

The Genomic, Transcriptomic, and Immunologic Landscape of HRAS Mutations in Solid Tumors
 , , , , , , , ,
, , , , , , , ,  , ,
, , 
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Cohort Information and Tumor Types
2.2. Next-Generation Sequencing-592 Gene Panel (NGS-592)/Whole Exome Sequencing (WES)
2.3. Identification of Genetic Variants
2.4. Whole Transcriptome Sequencing
2.5. Immune Signatures
2.6. Microsatellite Insability/Mismatch Repair Deficiency (MSI-h/MMR) Status
2.7. Tumor Mutational Burden (TMB)
2.8. Immunohistochemistry (IHC)
2.9. Clinical Outcomes
2.10. Statistics and Reproducibility
3. Results
3.1. Prevlance of HRASmt and Characteristics of Cohort
3.2. HRASmt Co-Alterations and Biomarkers
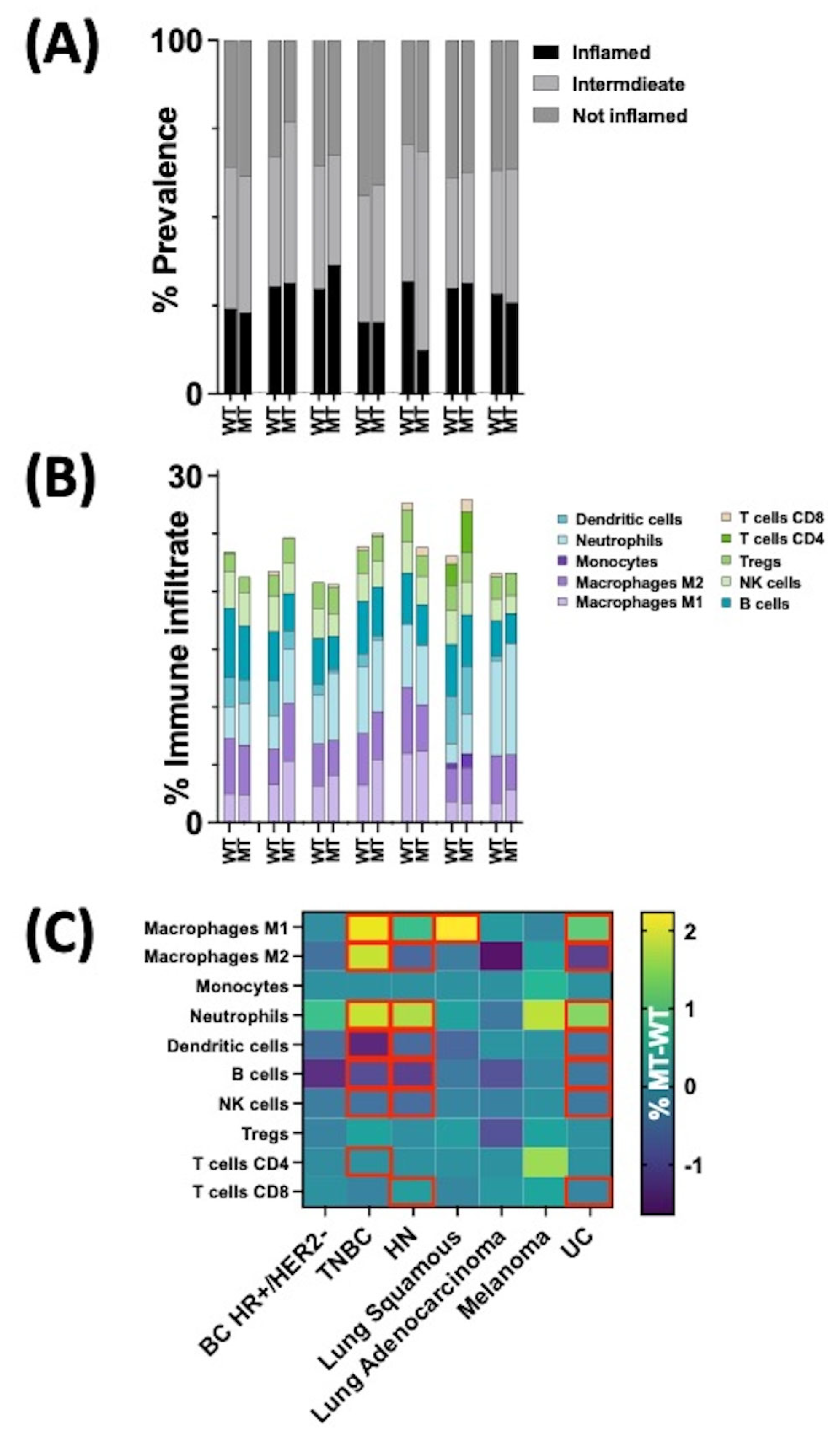
3.3. Tumor Microenvironment (TME) of HRASmt
3.4. Clinical Outcomes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A18V, A59T, E143K, G12A, G12C, G12D, G12N, G12R, G12S, G12V, G13C, G13D, G13K, G13N, G13R, G13V, G60D, I55M, K117N, Q150, Q61H, Q61K, Q61L, Q61R, R149fs, S189C, T2M, T50M, T58I, V45A |
| Q61 | G12 | G13 | Misc | N of Mutations | N of Mutant Tumors | |
|---|---|---|---|---|---|---|
| UC | 63 | 34 | 37 | 4 | 138 | 138 |
| BC HR+/HER2- | 9 | 5 | 1 | 2 | 17 | 17 |
| TNBC | 24 | 15 | 13 | 2 | 54 | 54 |
| NSCLC Squamous | 23 | 23 | 41 | 3 | 90 | 157 |
| NSCLC Adenocarcinoma | 18 | 3 | 12 | 1 | 34 | 50 |
| Melanoma | 16 | 10 | 22 | 7 | 55 | 55 |
| HNSCC | 25 | 41 | 42 | 3 | 112 | 110 |
References
- Marín-Ramos, N.I.; Ortega-Gutiérrez, S.; López-Rodríguez, M.L. Blocking Ras Inhibition as an Antitumor Strategy. Semin. Cancer Biol. 2019, 54, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS Oncogenes: Weaving a Tumorigenic Web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Vander Heiden, M.G.; McCormick, F. The Metabolic Landscape of RAS-Driven Cancers from Biology to Therapy. Nat. Cancer 2021, 2, 271–283. [Google Scholar] [CrossRef] [PubMed]
- De Langen, A.J.; Johnson, M.L.; Mazieres, J.; Dingemans, A.-M.C.; Mountzios, G.; Pless, M.; Wolf, J.; Schuler, M.; Lena, H.; Skoulidis, F.; et al. Sotorasib versus Docetaxel for Previously Treated Non-Small-Cell Lung Cancer with KRASG12C Mutation: A Randomised, Open-Label, Phase 3 Trial. Lancet 2023, 401, 733–746. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Riely, G.J.; Gadgeel, S.M.; Heist, R.S.; Ou, S.-H.I.; Pacheco, J.M.; Johnson, M.L.; Sabari, J.K.; Leventakos, K.; Yau, E.; et al. Adagrasib in Non–Small-Cell Lung Cancer Harboring a KRASG12C Mutation. N. Engl. J. Med. 2022, 387, 120–131. [Google Scholar] [CrossRef]
