

Drivers Underlying Metastasis and Relapse in Medulloblastoma and Targeting Strategies
 , , ,
, , ,
Abstract
Simple Summary
Abstract
1. Introduction
Metastatic Disease and Relapse Patterns in MB Subgroups
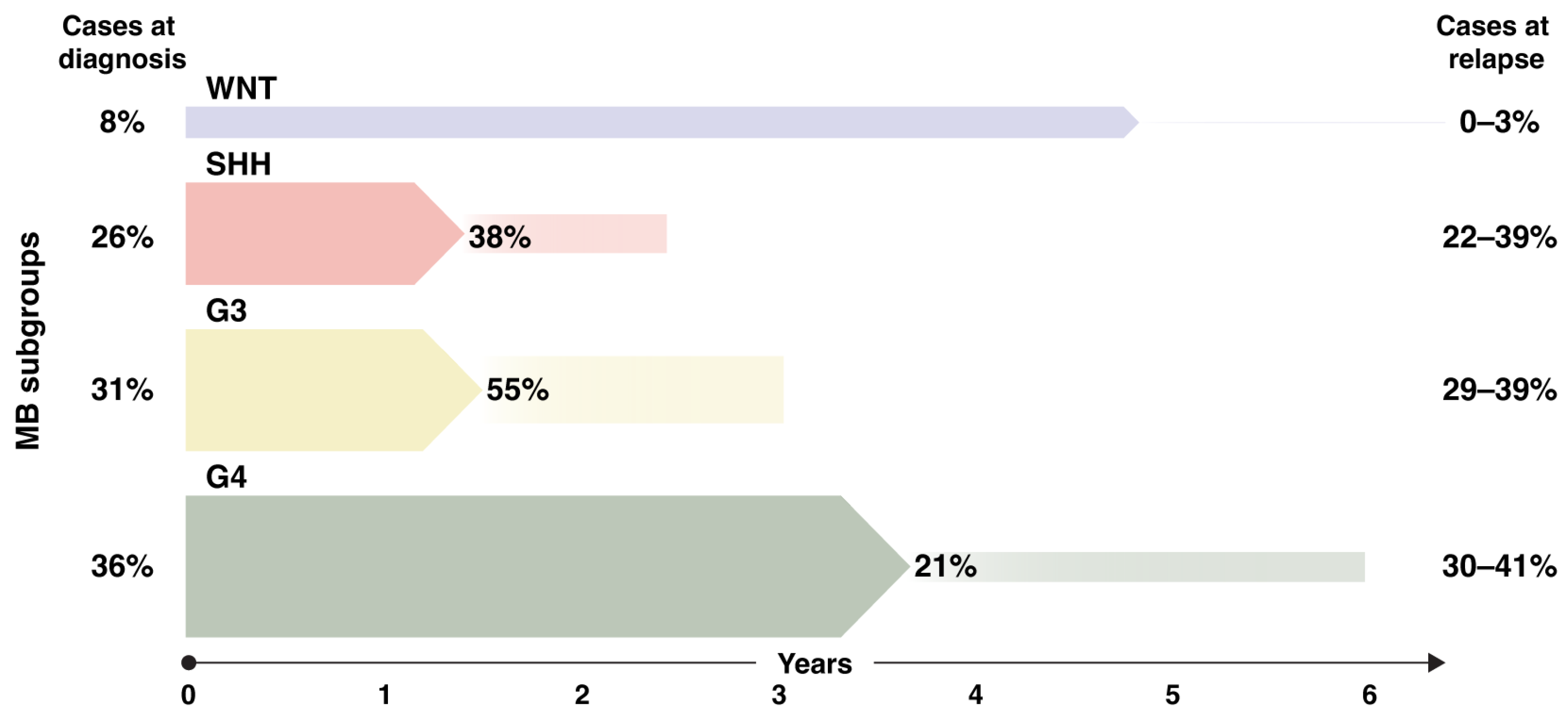
2. Biology of Medulloblastoma Metastasis and Relapse
2.1. Leptomeningeal Disease
2.2. Means to Follow Metastatic Cells and Tumor Relapse
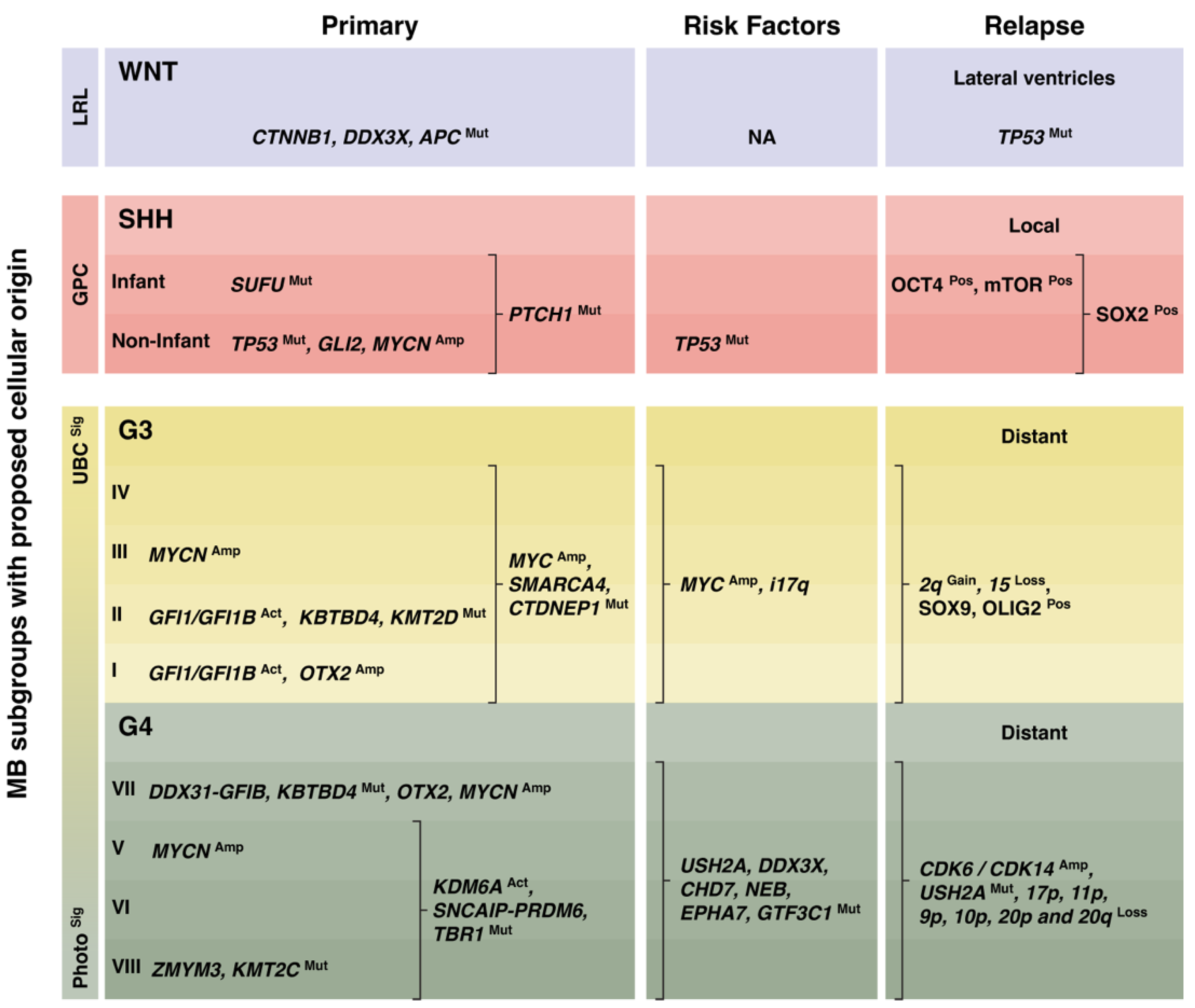
3. Molecular Signatures of Medulloblastoma Relapse
3.1. WNT Medulloblastoma
3.2. SHH Medulloblastoma
3.3. Group 3 Medulloblastoma
3.4. Group 4 Medulloblastoma
3.5. Group 3 and Group 4 Medulloblastoma Subtypes

{kind=link}
{kind=link}
| MB Model | Subgroup Resemblance | Model Type | Incidence | Latency (Months) | Metastasis/ LMD Rate | Refs. |
|---|---|---|---|---|---|---|
| mWnt-MB | Wnt | Orthotopic | 100% | average of 1.7 m | 0% | [57] |
| mWnt-MB, MycT58A | Wnt | Orthotopic | 100% | average of 0.5 m | 80% | [57] |
| Ptch1+/− | SHH | GEMM | 28%–39% | 6–8 m | 50% | [68] |
| Ptch1+/−, Math1-cre; LSL- Atoh1 | SHH | GEMM | 100% | 3–4 m | 100% | [70] |
| Ptch1+/−, Atoh1-creER;LSL- Atoh1 | SHH | GEMM | 100% | 6–7 m | 100% | [70] |
| Ptch1+/−, Math1-SB11/T2Onc transposon | SHH | GEMM | 97% | 2.5 m | 80% | [48] |
| RCAS Shh | SHH | Retroviral | 40% | 2–3 m | ~10% | [71] |
| RCAS Shh, Eras; Lhx1; Ccrk or Akt | SHH | Retroviral | 50–60% | 1–2 m | 30–45% | [71] |
| Heterzygous NeuroD2-SmoA1 | SHH | GEMM | 48% | 6 m | 0% | [73] |
| Homozygous NeuroD2-SmoA1 | SHH | GEMM | 94% | 1–2 m | 30% | [73] |
| NeuroD2-SmoA1, Math1-GFP | SHH | GEMM | 100% | 5.4 m | 28% | [88] |
| Atoh1-SmoM2 | SHH | GEMM | ~50% | 3–4 m | 15–30% | [74] |
| Atoh1-SmoM2, Kmt2dfl/+ or Kmt2dfl/fl | SHH | GEMM | 100% | 2 m | 65–100% | [74] |
| Trp53mut | SHH | GEMM | 0% | - | 0% | [48] |
| Trp53mut, Math1-SB11/T2Onc transposon | SHH | GEMM | 40% | average of 3 m | 100% | [48] |
| MYCN in iPSC-NES; hbNES | SHH | Orthotopic | 90–100% | 2–4; 4–6 m | 30–70%; 0–30% | [64] |
| MYCN + OCT4 in iPSC-NES; hbNES | SHH | Orthotopic | 100% | 1; 2 m | 100%; 50% | [64] |
| GTML (Glt1-tTA, TRE-MYCN) | Group 3 | GEMM | 75% | 2–6 m | ~10% | [78] |
| GTML, Trp53KI/KI | Group 3 | GEMM | 100% | 1.3–3.3 m | NA | [79] |
| GTS (MYCN/SOX9-driven) | Group 3 | GEMM | 100% | 1.5–4 m after dox loss | 50–60% | [80] |
| GMYC (Glt1-tTA, TRE-MYC) | Group 3 | GEMM | 62% | average of 4.3 m | ~5% | [82] |
| GMYC, ARF−/− | Group 3 | GEMM | 95% | average of 3.3 m | 50–60% | [82] |
4. Clinical Investigations with Implications for Relapsed/Metastatic Medulloblastoma
4.1. Targeting of SMO-Driven Medulloblastoma
4.2. Targeting of MYC-Driven Medulloblastoma
4.3. Immune-Therapies
5. Discussion and Future Ideas
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro-Oncology 2021, 23, iii1–iii105. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Bailey, S.; Clifford, S.C.; Doz, F.; Kool, M.; Dufour, C.; Vassal, G.; Milde, T.; et al. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. 2016, 131, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Zeltzer, P.M.; Boyett, J.M.; Finlay, J.L.; Albright, A.L.; Rorke, L.B.; Milstein, J.M.; Allen, J.C.; Stevens, K.R.; Stanley, P.; Li, H.; et al. Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: Conclusions from the children’s cancer group 921 randomized phase III study. J. Clin. Oncol. 1999, 17, 832. [Google Scholar] [CrossRef] [PubMed]
- Packer, R.J.; Rood, B.R.; MacDonald, T.J. Medulloblastoma: Present concepts of stratification into risk groups. Pediatr. Neurosurg. 2003, 39, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Grill, J.; Sainte-Rose, C.; Jouvet, A.; Gentet, J.-C.; Lejars, O.; Frappaz, D.; Doz, F.; Rialland, X.; Pichon, F.; Bertozzi, A.-I.; et al. Treatment of medulloblastoma with postoperative chemotherapy alone: An SFOP prospective trial in young children. Lancet Oncol. 2005, 6, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, S.; Bode, U.; Deinlein, F.; Ottensmeier, H.; Warmuth-Metz, M.; Soerensen, N.; Graf, N.; Emser, A.; Pietsch, T.; Wolff, J.E.; et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy Alone. N. Engl. J. Med. 2005, 352, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Mostoufi-Moab, S.; Grimberg, A. Pediatric brain tumor treatment: Growth consequences and their management. Pediatr. Endocrinol. Rev. 2010, 8, 6–17. [Google Scholar] [PubMed]
- Maciel, J.; Dias, D.; Cavaco, D.; Donato, S.; Pereira, M.C.; Simões-Pereira, J. Growth hormone deficiency and other endocrinopathies after childhood brain tumors: Results from a close follow-up in a cohort of 242 patients. J. Endocrinol. Investig. 2021, 44, 2367–2374. [Google Scholar] [CrossRef]
- Roddy, E.; Mueller, S. Late Effects of Treatment of Pediatric Central Nervous System Tumors. J. Child Neurol. 2016, 31, 237–254. [Google Scholar] [CrossRef]
