry Data. Eur. J. Immunol. 2016, 46, 34–43. [Google Scholar] [CrossRef]
- Toedling, J.; Rhein, P.; Ratei, R.; Karawajew, L.; Spang, R. Automated In-Silico Detection of Cell Populations in Flow Cytometry Readouts and Its Application to Leukemia Disease Monitoring. BMC Bioinform. 2006, 7, 282. [Google Scholar] [CrossRef]
- Dekker, S.E.; Rea, D.; Cayuela, J.-M.; Arnhardt, I.; Leonard, J.; Heuser, M. Using Measurable Residual Disease to Optimize Management of AML, ALL, and Chronic Myeloid Leukemia. Am. Soc. Clin. Oncol. Educ. Book. 2023, 43, e390010. [Google Scholar] [CrossRef]
- Woll, P.S.; Jacobsen, S.E.W. Stem Cell Concepts in Myelodysplastic Syndromes: Lessons and Challenges. J. Intern. Med. 2021, 289, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Ngai, L.L.; Veenstra, C.R.; Gradowska, P.; Kelder, A.; Scholten, W.; Snel, A.N.; Tettero, J.M.; Bachas, C.; Breems, D.; Fischer, T.; et al. Prognostic Value of Measurable Residual Disease in High-Risk Myelodysplastic Syndromes with Intensive Chemotherapy Treatment: Analysis of HOVON-SAKK Studies. Blood 2023, 142, 323. [Google Scholar] [CrossRef]
- Shadman, M.; Mawad, R.; Dean, C.; Chen, T.L.; Shannon-Dorcy, K.; Sandhu, V.; Hendrie, P.C.; Scott, B.L.; Walter, R.B.; Becker, P.S.; et al. Idarubicin, Cytarabine, and Pravastatin as Induction Therapy for Untreated Acute Myeloid Leukemia and High-Risk Myelodysplastic Syndrome. Am. J. Hematol. 2015, 90, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.Y.; Wei, Z.-L.; Xu, Y.J.; Shi, J.M.; Yi, H.; Lai, Y.R.; Jiang, E.L.; Wang, S.B.; Wu, T.; Gao, L.; et al. Poor Pretransplantation Minimal Residual Disease Clearance as an Independent Prognostic Risk Factor for Survival in Myelodysplastic Syndrome with Excess Blasts: A Multicenter, Retrospective Cohort Study. Cancer 2023, 129, 2013–2022. [Google Scholar] [CrossRef]
- Bader, P.; Niemeyer, C.; Weber, G.; Coliva, T.; Rossi, V.; Kreyenberg, H.; Gerecke, A.; Biondi, A. WT1 Gene Expression: Useful Marker for Minimal Residual Disease in Childhood Myelodysplastic Syndromes and Juvenile Myelo-Monocytic Leukemia? Eur. J. Haematol. 2004, 73, 25–28. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, L.; Wang, J.; Cao, Z.; Zhao, Z. The Wilms’ Tumor Gene-1 Is a Prognostic Factor in Myelodysplastic Syndrome: A Meta Analysis. Oncotarget 2018, 9, 16205–16212. [Google Scholar] [CrossRef]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Bève, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt Plus Azacitidine in TP53-Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Phase II Study by the Groupe Francophone Des Myélodysplasies (GFM). J. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef]
- Sallman, D.A.; Komrokji, R.S.; DeZern, A.E.; Sebert, M.; Garcia-Manero, G.; Rahmé, R.; Steensma, D.P.; Lehmann che, J.; Roboz, G.J.; Madelaine, I.; et al. Long Term Follow-up and Combined Phase 2 Results of Eprenetapopt (APR-246) and Azacitidine (AZA) in Patients with TP53 Mutant Myelodysplastic Syndromes (MDS) and Oligoblastic Acute Myeloid Leukemia (AML). Blood 2021, 138, 246. [Google Scholar] [CrossRef]
- Sallman, D.A.; Dezern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Mo, Q.; Sekeres, A.; Cluzeau, T.; Sweet, K.L.; List, A.F.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef]
- Dillon, L.W.; Gui, G.; Logan, B.R.; Fei, M.; Ghannam, J.; Li, Y.; Licon, A.; Alyea, E.P.; Bashey, A.; Devine, S.M.; et al. Impact of Conditioning Intensity and Genomics on Relapse After Allogeneic Transplantation for Patients with Myelodysplastic Syndrome. JCO Precis. Oncol. 2021, 5, 265–274. [Google Scholar] [CrossRef]