- Strickler, J.H.; Satake, H.; George, T.J.; Yaeger, R.; Hollebecque, A.; Garrido-Laguna, I.; Schuler, M.; Burns, T.F.; Coveler, A.L.; Falchook, G.S.; et al. Sotorasib in KRAS p.G12C–Mutated Advanced Pancreatic Cancer. N. Engl. J. Med. 2023, 388, 33–43. [Google Scholar] [CrossRef]
- Sebti, S.M.; Der, C.J. Searching for the Elusive Targets of Farnesyltransferase Inhibitors. Nat. Rev. Cancer 2003, 3, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Whyte, D.B.; Kirschmeier, P.; Hockenberry, T.N.; Nunez-Oliva, I.; James, L.; Catino, J.J.; Bishop, W.R.; Pai, J.K. K- and N-Ras Are Geranylgeranylated in Cells Treated with Farnesyl Protein Transferase Inhibitors. J. Biol. Chem. 1997, 272, 14459–14464. [Google Scholar] [CrossRef] [PubMed]
- Kodaz, H. Frequency of RAS Mutations (KRAS, NRAS, HRAS) in Human Solid Cancer. Eurasian J. Med. Oncol. 2017, 7, 22931. [Google Scholar] [CrossRef]
- Crona, J.; Delgado Verdugo, A.; Maharjan, R.; Stålberg, P.; Granberg, D.; Hellman, P.; Björklund, P. Somatic Mutations in H-RAS in Sporadic Pheochromocytoma and Paraganglioma Identified by Exome Sequencing. J. Clin. Endocrinol. Metab. 2013, 98, E1266–E1271. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.L.; Brana, I.; Haddad, R.; Bauman, J.; Bible, K.; Oosting, S.; Wong, D.J.; Ahn, M.-J.; Boni, V.; Even, C.; et al. Tipifarnib in Head and Neck Squamous Cell Carcinoma with HRAS Mutations. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 1856–1864. [Google Scholar] [CrossRef]
- Wei, S.; Krause, H.B.; Geynisman, D.M.; Elliott, A.; Kutikov, A.; Uzzo, R.G.; Pei, J.; Barata, P.; Carneiro, B.; Heath, E.; et al. Molecular Characterization of TFE3-Rearranged Renal Cell Carcinoma: A Comparative Study With Papillary and Clear Cell Renal Cell Carcinomas. Mod. Pathol. 2024, 37, 100404. [Google Scholar] [CrossRef]
- Finotello, F.; Mayer, C.; Plattner, C.; Laschober, G.; Rieder, D.; Hackl, H.; Krogsdam, A.; Loncova, Z.; Posch, W.; Wilflingseder, D.; et al. Molecular and Pharmacological Modulators of the Tumor Immune Contexture Revealed by Deconvolution of RNA-Seq Data. Genome Med. 2019, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Bao, R.; Stapor, D.; Luke, J.J. Molecular Correlates and Therapeutic Targets in T Cell-Inflamed versus Non-T Cell-Inflamed Tumors across Cancer Types. Genome Med. 2020, 12, 90. [Google Scholar] [CrossRef]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H.; et al. Association of Tumour Mutational Burden with Outcomes in Patients with Advanced Solid Tumours Treated with Pembrolizumab: Prospective Biomarker Analysis of the Multicohort, Open-Label, Phase 2 KEYNOTE-158 Study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Merino, D.M.; McShane, L.M.; Fabrizio, D.; Funari, V.; Chen, S.