- Olsson, I.T.; Perrin, S.; Lundgren, J.; Hjorth, L.; Johanson, A. Long-term cognitive sequelae after pediatric brain tumor related to medical risk factors, age, and sex. Pediatr. Neurol. 2014, 51, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Ris, M.D.; Walsh, K.; Wallace, D.; Armstrong, F.D.; Holmes, E.; Gajjar, A.; Zhou, T.; Packer, R.J. Intellectual and academic outcome following two chemotherapy regimens and radiotherapy for average-risk medulloblastoma: COG A9961. Pediatr. Blood Cancer 2013, 60, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Mulhern, R.K.; Palmer, S.L.; Merchant, T.E.; Wallace, D.; Kocak, M.; Brouwers, P.; Krull, K.; Chintagumpala, M.; Stargatt, R.; Ashley, D.M.; et al. Neurocognitive consequences of risk-adapted therapy for childhood medulloblastoma. J. Clin. Oncol. 2005, 23, 5511–5519. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.; André, N.; Gandola, L.; Massimino, M.; Wheatley, K.; Gates, S.; Homer, V.; Rutkowski, S.; Clifford, S.C. Clinical Trials in High-Risk Medulloblastoma: Evolution of the SIOP-Europe HR-MB Trial. Cancers 2022, 14, 374. [Google Scholar] [CrossRef] [PubMed]
- Gandola, L.; Massimino, M.; Cefalo, G.; Solero, C.; Spreafico, F.; Pecori, E.; Riva, D.; Collini, P.; Pignoli, E.; Giangaspero, F.; et al. Hyperfractionated accelerated radiotherapy in the Milan strategy for metastatic medulloblastoma. J. Clin. Oncol. 2009, 27, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Dufour, C.; Kieffer, V.; Varlet, P.; Raquin, M.A.; Dhermain, F.; Puget, S.; Valteau-Couanet, D.; Grill, J. Tandem high-dose chemotherapy and autologous stem cell rescue in children with newly diagnosed high-risk medulloblastoma or supratentorial primitive neuro-ectodermic tumors. Pediatr. Blood Cancer 2014, 61, 1398–1402. [Google Scholar] [CrossRef] [PubMed]
- Verlooy, J.; Mosseri, V.; Bracard, S.; Tubiana, A.L.; Kalifa, C.; Pichon, F.; Frappaz, D.; Chastagner, P.; Pagnier, A.; Bertozzi, A.-I.; et al. Treatment of high risk medulloblastomas in children above the age of 3 years: A SFOP study. Eur. J. Cancer 2006, 42, 3004–3014. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.E.; Bailey, C.C.; Robinson, K.J.; Weston, C.L.; Walker, D.A.; Ellison, D.; Ironside, J.; Pizer, B.L.; Lashford, L.S. Outcome for patients with metastatic (M2–3) medulloblastoma treated with SIOP/UKCCSG PNET-3 chemotherapy. Eur. J. Cancer 2005, 41, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Zapotocky, M.; Mata-Mbemba, D.; Sumerauer, D.; Liby, P.; Lassaletta, A.; Zamecnik, J.; Krskova, L.; Kyncl, M.; Stary, J.; Laughlin, S.; et al. Differential patterns of metastatic dissemination across medulloblastoma subgroups. J. Neurosurg. Pediatr. 2018, 21, 145–152. [Google Scholar] [CrossRef]
- Cavalli, F.M.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, A.S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737–754.e6. [Google Scholar] [CrossRef]
- Hovestadt, V.; Ayrault, O.; Swartling, F.J.; Robinson, G.W.; Pfister, S.M.; Northcott, P.A. Medulloblastomics revisited: Biological and clinical insights from thousands of patients. Nat. Rev. Cancer 2020, 20, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Nobre, L.; Zapotocky, M.; Khan, S.; Fukuoka, K.; Fonseca, A.; McKeown, T.; Sumerauer, D.; Vicha, A.; Grajkowska, W.A.; Trubicka, J.; et al. Pattern of Relapse and Treatment Response in WNT-Activated Medulloblastoma. Cell Rep. Med. 2020, 1, 23. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Northcott, P.A.; Taylor, M.D. FISH and chips: The recipe for improved prognostication and outcomes for children with medulloblastoma. Cancer Genet. 2011, 204, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Faria, C.C.; Perreault, S.; Cho, Y.-J.; Shih, D.J.; Luu, B.; Dubuc, A.M.; A Northcott, P.; et al. Recurrence patterns across medulloblastoma subgroups: An integrated clinical and molecular analysis. Lancet Oncol. 2013, 14, 1200–1207. [Google Scholar] [CrossRef]
- Kumar, R.; Smith, K.S.; Deng, M.; Terhune, C.; Robinson, G.W.; Orr, B.A.; Liu, A.P.Y.; Lin, T.; Billups, C.A.; Chintagumpala, M.; et al. Clinical Outcomes and Patient-Matched Molecular Composition of Relapsed Medulloblastoma. J. Clin. Oncol. 2021, 39, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.M.; Richardson, S.; Schwalbe, E.C.; Hicks, D.; Lindsey, J.C.; Crosier, S.; Rafiee, G.; Grabovska, Y.; Wharton, S.B.; Jacques, T.S.; et al. Time, pattern, and outcome of medulloblastoma relapse and their association with tumour biology at diagnosis and therapy: A multicentre cohort study. Lancet Child Adolesc. Health 2020, 4, 865–874. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Federico, A.; Schrimpf, D.; Sievers, P.; Sahm, F.; Koster, J.; Jones, D.T.W.; von Deimling, A.; Pfister, S.M.; Kool, M.; et al. Comparison of transcriptome profiles between medulloblastoma primary and recurrent tumors uncovers novel variance effects in relapses. Acta Neuropathol. Commun. 2023, 11, 7. [Google Scholar] [CrossRef]