- Eladl, E.; Tremblay-Lemay, R.; Rastgoo, N.; Musani, R.; Chen, W.; Liu, A.; Chang, H. Role of CD47 in Hematological Malignancies. J. Hematol. Oncol. 2020, 13, 96. [Google Scholar] [CrossRef]
- Sallman, D.A.; Al Malki, M.M.; Asch, A.S.; Wang, E.S.; Jurcic, J.G.; Bradley, T.J.; Flinn, I.W.; Pollyea, D.A.; Kambhampati, S.; Tanaka, T.N.; et al. Magrolimab in Combination with Azacitidine in Patients with Higher-Risk Myelodysplastic Syndromes: Final Results of a Phase Ib Study. J. Clin. Oncol. 2023, 41, 2815–2826. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Stevenson, K.E.; Caughey, B.; Lindsley, R.C.; Mar, B.G.; Stojanov, P.; Getz, G.; Steensma, D.P.; Ritz, J.; Soiffer, R.; et al. Somatic Mutations Predict Poor Outcome in Patients with Myelodysplastic Syndrome after Hematopoietic Stem-Cell Transplantation. J. Clin. Oncol. 2014, 32, 2691–2698. [Google Scholar] [CrossRef] [PubMed]
- Kharfan-Dabaja, M.A.; Komrokji, R.S.; Zhang, Q.; Kumar, A.; Tsalatsanis, A.; Perkins, J.; Nishihori, T.; Field, T.; Al Ali, N.; Mishra, A.; et al. TP53 and IDH2 Somatic Mutations Are Associated with Inferior Overall Survival After Allogeneic Hematopoietic Cell Transplantation for Myelodysplastic Syndrome. Clin. Lymphoma Myeloma Leuk. 2017, 17, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Yeh, P.; Dickinson, M.; Ftouni, S.; Hunter, T.; Sinha, D.; Wong, S.Q.; Agarwal, R.; Vedururu, R.; Doig, K.; Fong, C.Y.; et al. Molecular Disease Monitoring Using Circulating Tumor DNA in Myelodysplastic Syndromes. Blood 2017, 129, 1685–1690. [Google Scholar] [CrossRef] [PubMed]
- Hunter, A.M.; Komrokji, R.S.; Yun, S.; Al Ali, N.; Chan, O.; Song, J.; Hussaini, M.; Talati, C.; Sweet, K.L.; Lancet, J.E.; et al. Baseline and Serial Molecular Profiling Predicts Outcomes with Hypomethylating Agents in Myelodysplastic Syndromes. Blood Adv. 2021, 5, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Festuccia, M.; Deeg, H.J.; Gooley, T.A.; Baker, K.; Wood, B.L.; Fang, M.; Sandmaier, B.M.; Scott, B.L. Minimal Identifiable Disease and the Role of Conditioning Intensity in Hematopoietic Cell Transplantation for Myelodysplastic Syndrome and Acute Myelogenous Leukemia Evolving from Myelodysplastic Syndrome. Biol. Blood Marrow Transplant. 2016, 22, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Craddock, C.; Jackson, A.; Loke, J.; Siddique, S.; Hodgkinson, A.; Mason, J.; Andrew, G.; Nagra, S.; Malladi, R.; Peniket, A.; et al. Augmented Reduced-Intensity Regimen Does Not Improve Postallogeneic Transplant Outcomes in Acute Myeloid Leukemia. J. Clin. Oncol. 2020, 39, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Bernal, T.; Diez-Campelo, M.; Godoy, V.; Rojas, S.; Colado, E.; Alcoceba, M.; González, M.; Vidriales, B.; Sánchez-Guijo, F.M.; López-Corral, L.; et al. Role of Minimal Residual Disease and Chimerism after Reduced-Intensity and Myeloablative Allo-Transplantation in Acute Myeloid Leukemia and High-Risk Myelodysplastic Syndrome. Leuk. Res. 2014, 38, 551–556. [Google Scholar] [CrossRef]
- Mo, X.D.; Qin, Y.Z.; Zhang, X.H.; Xu, L.P.; Wang, Y.; Yan, C.H.; Chen, H.; Chen, Y.H.; Han, W.; Wang, F.R.; et al. Minimal Residual Disease Monitoring and Preemptive Immunotherapy in Myelodysplastic Syndrome after Allogeneic Hematopoietic Stem Cell Transplantation. Ann. Hematol. 2016, 95, 1233–1240. [Google Scholar] [CrossRef]