-J.; White, J.R.; Wenz, P.; Baden, J.; Barrett, J.C.; Chaudhary, R.; et al. Establishing Guidelines to Harmonize Tumor Mutational Burden (TMB): In Silico Assessment of Variation in TMB Quantification across Diagnostic Platforms: Phase I of the Friends of Cancer Research TMB Harmonization Project. J. Immunother. Cancer 2020, 8, e000147. [Google Scholar] [CrossRef]
- Zimmer, K.; Kocher, F.; Untergasser, G.; Kircher, B.; Amann, A.; Baca, Y.; Xiu, J.; Korn, W.M.; Berger, M.D.; Lenz, H.-J.; et al. PBRM1 Mutations Might Render a Subtype of Biliary Tract Cancers Sensitive to Drugs Targeting the DNA Damage Repair System. NPJ Precis. Oncol. 2023, 7, 64. [Google Scholar] [CrossRef] [PubMed]
- Necchi, A.; Madison, R.; Pal, S.K.; Ross, J.S.; Agarwal, N.; Sonpavde, G.; Joshi, M.; Yin, M.; Miller, V.A.; Grivas, P.; et al. Comprehensive Genomic Profiling of Upper-Tract and Bladder Urothelial Carcinoma. Eur. Urol. Focus 2021, 7, 1339–1346. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network Genome Sequencing Centre. Comprehensive Genomic Characterization of Head and Neck Squamous Cell Carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Coleman, N.; Marcelo, K.L.; Hopkins, J.F.; Khan, N.I.; Du, R.; Hong, L.; Park, E.; Balsara, B.; Leoni, M.; Pickering, C.; et al. HRAS Mutations Define a Distinct Subgroup in Head and Neck Squamous Cell Carcinoma. JCO Precis. Oncol. 2023, 7, e2200211. [Google Scholar] [CrossRef]
- Wan, X.; Liu, R.; Li, Z. The Prognostic Value of HRAS mRNA Expression in Cutaneous Melanoma. BioMed Res. Int. 2017, 2017, 5356737. [Google Scholar] [CrossRef]
- Mathiot, L.; Herbreteau, G.; Robin, S.; Fenat, C.; Bennouna, J.; Blanquart, C.; Denis, M.; Pons-Tostivint, E. HRAS Q61L Mutation as a Possible Target for Non-Small Cell Lung Cancer: Case Series and Review of Literature. Curr. Oncol. 2022, 29, 3748–3758. [Google Scholar] [CrossRef] [PubMed]
- Pareja, F.; Toss, M.S.; Geyer, F.C.; da Silva, E.M.; Vahdatinia, M.; Sebastiao, A.P.M.; Selenica, P.; Szatrowski, A.; Edelweiss, M.; Wen, H.Y.; et al. Immunohistochemical Assessment of HRAS Q61R Mutations in Breast Adenomyoepitheliomas. Histopathology 2020, 76, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, L.G. C-Src Activation by ErbB2 Leads to Attachment-Independent Growth of Human Breast Epithelial Cells. Biochem. Biophys. Res. Commun. 1998, 250, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Graff, S.L.; Yan, F.; Abdou, Y. Newly Approved and Emerging Agents in HER2-Positive Metastatic Breast Cancer. Clin. Breast Cancer 2023, 23, e380–e393. [Google Scholar] [CrossRef] [PubMed]