- Deng, M.Y.; Sturm, D.; Pfaff, E.; Sill, M.; Stichel, D.; Balasubramanian, G.P.; Tippelt, S.; Kramm, C.; Donson, A.M.; Green, A.L.; et al. Radiation-induced gliomas represent H3-/IDH-wild type pediatric gliomas with recurrent PDGFRA amplification and loss of CDKN2A/B. Nat. Commun. 2021, 12, 5530. [Google Scholar] [CrossRef]
- Liu, E.K.; Oh, C.; Kondziolka, D.; Sulman, E.P. Risk of Second Primary Neoplasms of the Central Nervous System. Adv. Radiat. Oncol. 2022, 7, 100969. [Google Scholar] [CrossRef]
- Chung, C.S.; Yock, T.I.; Nelson, K.; Xu, Y.; Keating, N.L.; Tarbell, N.J. Incidence of second malignancies among patients treated with proton versus photon radiation. Int. J. Radiat. Oncol. 2013, 87, 46–52. [Google Scholar] [CrossRef]
- de González, A.B.; Gibson, T.M.; Lee, C.; Albert, P.S.; Griffin, K.T.; Kitahara, C.M.; Liu, D.; Mille, M.M.; Shin, J.; Bajaj, B.V.; et al. The Pediatric Proton and Photon Therapy Comparison Cohort: Study Design for a Multicenter Retrospective Cohort to Investigate Subsequent Cancers after Pediatric Radiation Therapy. Adv. Radiat. Oncol. 2023, 8, 101273. [Google Scholar] [CrossRef]
- Schwalbe, E.C.; Lindsey, J.C.; Nakjang, S.; Crosier, S.; Smith, A.J.; Hicks, D.; Rafiee, G.; Hill, R.M.; Iliasova, A.; Stone, T.; et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: A cohort study. Lancet Oncol. 2017, 18, 958–971. [Google Scholar] [CrossRef]
- Nguyen, A.; Nguyen, A.; Dada, O.T.; Desai, P.D.; Ricci, J.C.; Godbole, N.B.; Pierre, K.; Lucke-Wold, B. Leptomeningeal Metastasis: A Review of the Pathophysiology, Diagnostic Methodology, and Therapeutic Landscape. Curr. Oncol. 2023, 30, 5906–5931. [Google Scholar] [CrossRef] [PubMed]
- Garzia, L.; Kijima, N.; Morrissy, A.S.; De Antonellis, P.; Guerreiro-Stucklin, A.; Holgado, B.L.; Wu, X.; Wang, X.; Parsons, M.; Zayne, K.; et al. A Hematogenous Route for Medulloblastoma Leptomeningeal Metastases. Cell 2018, 172, 1050–1062.e14. [Google Scholar] [CrossRef] [PubMed]
- Jankovska, E.; Svitek, M.; Holada, K.; Petrak, J. Affinity depletion versus relative protein enrichment: A side-by-side comparison of two major strategies for increasing human cerebrospinal fluid proteome coverage. Clin. Proteom. 2019, 16, 9. [Google Scholar] [CrossRef] [PubMed]
- Margarido, A.S.; Uceda-Castro, R.; Hahn, K.; de Bruijn, R.; Kester, L.; Hofland, I.; Lohuis, J.; Seinstra, D.; Broeks, A.; Jonkers, J.; et al. Epithelial-to-Mesenchymal Transition Drives Invasiveness of Breast Cancer Brain Metastases. Cancers 2022, 14, 3115. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, U.D.; Joseph, J.V.; Kruyt, F.A.E. EMT- and MET-related processes in nonepithelial tumors: Importance for disease progression, prognosis, and therapeutic opportunities. Mol. Oncol. 2017, 11, 860–877. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Xu, Q.; Cui, Y.; Li, N.; Bian, X.; Lv, S. Medulloblastoma stem cells: Promising targets in medulloblastoma therapy. Cancer Sci. 2016, 107, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Aouad, P.; Zhang, Y.; De Martino, F.; Stibolt, C.; Ali, S.; Ambrosini, G.; Mani, S.A.; Maggs, K.; Quinn, H.M.; Sflomos, G.; et al. Epithelial-mesenchymal plasticity determines estrogen receptor positive breast cancer dormancy and epithelial reconversion drives recurrence. Nat. Commun. 2022, 13, 4975. [Google Scholar] [CrossRef]
- Fouladi, M.; Gajjar, A.; Boyett, J.M.; Walter, A.W.; Thompson, S.J.; Merchant, T.E.; Jenkins, J.J.; Langston, J.W.; Liu, A.; Kun, L.E.; et al. Comparison of CSF cytology and spinal magnetic resonance imaging in the detection of leptomeningeal disease in pediatric medulloblastoma or primitive neuroectodermal tumor. J. Clin. Oncol. 1999, 17, 3234–3237. [Google Scholar] [CrossRef]
- Liu, A.P.; Smith, K.S.; Kumar, R.; Paul, L.; Bihannic, L.; Lin, T.; Maass, K.K.; Pajtler, K.W.; Chintagumpala, M.; Su, J.M.; et al. Serial assessment of measurable residual disease in medulloblastoma liquid biopsies. Cancer Cell 2021, 39, 1519–1530.e4. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Lehner, K.R.; Jiang, K.; Rincon-Torroella, J.; Perera, R.; Bettegowda, C. Cerebrospinal Fluid biomarkers in pediatric brain tumors: A systematic review. Neoplasia 2023, 35, 100852. [Google Scholar] [CrossRef] [PubMed]
- Arthur, C.; Jylhä, C.; de Ståhl, T.D.; Shamikh, A.; Sandgren, J.; Rosenquist, R.; Nordenskjöld, M.; Harila, A.; Barbany, G.; Sandvik, U.; et al. Simultaneous Ultra-Sensitive Detection of Structural and Single Nucleotide Variants Using Multiplex Droplet Digital PCR in Liquid Biopsies from Children with Medulloblastoma. Cancers 2023, 15, 1972. [Google Scholar] [CrossRef] [PubMed]
- Schuhmann, M.; Zucht, H.; Nassimi, R.; Heine, G.; Schneekloth, C.; Stuerenburg, H.; Selle, H. Peptide screening of cerebrospinal fluid in patients with glioblastoma multiforme. Eur. J. Surg. Oncol. 2010, 36, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Escudero, L.; Llort, A.; Arias, A.; Diaz-Navarro, A.; Martínez-Ricarte, F.; Rubio-Perez, C.; Mayor, R.; Caratù, G.; Martínez-Sáez, E.; Vázquez-Méndez, É.; et al. Circulating tumour DNA from the cerebrospinal fluid allows the characterisation and monitoring of medulloblastoma. Nat. Commun. 2020, 11, 5736. [Google Scholar] [CrossRef]