- Duncavage, E.J.; Jacoby, M.A.; Chang, G.S.; Miller, C.A.; Edwin, N.; Shao, J.; Elliott, K.; Robinson, J.; Abel, H.; Fulton, R.S.; et al. Mutation Clearance after Transplantation for Myelodysplastic Syndrome. N. Engl. J. Med. 2018, 379, 1028–1041. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.; Geyer, S.M.; Komrokji, R.S.; Al Ali, N.H.; Song, J.; Hussaini, M.; Sweet, K.L.; Lancet, J.E.; List, A.F.; Padron, E.; et al. Prognostic Significance of Serial Molecular Annotation in Myelodysplastic Syndromes (MDS) and Secondary Acute Myeloid Leukemia (SAML). Leukemia 2021, 35, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Yokoyama, K.; Shimizu, E.; Yusa, N.; Kondoh, K.; Ogawa, M.; Takei, T.; Kobayashi, A.; Ito, M.; Isobe, M.; et al. Prognostic Impact of Circulating Tumor DNA Status Post-Allogeneic Hematopoietic Stem Cell Transplantation in AML and MDS. Blood 2019, 133, 2682–2695. [Google Scholar] [CrossRef] [PubMed]
- Loke, J.; McCarthy, N.; Jackson, A.; Siddique, S.; Hodgkinson, A.; Mason, J.; Crawley, C.; Gilleece, M.; Peniket, A.; Protheroe, R.; et al. Posttransplant MRD and T-Cell Chimerism Status Predict Outcomes in Patients Who Received Allografts for AML/MDS. Blood Adv. 2023, 7, 3666–3676. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, M.A.; Duncavage, E.J.; Chang, G.S.; Miller, C.A.; Shao, J.; Elliott, K.; Robinson, J.; Fulton, R.S.; Fronick, C.C.; O’Laughlin, M.; et al. Subclones Dominate at MDS Progression Following Allogeneic Hematopoietic Cell Transplant. JCI Insight 2018, 3, e98962. [Google Scholar] [CrossRef]
- Platzbecker, U.; Wermke, M.; Radke, J.; Oelschlaegel, U.; Seltmann, F.; Kiani, A.; Klut, I.M.; Knoth, H.; Röllig, C.; Schetelig, J.; et al. Azacitidine for Treatment of Imminent Relapse in MDS or AML Patients after Allogeneic HSCT: Results of the RELAZA Trial. Leukemia 2012, 26, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Platzbecker, U.; Middeke, J.M.; Sockel, K.; Herbst, R.; Wolf, D.; Baldus, C.D.; Oelschlägel, U.; Mütherig, A.; Fransecky, L.; Noppeney, R.; et al. Measurable Residual Disease-Guided Treatment with Azacitidine to Prevent Haematological Relapse in Patients with Myelodysplastic Syndrome and Acute Myeloid Leukaemia (RELAZA2): An Open-Label, Multicentre, Phase 2 Trial. Lancet Oncol. 2018, 19, 1668–1679. [Google Scholar] [CrossRef] [PubMed]
- Ogbue, O.; Unlu, S.; Ibodeng, G.-O.; Singh, A.; Durmaz, A.; Visconte, V.; Molina, J.C. Single-Cell Next-Generation Sequencing to Monitor Hematopoietic Stem-Cell Transplantation: Current Applications and Future Perspectives. Cancers 2023, 15, 2477. [Google Scholar] [CrossRef]
- Shapiro, R.M.; Kim, D.D.H. Next-Generation Sequencing-Based Minimal Residual Disease Monitoring in Patients Receiving Allogeneic Hematopoietic Stem Cell Transplantation for Acute Myeloid Leukemia or Myelodysplastic Syndrome. Curr. Opin. Hematol. 2018, 25, 425–432. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Deeb, G.; Deeb, K.K.; Vale, C.; Peker Barclift, D.; Papadantonakis, N. Measurable (Minimal) Residual Disease in Myelodysplastic Neoplasms (MDS): Current State and Perspectives. Cancers 2024, 16, 1503. https://doi.org/10.3390/cancers16081503
Zhang L, Deeb G, Deeb KK, Vale C, Peker Barclift D, Papadantonakis N. Measurable (Minimal) Residual Disease in Myelodysplastic Neoplasms (MDS): Current State and Perspectives. Cancers. 2024; 16(8):1503. https://doi.org/10.3390/cancers16081503
Chicago/Turabian StyleZhang, Linsheng, George Deeb, Kristin K. Deeb, Colin Vale, Deniz Peker Barclift, and Nikolaos Papadantonakis. 2024. "Measurable (Minimal) Residual Disease in Myelodysplastic Neoplasms (MDS): Current State and Perspectives" Cancers 16, no. 8: 1503. https://doi.org/10.3390/cancers16081503