- Bièche, I.; Coussy, F.; El-Botty, R.; Vacher, S.; Château-Joubert, S.; Dahmani, A.; Montaudon, E.; Reyes, C.; Gentien, D.; Reyal, F.; et al. HRAS Is a Therapeutic Target in Malignant Chemo-Resistant Adenomyoepithelioma of the Breast. J. Hematol. Oncol. 2021, 14, 143. [Google Scholar] [CrossRef] [PubMed]
- Pązik, M.; Michalska, K.; Żebrowska-Nawrocka, M.; Zawadzka, I.; Łochowski, M.; Balcerczak, E. Clinical Significance of HRAS and KRAS Genes Expression in Patients with Non-Small-Cell Lung Cancer—Preliminary Findings. BMC Cancer 2021, 21, 130. [Google Scholar] [CrossRef] [PubMed]
- Sfakianos, J.P.; Cha, E.K.; Iyer, G.; Scott, S.N.; Zabor, E.C.; Shah, R.H.; Ren, Q.; Bagrodia, A.; Kim, P.H.; Hakimi, A.A.; et al. Genomic Characterization of Upper Tract Urothelial Carcinoma. Eur. Urol. 2015, 68, 970–977. [Google Scholar] [CrossRef]
- Jung, J.; Liao, H.; Coker, S.A.; Liang, H.; Hancock, J.F.; Denicourt, C.; Venkatachalam, K. P53 Mitigates the Effects of Oncogenic HRAS in Urothelial Cells via the Repression of MCOLN1. iScience 2021, 24, 102701. [Google Scholar] [CrossRef]
- Scheffler, M.; Ihle, M.A.; Hein, R.; Merkelbach-Bruse, S.; Scheel, A.H.; Siemanowski, J.; Brägelmann, J.; Kron, A.; Abedpour, N.; Ueckeroth, F.; et al. K-Ras Mutation Subtypes in NSCLC and Associated Co-Occuring Mutations in Other Oncogenic Pathways. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2019, 14, 606–616. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef]
- Isnaldi, E.; Garuti, A.; Cirmena, G.; Scabini, S.; Rimini, E.; Ferrando, L.; Lia, M.; Murialdo, R.; Tixi, L.; Carminati, E.; et al. Clinico-Pathological Associations and Concomitant Mutations of the RAS/RAF Pathway in Metastatic Colorectal Cancer. J. Transl. Med. 2019, 17, 137. [Google Scholar] [CrossRef]
- Boulalas, I.; Zaravinos, A.; Karyotis, I.; Delakas, D.; Spandidos, D.A. Activation of RAS Family Genes in Urothelial Carcinoma. J. Urol. 2009, 181, 2312–2319. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Guo, T.; Niu, D.; Zhu, Y.; Ren, W.; Yao, Q.; Huang, X.; Feng, Q.; Wang, T.; Ma, X.; et al. Clinical Significance and Interrelations of PD-L1 Expression, Ki-67 Index, and Molecular Alterations in Sporadic Medullary Thyroid Carcinoma from a Chinese Population. Virchows Arch. 2022, 481, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Lyu, H.; Li, M.; Jiang, Z.; Liu, Z.; Wang, X. Correlate the TP53 Mutation and the HRAS Mutation with Immune Signatures