- Harris, P.; Diouf, A.; Guilbert, F.; Ameur, F.; Letourneau-Guillon, L.; Ménard, C.; Masucci, L.; Bélair, M.; Roberge, D. Diagnostic Reliability of Leptomeningeal Disease Using Magnetic Resonance Imaging. Cureus 2019, 11, e4416. [Google Scholar] [CrossRef]
- Wu, X.; Northcott, P.A.; Dubuc, A.; Dupuy, A.J.; Shih, D.J.H.; Witt, H.; Croul, S.; Bouffet, E.; Fults, D.W.; Eberhart, C.G.; et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature 2012, 482, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Van Ommeren, R.; Garzia, L.; Holgado, B.L.; Ramaswamy, V.; Taylor, M.D. The molecular biology of medulloblastoma metastasis. Brain Pathol. 2020, 30, 691–702. [Google Scholar] [CrossRef]
- Li, M.; Deng, Y.; Zhang, W. Molecular Determinants of Medulloblastoma Metastasis and Leptomeningeal Dissemination. Mol. Cancer Res. 2021, 19, 743–752. [Google Scholar] [CrossRef]
- Wang, X.; Dubuc, A.M.; Ramaswamy, V.; Mack, S.; Gendoo, D.M.A.; Remke, M.; Wu, X.; Garzia, L.; Luu, B.; Cavalli, F.; et al. Medulloblastoma subgroups remain stable across primary and metastatic compartments. Acta Neuropathol. 2015, 129, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Hendrikse, L.D.; Haldipur, P.; Saulnier, O.; Millman, J.; Sjoboen, A.H.; Erickson, A.W.; Ong, W.; Gordon, V.; Coudière-Morrison, L.; Mercier, A.L.; et al. Failure of human rhombic lip differentiation underlies medulloblastoma formation. Nature 2022, 609, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.S.; Bihannic, L.; Gudenas, B.L.; Haldipur, P.; Tao, R.; Gao, Q.; Li, Y.; Aldinger, K.A.; Iskusnykh, I.Y.; Chizhikov, V.V.; et al. Unified rhombic lip origins of group 3 and group 4 medulloblastoma. Nature 2022, 609, 1012–1020. [Google Scholar] [CrossRef]
- Richardson, S.; Hill, R.M.; Kui, C.; Lindsey, J.C.; Grabovksa, Y.; Keeling, C.; Pease, L.; Bashton, M.; Crosier, S.; Vinci, M.; et al. Emergence and maintenance of actionable genetic drivers at medulloblastoma relapse. Neuro-Oncology 2022, 24, 153–165. [Google Scholar] [CrossRef]
- Shih, D.J.; Northcott, P.A.; Remke, M.; Korshunov, A.; Ramaswamy, V.; Kool, M.; Luu, B.; Yao, Y.; Wang, X.; Dubuc, A.M.; et al. Cytogenetic prognostication within medulloblastoma subgroups. J. Clin. Oncol. 2014, 32, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Phoenix, T.N.; Patmore, D.M.; Boop, S.; Boulos, N.; Jacus, M.O.; Patel, Y.T.; Roussel, M.F.; Finkelstein, D.; Goumnerova, L.; Perreault, S.; et al. Medulloblastoma Genotype Dictates Blood Brain Barrier Phenotype. Cancer Cell 2016, 29, 508–522. [Google Scholar] [CrossRef] [PubMed]
- Hartley, R.; Phoenix, T.N. MYC promotes aggressive growth and metastasis of a WNT-medulloblastoma mouse model. Dev. Neurosci. 2023, 129, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Achiha, T.; Kijima, N.; Kodama, Y.; Kagawa, N.; Kinoshita, M.; Fujimoto, Y.; Nonaka, M.; Fukai, J.; Inoue, A.; Nishida, N.; et al. Activated leukocyte cell adhesion molecule expression correlates with the WNT subgroup in medulloblastoma and is involved in regulating tumor cell proliferation and invasion. PLoS ONE 2020, 15, e0243272. [Google Scholar] [CrossRef]
- Zhukova, N.; Ramaswamy, V.; Remke, M.; Pfaff, E.; Shih, D.J.; Martin, D.C.; Castelo-Branco, P.; Baskin, B.; Ray, P.N.; Bouffet, E.; et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J. Clin. Oncol. 2013, 31, 2927–2935. [Google Scholar] [CrossRef]
- Rausch, T.; Jones, D.T.; Zapatka, M.; Stütz, A.M.; Zichner, T.; Weischenfeldt, J.; Jäger, N.; Remke, M.; Shih, D.; Northcott, P.A.; et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012, 148, 59–71. [Google Scholar] [CrossRef]
- Brada, M.; Hoang-Xuan, K.; Rampling, R.; Dietrich, P.-Y.; Dirix, L.Y.; Macdonald, D.; Heimans, J.J.; Zonnenberg, B.A.; Bravo-Marques, J.M.; Henriksson, R.; et al. Multicenter phase II trial of temozolomide in patients with glioblastoma multiforme at first relapse. Ann. Oncol. 2001, 12, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, R.J.; Clifford, S.C. PDGFRB is overexpressed in metastatic medulloblastoma. Nat. Genet. 2003, 35, 197–198. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Santi, M.; Rushing, E.J.; Cornelison, R.; MacDonald, T.J. ERK activation of p21 activated kinase-1 (Pak1) is critical for medulloblastoma cell migration. Clin. Exp. Metastasis 2010, 27, 481–491. [Google Scholar] [CrossRef]
- Čančer, M.; Hutter, S.; Holmberg, K.O.; Rosén, G.; Sundström, A.; Tailor, J.; Bergström, T.; Garancher, A.; Essand, M.; Wechsler-Reya, R.J.; et al. Humanized Stem Cell Models of Pediatric Medulloblastoma Reveal an Oct4/mTOR Axis that Promotes Malignancy. Cell Stem Cell 2019, 25, 855–870.e11. [Google Scholar] [CrossRef] [PubMed]
