
Metabolic-Modulating Effects of Radiation: Undetectable Yet Deadly—A Review on Radiotherapy

 ,
,  ,
,  and
and
Simple Summary
Abstract
1. Introduction
2. Radiation-Induced Metabolic Reprogramming of Glucides
3. Radiation-Induced Metabolic Reprogramming of Lipids
4. Radiation-Induced Metabolic Reprogramming of Amino Acids
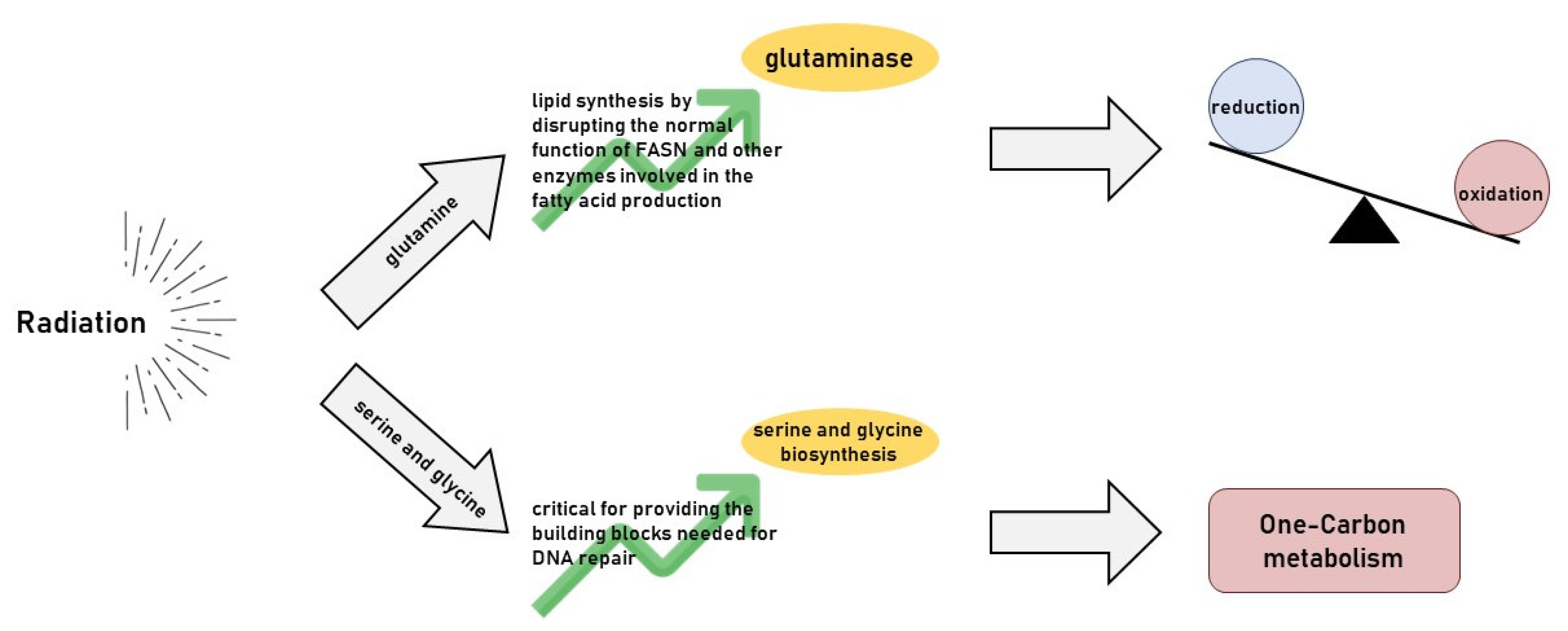
5. Radiotherapy’s Impact on Glutamine Metabolism
6. Radiotherapy’s Impact on Serine and Glycine Metabolism
7. Radiotherapy’s Effect on Arginine Metabolism
8. Radiotherapy’s Interaction with Asparagine Metabolism
9. Radiotherapy’s Interaction with Branched-Chain Amino Acid (BCAA)
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lomax, M.E.; Folkes, L.K.; O’Neill, P. Biological Consequences of Radiation-Induced DNA Damage: Relevance to Radiotherapy. Clin. Oncol. 2013, 25, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Mak, T.W. Pathways of Apoptotic and Non-Apoptotic Death in Tumour Cells. Nat. Rev. Cancer 2004, 4, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J. Cell. Physiol. 2016, 231, 3. [Google Scholar] [CrossRef] [PubMed]
- Bortfeld, T.; Jeraj, R. The Physical Basis and Future of Radiation Therapy. Br. J. Radiol. 2011, 84, 485. [Google Scholar] [CrossRef]
- Otto, K. Volumetric Modulated Arc Therapy: IMRT in a Single Gantry Arc. Med. Phys. 2008, 35, 310–317. [Google Scholar] [CrossRef]
- Teoh, M.; Clark, C.H.; Wood, K.; Whitaker, S.; Nisbet, A. Volumetric Modulated Arc Therapy: A Review of Current Literature and Clinical Use in Practice. Br. J. Radiol. 2011, 84, 967–996. [Google Scholar] [CrossRef]
- Sterzing, F.; Engenhart-Cabillic, R.; Flentje, M.; Debus, J. Image-Guided Radiotherapy: A New Dimension in Radiation Oncology. Dtsch. Ärzteblatt Int. 2011, 108, 274. [Google Scholar] [CrossRef]
- Schreibmann, E.; Thorndyke, B.; Li, T.; Wang, J.; Xing, L. Four-Dimensional Image Registration for Image-Guided Radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2008, 71, 578–586. [Google Scholar] [CrossRef]
- Chandra, R.A.; Keane, F.K.; Voncken, F.E.M.; Thomas, C.R. Contemporary Radiotherapy: Present and Future. Lancet 2021, 398, 171–184. [Google Scholar] [CrossRef]
- Fiorica, F. Postoperative Radiotherapy and Lung Cancer in Stage III: Helpful or Harmful. J. Thorac. Dis. 2017, 9, E277–E278. [Google Scholar] [CrossRef]