in Head and Neck Squamous Cell Cancer. Comput. Struct. Biotechnol. J. 2019, 17, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor Mutational Load Predicts Survival after Immunotherapy across Multiple Cancer Types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Hanna, G.J.; Guenette, J.P.; Chau, N.G.; Sayehli, C.M.; Wilhelm, C.; Metcalf, R.; Wong, D.J.; Brose, M.; Razaq, M.; Pérez-Ruiz, E.; et al. Tipifarnib in Recurrent, Metastatic HRAS-mutant Salivary Gland Cancer. Cancer 2020, 126, 3972–3981. [Google Scholar] [CrossRef] [PubMed]
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.-R.; Cupissol, D.; et al. Platinum-Based Chemotherapy plus Cetuximab in Head and Neck Cancer. N. Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Odeniyide, P.; Yohe, M.E.; Pollard, K.; Vaseva, A.V.; Calizo, A.; Zhang, L.; Rodriguez, F.J.; Gross, J.M.; Allen, A.N.; Wan, X.; et al. Targeting Farnesylation as a Novel Therapeutic Approach in HRAS-Mutant Rhabdomyosarcoma. Oncogene 2022, 41, 2973–2983. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Chung, W.; Lee, H.-O.; Jeong, D.E.; Jo, A.; Lim, J.E.; Hong, J.H.; Nam, D.-H.; Jeong, B.C.; Park, S.H.; et al. Single-Cell RNA Sequencing Reveals the Tumor Microenvironment and Facilitates Strategic Choices to Circumvent Treatment Failure in a Chemorefractory Bladder Cancer Patient. Genome Med. 2020, 12, 47. [Google Scholar] [CrossRef]
- Lopes-Ventura, S.; Pojo, M.; Matias, A.T.; Moura, M.M.; Marques, I.J.; Leite, V.; Cavaco, B.M. The Efficacy of HRAS and CDK4/6 Inhibitors in Anaplastic Thyroid Cancer Cell Lines. J. Endocrinol. Investig. 2019, 42, 527–540. [Google Scholar] [CrossRef]
- Untch, B.R.; Dos Anjos, V.; Garcia-Rendueles, M.E.R.; Knauf, J.A.; Krishnamoorthy, G.P.; Saqcena, M.; Bhanot, U.K.; Socci, N.D.; Ho, A.L.; Ghossein, R.; et al. Tipifarnib Inhibits HRAS-Driven Dedifferentiated Thyroid Cancers. Cancer Res. 2018, 78, 4642–4657. [Google Scholar] [CrossRef]
- Rieke, D.T.; Schröder, S.; Schafhausen, P.; Blanc, E.; Zuljan, E.; von der Emde, B.; Beule, D.; Keller, U.; Keilholz, U.; Klinghammer, K. Targeted Treatment in a Case Series of AR+, HRAS/PIK3CA Co-Mutated Salivary Duct Carcinoma. Front. Oncol. 2023, 13, 1107134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.S.; Nagasaka, M. Spotlight on Sotorasib (AMG 510) for KRASG12C Positive Non-Small Cell Lung Cancer. Lung Cancer Targets Ther. 2021, 12, 115–122. [Google Scholar] [CrossRef]
- Liu, J.; Kang, R.; Tang, D. The KRAS-G12C Inhibitor: Activity and Resistance. Cancer Gene Ther. 2022, 29, 875–878. [Google Scholar] [CrossRef]