- Malawsky, D.S.; Dismuke, T.; Liu, H.; Castellino, E.; Brenman, J.; Dasgupta, B.; Tikunov, A.; Gershon, T.R. Chronic AMPK inactivation slows SHH medulloblastoma progression by inhibiting mTORC1 signaling and depleting tumor stem cells. iScience 2023, 26, 108443. [Google Scholar] [CrossRef]
- Cai, J.; Wang, Y.; Wang, X.; Ai, Z.; Li, T.; Pu, X.; Yang, X.; Yao, Y.; He, J.; Cheng, S.Y.; et al. AMPK attenuates SHH subgroup medulloblastoma growth and metastasis by inhibiting NF-κB activation. Cell Biosci. 2023, 13, 15. [Google Scholar] [CrossRef]
- Vanner, R.J.; Remke, M.; Gallo, M.; Selvadurai, H.J.; Coutinho, F.; Lee, L.; Kushida, M.; Head, R.; Morrissy, S.; Zhu, X.; et al. Quiescent Sox2+ cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell 2014, 26, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Raffel, C.; Jenkins, R.B.; Frederick, L.; Hebrink, D.; Alderete, B.; Fults, D.W.; James, C.D. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997, 57, 842–845. [Google Scholar] [PubMed]
- Flora, A.; Klisch, T.J.; Schuster, G.; Zoghbi, H.Y. Deletion of Atoh1 disrupts sonic hedgehog signaling in the developing cerebellum and prevents medulloblastoma. Science 2009, 326, 1424–1427. [Google Scholar] [CrossRef]
- Grausam, K.B.; Dooyema, S.D.; Bihannic, L.; Premathilake, H.; Morrissy, A.S.; Forget, A.; Schaefer, A.M.; Gundelach, J.H.; Macura, S.; Maher, D.M.; et al. ATOH1 Promotes Leptomeningeal Dissemination and Metastasis of Sonic Hedgehog Subgroup Medulloblastomas. Cancer Res. 2017, 77, 3766–3777. [Google Scholar] [CrossRef]
- Mumert, M.; Dubuc, A.; Wu, X.; Northcott, P.A.; Chin, S.S.; Pedone, C.A.; Taylor, M.D.; Fults, D.W. Functional genomics identifies drivers of medulloblastoma dissemination. Cancer Res. 2012, 72, 4944–4953. [Google Scholar] [CrossRef] [PubMed]
- Hallahan, A.R.; Pritchard, J.I.; Hansen, S.; Benson, M.; Stoeck, J.; Hatton, B.A.; Russell, T.L.; Ellenbogen, R.G.; Bernstein, I.D.; Beachy, P.A.; et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res. 2004, 64, 7794–7800. [Google Scholar] [CrossRef] [PubMed]
- Hatton, B.A.; Villavicencio, E.H.; Tsuchiya, K.D.; Pritchard, J.I.; Ditzler, S.; Pullar, B.; Hansen, S.; Knoblaugh, S.E.; Lee, D.; Eberhart, C.G.; et al. The Smo/Smo model: Hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res. 2008, 68, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Sanghrajka, R.M.; Koche, R.; Medrano, H.; El Nagar, S.; Stephen, D.N.; Lao, Z.; Bayin, N.S.; Ge, K.; Joyner, A.L. KMT2D suppresses Sonic hedgehog-driven medulloblastoma progression and metastasis. iScience 2023, 26, 107831. [Google Scholar] [CrossRef]
- Cho, Y.-J.; Tsherniak, A.; Tamayo, P.; Santagata, S.; Ligon, A.; Greulich, H.; Berhoukim, R.; Amani, V.; Goumnerova, L.; Eberhart, C.G.; et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J. Clin. Oncol. 2011, 29, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Tamayo, P.; Cho, Y.-J.; Tsherniak, A.; Greulich, H.; Ambrogio, L.; Meeteren, N.S.-V.; Zhou, T.; Buxton, A.; Kool, M.; Meyerson, M.; et al. Predicting relapse in patients with medulloblastoma by integrating evidence from clinical and genomic features. J. Clin. Oncol. 2011, 29, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.L.; Schwalbe, E.C.; Cole, M.; Lu, Y.; Lusher, M.E.; Megahed, H.; O’toole, K.; Nicholson, S.L.; Bognar, L.; Garami, M.; et al. MYC family amplification and clinical risk-factors interact to predict an extremely poor prognosis in childhood medulloblastoma. Acta Neuropathol. 2012, 123, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Swartling, F.J.; Grimmer, M.R.; Hackett, C.S.; Northcott, P.A.; Fan, Q.-W.; Goldenberg, D.D.; Lau, J.; Masic, S.; Nguyen, K.; Yakovenko, S.; et al. Pleiotropic role for MYCN in medulloblastoma. Genes Dev. 2010, 24, 1059–1072. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.M.; Kuijper, S.; Lindsey, J.C.; Petrie, K.; Schwalbe, E.C.; Barker, K.; Boult, J.K.; Williamson, D.; Ahmad, Z.; Hallsworth, A.; et al. Combined MYC and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell 2015, 27, 72–84. [Google Scholar] [CrossRef]
- Borgenvik, A.; Holmberg, K.O.; Bolin, S.; Zhao, M.; Savov, V.; Rosén, G.; Hutter, S.; Garancher, A.; Rahmanto, A.S.; Bergström, T.; et al. Dormant SOX9-Positive Cells Facilitate MYC-Driven Recurrence of Medulloblastoma. Cancer Res. 2022, 82, 4586–4603. [Google Scholar] [CrossRef]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Gröbner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Mainwaring, O.J.; Weishaupt, H.; Zhao, M.; Rosén, G.; Borgenvik, A.; Breinschmid, L.; Verbaan, A.D.; Richardson, S.; Thompson, D.; Clifford, S.C.; et al. ARF suppression by MYC but not MYCN confers increased malignancy of aggressive pediatric brain tumors. Nat. Commun. 2023, 14, 1221. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Murad, N.; Malawsky, D.; Tao, R.; Rivero-Hinojosa, S.; Holdhof, D.; Schüller, U.; Zhang, P.; Lazarski, C.; Rood, B.R.; et al. OLIG2 Is a Determinant for the Relapse of MYC-Amplified Medulloblastoma. Clin. Cancer Res. 2022, 28, 4278–4291. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.A.; Wang, X.; Nitta, R.T.; Gholamin, S.; Theruvath, J.; Hutter, G.; Azad, T.D.; Wadi, L.; Bolin, S.; Ramaswamy, V.; et al. Notch1 regulates the initiation of metastasis and self-renewal of Group 3 medulloblastoma. Nat. Commun. 2018, 9, 4121. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.; Schlanstein, M.; Northcott, P.A.; Cho, Y.-J.; Koster, J.; Schouten-van Meeteren, A.; Van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Sharma, T.; Schwalbe, E.C.; Williamson, D.; Sill, M.; Hovestadt, V.; Mynarek, M.; Rutkowski, S.; Robinson, G.W.; Gajjar, A.; Cavalli, F.; et al. Second-generation molecular subgrouping of medulloblastoma: An international meta-analysis of Group 3 and Group 4 subtypes. Acta Neuropathol. 2019, 138, 309–326. [Google Scholar] [CrossRef] [PubMed]
- Bakhshinyan, D.; Adile, A.A.; Liu, J.; Gwynne, W.D.; Suk, Y.; Custers, S.; Burns, I.; Singh, M.; McFarlane, N.; Subapanditha, M.K.; et al. Temporal profiling of therapy resistance in human medulloblastoma identifies novel targetable drivers of recurrence. Sci. Adv. 2021, 7, eabi5568. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Liu, J.; Eldridge, R.C.; Gaul, D.A.; Paine, M.R.L.; Uppal, K.; MacDonald, T.J.; Fernández, F.M. Lipidome signatures of metastasis in a transgenic mouse model of sonic hedgehog medulloblastoma. Anal. Bioanal. Chem. 2020, 412, 7017–7027. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, P.M.M.; Ariza, C.B.; Ishibashi, C.M.; Fujita, T.C.; Banin-Hirata, B.K.; Oda, J.M.M.; Watanabe, M.A.E. Role of CXCL12 and CXCR4 in normal cerebellar development and medulloblastoma. Int. J. Cancer 2016, 138, 10–13. [Google Scholar] [CrossRef]
- Fiscella, M.; Zhang, H.; Fan, S.; Sakaguchi, K.; Shen, S.; Mercer, W.E.; Woude, G.F.V.; O’connor, P.M.; Appella, E. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc. Natl. Acad. Sci. USA 1997, 94, 6048–6053. [Google Scholar] [CrossRef]
- Buss, M.C.; Remke, M.; Lee, J.; Gandhi, K.; Schniederjan, M.J.; Kool, M.; Northcott, P.A.; Pfister, S.S.; Taylor, M.D.; Castellino, R.C. The WIP1 oncogene promotes progression and invasion of aggressive medulloblastoma variants. Oncogene 2015, 34, 1126–1140. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.W.; Orr, B.A.; Wu, G.; Gururangan, S.; Lin, T.; Qaddoumi, I.; Packer, R.J.; Goldman, S.; Prados, M.D.; Desjardins, A.; et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog–Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. 2015, 33, 2646–2654. [Google Scholar] [CrossRef] [PubMed]
- Kieran, M.W.; Chisholm, J.; Casanova, M.; A Brandes, A.; Aerts, I.; Bouffet, E.; Bailey, S.; Leary, S.; MacDonald, T.J.; Mechinaud, F.; et al. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro-Oncology 2017, 19, 1542–1552. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Jones, D.T.W.; Jaeger, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Piro, R.M.; Esparza, L.A.; Markant, S.L.; Remke, M.; et al. Genome sequencing of shh medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 2014, 25, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.W.; Kaste, S.C.; Chemaitilly, W.; Bowers, D.C.; Laughton, S.; Smith, A.; Gottardo, N.G.; Partap, S.; Bendel, A.; Wright, K.D.; et al. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget 2017, 8, 69295–69302. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Li, H.; Kuang, L.; Zhao, Z.; He, W.; Liu, C.; Wang, Y.; Cheng, S.Y.; Chen, W. Identification of a potent antagonist of smoothened in hedgehog signaling. Cell Biosci. 2021, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-B.; He, L.-M.; Sun, M.; Luo, W.-J.; Lin, Z.-C.; Qiu, Z.-P.; Zhang, Y.-L.; Hu, A.; Luo, J.; Qiu, W.-W.; et al. A sterol analog inhibits hedgehog pathway by blocking cholesterylation of smoothened. Cell Chem. Biol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Blanco, J.; Li, B.; Long, J.; Shen, C.; Yang, F.; Orton, D.; Collins, S.; Kasahara, N.; Ayad, N.G.; McCrea, H.J.; et al. A CK1α Activator Penetrates the Brain and Shows Efficacy Against Drug-resistant Metastatic Medulloblastoma. Clin. Cancer Res. 2019, 25, 1379–1388. [Google Scholar] [CrossRef]
- Zhao, X.; Ponomaryov, T.; Ornell, K.J.; Zhou, P.; Dabral, S.K.; Pak, E.; Li, W.; Atwood, S.X.; Whitson, R.J.; Chang, A.L.S.; et al. RAS/MAPK Activation Drives Resistance to Smo Inhibition, Metastasis, and Tumor Evolution in Shh Pathway–Dependent Tumors. Cancer Res. 2015, 75, 3623–3635. [Google Scholar] [CrossRef]
- Dubuc, A.M.; Remke, M.; Korshunov, A.; Northcott, P.A.; Zhan, S.H.; Mendez-Lago, M.; Kool, M.; Jones, D.T.W.; Unterberger, A.; Morrissy, A.S.; et al. Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta Neuropathol. 2013, 125, 373–384. [Google Scholar] [CrossRef]