- Fiorica, F.; Belluomini, L.; Stefanelli, A.; Santini, A.; Urbini, B.; Giorgi, C.; Frassoldati, A. Immune Checkpoint Inhibitor Nivolumab and Radiotherapy in Pretreated Lung Cancer Patients: Efficacy and Safety of Combination. Am. J. Clin. Oncol. 2018, 41, 1101–1105. [Google Scholar] [CrossRef] [PubMed]
- Fiorica, F.; Tebano, U.; Gabbani, M.; Perrone, M.; Missiroli, S.; Berretta, M.; Giuliani, J.; Bonetti, A.; Remo, A.; Pigozzi, E.; et al. Beyond Abscopal Effect: A Meta-Analysis of Immune Checkpoint Inhibitors and Radiotherapy in Advanced Non-Small Cell Lung Cancer. Cancers 2021, 13, 2352. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, W.; Zhao, Z.; Lv, J.; Chen, J.; Yan, X.; Lin, X.; Zhang, J.; Wang, B.; Gao, S.; et al. Current Views on Mechanisms of the FLASH Effect in Cancer Radiotherapy. Natl. Sci. Rev. 2024, 11, nwae350. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.C.L.; Ruda, H.E. Mechanisms of Action in FLASH Radiotherapy: A Comprehensive Review of Physicochemical and Biological Processes on Cancerous and Normal Cells. Cells 2024, 13, 835. [Google Scholar] [CrossRef]
- Chen, Q.; Fang, C.; Xia, F.; Wang, Q.; Li, F.; Ling, D. Metal Nanoparticles for Cancer Therapy: Precision Targeting of DNA Damage. Acta Pharm. Sin. B 2024, 14, 1132–1149. [Google Scholar] [CrossRef]
- Chow, J.C.L. Biophysical Insights into Nanomaterial-Induced DNA Damage: Mechanisms, Challenges, and Future Directions. AIMSBPOA 2024, 11, 340–369. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519. [Google Scholar] [CrossRef]
- Gong, B.; Chen, Q.; Almasan, A. Ionizing Radiation Stimulates Mitochondrial Gene Expression and Activity. Radiat. Res. 1998, 150, 505–512. [Google Scholar] [CrossRef]
- Lafargue, A.; Degorre, C.; Corre, I.; Alves-Guerra, M.-C.; Gaugler, M.-H.; Vallette, F.; Pecqueur, C.; Paris, F. Ionizing Radiation Induces Long-Term Senescence in Endothelial Cells through Mitochondrial Respiratory Complex II Dysfunction and Superoxide Generation. Free Radic. Biol. Med. 2017, 108, 750–759. [Google Scholar] [CrossRef]