- Lito, P.; Solomon, M.; Li, L.-S.; Hansen, R.; Rosen, N. Allele-Specific Inhibitors Inactivate Mutant KRAS G12C by a Trapping Mechanism. Science 2016, 351, 604–608. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.-S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The Clinical KRAS(G12C) Inhibitor AMG 510 Drives Anti-Tumour Immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef]
- Gilardi, M.; Wang, Z.; Proietto, M.; Chillà, A.; Calleja-Valera, J.L.; Goto, Y.; Vanoni, M.; Janes, M.R.; Mikulski, Z.; Gualberto, A.; et al. Tipifarnib as a Precision Therapy for HRAS-Mutant Head and Neck Squamous Cell Carcinomas. Mol. Cancer Ther. 2020, 19, 1784–1796. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, M.K.; Curioni-Fontecedro, A.; Samaras, P.; Atrott, K.; Cosin-Roger, J.; Lang, S.; Scharl, M.; Rogler, G. Mutant HRAS as Novel Target for MEK and mTOR Inhibitors. Oncotarget 2015, 6, 42183–42196. [Google Scholar] [CrossRef]
- Tidyman, W.E.; Goodwin, A.F.; Maeda, Y.; Klein, O.D.; Rauen, K.A. MEK-Inhibitor-Mediated Rescue of Skeletal Myopathy Caused by Activating Hras Mutation in a Costello Syndrome Mouse Model. Dis. Model. Mech. 2022, 15, dmm049166. [Google Scholar] [CrossRef]
- Catalano, A.; Adlesic, M.; Kaltenbacher, T.; Klar, R.F.U.; Albers, J.; Seidel, P.; Brandt, L.P.; Hejhal, T.; Busenhart, P.; Röhner, N.; et al. Sensitivity and Resistance of Oncogenic RAS-Driven Tumors to Dual MEK and ERK Inhibition. Cancers 2021, 13, 1852. [Google Scholar] [CrossRef]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A.; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in Postmenopausal Hormone-Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Y.; Liu, W.T.; Wu, Z.F.; Chen, C.; Liu, J.Y.; Wu, G.N.; Yao, X.Q.; Liu, F.K.; Li, G. Identification of HRAS as Cancer-Promoting Gene in Gastric Carcinoma Cell Aggressiveness. Am. J. Cancer Res. 2016, 6, 1935–1948. [Google Scholar] [PubMed]





| UC | HRASmt | HRASwt | p-Value |
|---|---|---|---|
| Count (N) | 138 | 4467 | NA |
| Median Age [range] | 70 [32–>89] | 72 [18–>89] | 0.001 |
| Male | 66.6% (92/138) | 72.2% (3223/4467) | 0.158 |
| Female | 33.3% (46/138) | 27.8% (1244/4467) | |
| BC | |||
| Count (N) | 80 | 15763 | NA |
| Median Age [range] | 68 [41–>89] | 60 [19–>89] | 0.000 |
| Male | 1.3% (1/80) | 1.2% (191/15,763) | 0.624 |
| Female | 98.8% (79/80) | 98.8% (15,572/15,763) | |
| HR+/HER2- | 17 | 8057 | <0.001 |
| TNBC | 54 | 4403 | |
| HR+/HER2+ | 0 | 688 | |
| HR-/HER2+ | 0 | 522 | |
| NSCLC | |||
| Count (N) | 150 | 34,160 | NA |
| Median Age [range] | 72 [46–>89] (150) | 69 [0–>89] (34,160) | 0.004 |