- A Northcott, P.; Nakahara, Y.; Wu, X.; Feuk, L.; Ellison, D.W.; Croul, S.; Mack, S.; Kongkham, P.N.; Peacock, J.; Dubuc, A.; et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat. Genet. 2009, 41, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Bandopadhayay, P.; Bergthold, G.; Nguyen, B.; Schubert, S.; Gholamin, S.; Tang, Y.; Bolin, S.; Schumacher, S.E.; Zeid, R.; Masoud, S.; et al. BET Bromodomain Inhibition of MYC-Amplified Medulloblastoma. Clin. Cancer Res. 2014, 20, 912–925. [Google Scholar] [CrossRef] [PubMed]
- Stratikopoulos, E.E.; Dendy, M.; Szabolcs, M.; Khaykin, A.J.; Lefebvre, C.; Zhou, M.-M.; Parsons, R. Kinase and BET Inhibitors Together Clamp Inhibition of PI3K Signaling and Overcome Resistance to Therapy. Cancer Cell 2015, 27, 837–851. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Liu, K.-W.; Wang, J.; Garancher, A.; Tao, R.; Esparza, L.A.; Maier, D.L.; Udaka, Y.T.; Murad, N.; Morrissy, S.; et al. HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC- Driven Medulloblastoma. Cancer Cell 2016, 29, 311–323. [Google Scholar] [CrossRef]
- Singh, A.R.; Joshi, S.; Zulcic, M.; Alcaraz, M.; Garlich, J.R.; Morales, G.A.; Cho, Y.J.; Bao, L.; Levy, M.L.; Newbury, R.; et al. PI-3K Inhibitors Preferentially Target CD15+ Cancer Stem Cell Population in SHH Driven Medulloblastoma. PLoS ONE 2016, 11, e0150836. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, M.; Craveiro, R.B.; Holst, M.I.; Pietsch, T.; Dilloo, D. The PI3K inhibitor GDC-0941 displays promising in vitro and in vivo efficacy for targeted medulloblastoma therapy. Oncotarget 2015, 6, 802–813. [Google Scholar] [CrossRef]
- Sholler, G.L.S.; Gerner, E.W.; Bergendahl, G.; MacArthur, R.B.; VanderWerff, A.; Ashikaga, T.; Bond, J.P.; Ferguson, W.; Roberts, W.; Wada, R.K.; et al. A Phase I Trial of DFMO Targeting Polyamine Addiction in Patients with Relapsed/Refractory Neuroblastoma. PLoS ONE 2015, 10, e0127246. [Google Scholar] [CrossRef] [PubMed]
- Wallick, C.J.; Gamper, I.; Thorne, M.; Feith, D.J.; Takasaki, K.Y.; Wilson, S.M.; A Seki, J.; E Pegg, A.; Byus, C.V.; Bachmann, A.S. Key role for p27Kip1, retinoblastoma protein Rb, and MYCN in polyamine inhibitor-induced G1 cell cycle arrest in MYCN-amplified human neuroblastoma cells. Oncogene 2005, 24, 5606–5618. [Google Scholar] [CrossRef] [PubMed]
- Nakkina, S.P.; Gitto, S.B.; Beardsley, J.M.; Pandey, V.; Rohr, M.W.; Parikh, J.G.; Phanstiel, O.; Altomare, D.A. DFMO Improves Survival and Increases Immune Cell Infiltration in Association with MYC Downregulation in the Pancreatic Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 13175. [Google Scholar] [CrossRef]
- Borgenvik, A.; Čančer, M.; Hutter, S.; Swartling, F.J. Targeting MYCN in Molecularly Defined Malignant Brain Tumors. Front. Oncol. 2020, 10, 626751. [Google Scholar] [CrossRef]
- Bandopadhayay, P.; Piccioni, F.; O’rourke, R.; Ho, P.; Gonzalez, E.M.; Buchan, G.; Qian, K.; Gionet, G.; Girard, E.; Coxon, M.; et al. Neuronal differentiation and cell-cycle programs mediate response to BET-bromodomain inhibition in MYC-driven medulloblastoma. Nat. Commun. 2019, 10, 2400. [Google Scholar] [CrossRef] [PubMed]
- Sangar, M.L.C.; Genovesi, L.A.; Nakamoto, M.W.; Davis, M.J.; Knobluagh, S.E.; Ji, P.; Millar, A.; Wainwright, B.J.; Olson, J.M. Inhibition of CDK4/6 by Palbociclib Significantly Extends Survival in Medulloblastoma Patient-Derived Xenograft Mouse Models. Clin. Cancer Res. 2017, 23, 5802–5813. [Google Scholar] [CrossRef]
- Vo, T.; Balderson, B.; Jones, K.; Ni, G.; Crawford, J.; Millar, A.; Tolson, E.; Singleton, M.; Kojic, M.; Robertson, T.; et al. Spatial transcriptomic analysis of Sonic hedgehog medulloblastoma identifies that the loss of heterogeneity and promotion of differentiation underlies the response to CDK4/6 inhibition. Genome Med. 2023, 15, 29. [Google Scholar] [CrossRef] [PubMed]
- Frame, S.; Saladino, C.; MacKay, C.; Atrash, B.; Sheldrake, P.; McDonald, E.; Clarke, P.A.; Workman, P.; Blake, D.; Zheleva, D. Fadraciclib (CYC065), a novel CDK inhibitor, targets key pro-survival and oncogenic pathways in cancer. PLoS ONE 2020, 15, e0234103. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Bhakat, R.; Kling, M.J.; Coulter, D.W.; Chaturvedi, N.K.; Ray, S.; Joshi, S.S. Targeting cyclin-dependent kinase 9 sensitizes medulloblastoma cells to chemotherapy. Biochem. Biophys. Res. Commun. 2019, 520, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Peyrl, A.; Chocholous, M.; Kieran, M.W.; Azizi, A.A.; Prucker, C.; Czech, T.; Dieckmann, K.; Schmook, M.; Haberler, C.; Leiss, U.; et al. Antiangiogenic metronomic therapy for children with recurrent embryonal brain tumors. Pediatr. Blood Cancer 2012, 59, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Peyrl, A.; Chocholous, M.; Sabel, M.; Lassaletta, A.; Sterba, J.; Leblond, P.; Nysom, K.; Torsvik, I.; Chi, S.N.; Perwein, T.; et al. Sustained Survival Benefit in Recurrent Medulloblastoma by a Metronomic Antiangiogenic Regimen. JAMA Oncol. 2023, 9, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