| Male | 46% (69/150) | 50.0% (17,107/34,160) | 0.319 |
| Female | 54.0% (81/150) | 49.9% (17,053/34,160) | |
| Adenocarcinoma | 22.7% (34/150) | 57.1% (19,513/34,160) | 0.002 |
| Squamous | 60.0% (90/150) | 22.0% (7505/34,160) | |
| Other | 17.3% (26/150) | 20.9% (7142/34,160) | |
| Melanoma | |||
| Count (N) | 55 | 5162 | NA |
| Median Age [range] | 71 [39–>89] (55) | 67 [0–>89] (5162) | 0.043 |
| Male | 71% (39/55) | 62% (3211/5162) | 0.231 |
| Female | 29.1% (16/55) | 37.8% (1951/5162) | |
| HNSCC | |||
| Count (N) | 110 | 3797 | |
| Median Age [range] | 69 [33–>89] (110) | 64 [15–>89] (3797) | 0.001 |
| Male | 62.7% (69/110) | 77% (2922/3797) | 0.001 |
| Female | 37.3% (41/110) | 23.0% (875/3797) |
| HR | CI | CI | p | N HRASWT | N HRASMT | |
|---|---|---|---|---|---|---|
| BC HR+/HER2- | 1.417 | 0.589 | 3.407 | 0.434 | 5353 | 7 |
| TNBC | 0.903 | 0.593 | 1.374 | 0.632 | 3230 | 35 |
| HN | 1.564 | 1.158 | 2.11 | 0.003 | 2330 | 56 |
| Lung Squamous | 0.971 | 0.731 | 1.291 | 0.842 | 5893 | 65 |
| Lung Adenocarcinoma | 1.121 | 0.715 | 1.759 | 0.618 | 15,451 | 26 |
| Melanoma | 0.959 | 0.636 | 1.447 | 0.842 | 3990 | 42 |
| UC | 1.143 | 0.917 | 1.424 | 0.234 | 3598 | 117 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kareff, S.A.; Trabolsi, A.; Krause, H.B.; Samec, T.; Elliott, A.; Rodriguez, E.; Olazagasti, C.; Watson, D.C.; Bustos, M.A.; Hoon, D.S.B.; et al. The Genomic, Transcriptomic, and Immunologic Landscape of HRAS Mutations in Solid Tumors. Cancers 2024, 16, 1572. https://doi.org/10.3390/cancers16081572
Kareff SA, Trabolsi A, Krause HB, Samec T, Elliott A, Rodriguez E, Olazagasti C, Watson DC, Bustos MA, Hoon DSB, et al. The Genomic, Transcriptomic, and Immunologic Landscape of HRAS Mutations in Solid Tumors. Cancers. 2024; 16(8):1572. https://doi.org/10.3390/cancers16081572
Chicago/Turabian StyleKareff, Samuel A., Asaad Trabolsi, Harris B. Krause, Timothy Samec, Andrew Elliott, Estelamari Rodriguez, Coral Olazagasti, Dionysios C. Watson, Matias A. Bustos, Dave S. B. Hoon, and et al. 2024. "The Genomic, Transcriptomic, and Immunologic Landscape of HRAS Mutations in Solid Tumors" Cancers 16, no. 8: 1572. https://doi.org/10.3390/cancers16081572
APA StyleKareff, S. A., Trabolsi, A., Krause, H. B., Samec, T., Elliott, A., Rodriguez, E., Olazagasti, C., Watson, D. C., Bustos, M. A., Hoon, D. S. B., Graff, S. L., Antonarakis, E. S., Goel, S., Sledge, G., & Lopes, G. (2024). The Genomic, Transcriptomic, and Immunologic Landscape of HRAS Mutations in Solid Tumors. Cancers, 16(8), 1572. https://doi.org/10.3390/cancers16081572