- Slavc, I.; Mayr, L.; Stepien, N.; Gojo, J.; Lippolis, M.A.; Azizi, A.A.; Chocholous, M.; Baumgartner, A.; Hedrich, C.S.; Holm, S.; et al. Improved Long-Term Survival of Patients with Recurrent Medulloblastoma Treated with a “MEMMAT-like” Metronomic Antiangiogenic Approach. Cancers 2022, 14, 5128. [Google Scholar] [CrossRef]
- Levy, A.S.; Krailo, M.; Chi, S.; Villaluna, D.; Springer, L.; Williams-Hughes, C.; Fouladi, M.; Gajjar, A. Temozolomide with irinotecan versus temozolomide, irinotecan plus bevacizumab for recurrent medulloblastoma of childhood: Report of a COG randomized Phase II screening trial. Pediatr. Blood Cancer 2021, 68, e29031. [Google Scholar] [CrossRef]
- Muller, A.J.; DuHadaway, J.B.; Donover, P.S.; Sutanto-Ward, E.; Prendergast, G.C. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat. Med. 2005, 11, 312–319. [Google Scholar] [CrossRef]
- Johnson, T.S.; MacDonald, T.J.; Pacholczyk, R.; Aguilera, D.; Al-Basheer, A.; Bajaj, M.; Bandopadhayay, P.; Berrong, Z.; Bouffet, E.; Castellino, R.C.; et al. Indoximod-based chemo-immunotherapy for pediatric brain tumors: A first-in-children phase I trial. Neuro-Oncology 2023, 26, 348–361. [Google Scholar] [CrossRef] [PubMed]
- Pizem, J.; Cör, A.; Zadravec-Zaletel, L.; Popovic, M. Survivin is a negative prognostic marker in medulloblastoma. Neuropathol. Appl. Neurobiol. 2005, 31, 422–428. [Google Scholar] [CrossRef]
- Ahluwalia, M.S.; Reardon, D.A.; Abad, A.P.; Curry, W.T.; Wong, E.T.; Figel, S.A.; Mechtler, L.L.; Peereboom, D.M.; Hutson, A.D.; Withers, H.G.; et al. Phase IIa Study of SurVaxM Plus Adjuvant Temozolomide for Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2023, 41, 1453–1465. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Petrecca, K.; Walbert, T.; Butowski, N.; Salacz, M.; Perry, J.; Damek, D.; Bota, D.; Bettegowda, C.; Zhu, J.-J.; et al. Effect of Vocimagene Amiretrorepvec in Combination With Flucytosine vs Standard of Care on Survival Following Tumor Resection in Patients With Recurrent High-Grade Glioma: A Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1936–1946. [Google Scholar] [CrossRef]
- Lentz, T.B.; Gray, S.J.; Samulski, R.J. Viral vectors for gene delivery to the central nervous system. Neurobiol. Dis. 2012, 48, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Vitanza, N.A.; Johnson, A.J.; Wilson, A.L.; Brown, C.; Yokoyama, J.K.; Künkele, A.; Chang, C.A.; Rawlings-Rhea, S.; Huang, W.; Seidel, K.; et al. Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: An interim analysis. Nat. Med. 2021, 27, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef]
- Donovan, L.K.; Delaidelli, A.; Joseph, S.K.; Bielamowicz, K.; Fousek, K.; Holgado, B.L.; Manno, A.; Srikanthan, D.; Gad, A.Z.; Van Ommeren, R.; et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat. Med. 2020, 26, 720–731. [Google Scholar] [CrossRef]
- Flores, C.; Wildes, T.; Dean, B.D.; Moore, G.; Drake, J.; Abraham, R.; Gil, J.; Yegorov, O.; Yang, C.; Dean, J.; et al. Massive clonal expansion of medulloblastoma-specific T cells during adoptive cellular therapy. Sci. Adv. 2019, 5, eaav9879. [Google Scholar] [CrossRef]
- Pham, C.D.; Flores, C.; Yang, C.; Pinheiro, E.M.; Yearley, J.H.; Sayour, E.J.; Pei, Y.; Moore, C.; McLendon, R.E.; Huang, J.; et al. Differential Immune Microenvironments and Response to Immune Checkpoint Blockade among Molecular Subtypes of Murine Medulloblastoma. Clin. Cancer Res. 2016, 22, 582–595. [Google Scholar] [CrossRef]
- Pérez-Larraya, J.G.; Garcia-Moure, M.; Labiano, S.; Patiño-García, A.; Dobbs, J.; Gonzalez-Huarriz, M.; Zalacain, M.; Marrodan, L.; Martinez-Velez, N.; Puigdelloses, M.; et al. Oncolytic DNX-2401 Virus for Pediatric Diffuse Intrinsic Pontine Glioma. N. Engl. J. Med. 2022, 386, 2471–2481. [Google Scholar] [CrossRef] [PubMed]
- Fine, H.A. Glioblastoma: Not Just Another Cancer. Cancer Discov. 2024, 14, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Vera-Ramirez, L.; Vodnala, S.K.; Nini, R.; Hunter, K.W.; Green, J.E. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun. 2018, 9, 1944. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holmberg, K.O.; Borgenvik, A.; Zhao, M.; Giraud, G.; Swartling, F.J. Drivers Underlying Metastasis and Relapse in Medulloblastoma and Targeting Strategies. Cancers 2024, 16, 1752. https://doi.org/10.3390/cancers16091752
Holmberg KO, Borgenvik A, Zhao M, Giraud G, Swartling FJ. Drivers Underlying Metastasis and Relapse in Medulloblastoma and Targeting Strategies. Cancers. 2024; 16(9):1752. https://doi.org/10.3390/cancers16091752
Chicago/Turabian StyleHolmberg, Karl O., Anna Borgenvik, Miao Zhao, Géraldine Giraud, and Fredrik J. Swartling. 2024. "Drivers Underlying Metastasis and Relapse in Medulloblastoma and Targeting Strategies" Cancers 16, no. 9: 1752. https://doi.org/10.3390/cancers16091752
APA StyleHolmberg, K. O., Borgenvik, A., Zhao, M., Giraud, G., & Swartling, F. J. (2024). Drivers Underlying Metastasis and Relapse in Medulloblastoma and Targeting Strategies. Cancers, 16(9), 1752. https://doi.org/10.3390/cancers16091752