- Dwarakanath, B.S.; Jain, V.K. Modification of the Radiation Induced Damage by 2-Deoxy-D-Glucose in Organ Cultures of Human Cerebral Gliomas. Int. J. Radiat. Oncol. Biol. Phys. 1987, 13, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Banerji, A.K.; Dwarakanath, B.S.; Tripathi, R.P.; Gupta, J.P.; Mathew, T.L.; Ravindranath, T.; Jain, V. Optimizing Cancer Radiotherapy with 2-Deoxy-d-Glucose Dose Escalation Studies in Patients with Glioblastoma Multiforme. Strahlenther. Onkol. 2005, 181, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Dwarakanath, B.S.; Singh, D.; Banerji, A.K.; Sarin, R.; Venkataramana, N.K.; Jalali, R.; Vishwanath, P.N.; Mohanti, B.K.; Tripathi, R.P.; Kalia, V.K.; et al. Clinical Studies for Improving Radiotherapy with 2-Deoxy-D-Glucose: Present Status and Future Prospects. J. Cancer Res. Ther. 2009, 5, S21. [Google Scholar] [CrossRef] [PubMed]
- Magno, L.; Terraneo, F.; Bertoni, F.; Tordiglione, M.; Bardelli, D.; Rosignoli, M.T.; Ciottoli, G.B. Double-Blind Randomized Study of Lonidamine and Radiotherapy in Head and Neck Cancer. Int. J. Radiat. Oncol. Biol. Phys. 1994, 29, 45–55. [Google Scholar] [CrossRef]
- Zerp, S.F.; Stoter, T.R.; Hoebers, F.J.P.; van den Brekel, M.W.M.; Dubbelman, R.; Kuipers, G.K.; Lafleur, M.V.M.; Slotman, B.J.; Verheij, M. Targeting Anti-Apoptotic Bcl-2 by AT-101 to Increase Radiation Efficacy: Data from in Vitro and Clinical Pharmacokinetic Studies in Head and Neck Cancer. Radiat. Oncol. 2015, 10, 158. [Google Scholar] [CrossRef]
- Song, S.; Chen, Q.; Li, Y.; Lei, G.; Scott, A.; Huo, L.; Li, C.Y.; Estrella, J.S.; Correa, A.; Pizzi, M.P.; et al. Targeting Cancer Stem Cells with a Pan-BCL-2 Inhibitor in Preclinical and Clinical Settings in Patients with Gastroesophageal Carcinoma. Gut 2021, 70, 2238–2248. [Google Scholar] [CrossRef]
- Kawada, K.; Iwamoto, M.; Sakai, Y. Mechanisms Underlying18 F-Fluorodeoxyglucose Accumulation in Colorectal Cancer. WJR 2016, 8, 880. [Google Scholar] [CrossRef]
- Zhao, Z.; Mei, Y.; Wang, Z.; He, W. The Effect of Oxidative Phosphorylation on Cancer Drug Resistance. Cancers 2022, 15, 62. [Google Scholar] [CrossRef]
- Rao, M.; Gao, C.; Guo, M.; Law, B.Y.K.; Xu, Y. Effects of Metformin Treatment on Radiotherapy Efficacy in Patients with Cancer and Diabetes: A Systematic Review and Meta-Analysis. Cancer Manag. Res. 2018, 10, 4881. [Google Scholar] [CrossRef]
- Liu, K.X.; Everdell, E.; Pal, S.; Haas-Kogan, D.A.; Milligan, M.G. Harnessing Lactate Metabolism for Radiosensitization. Front. Oncol. 2021, 11, 672339. [Google Scholar] [CrossRef]
- Zhang, L.; Bailleul, J.; Yazal, T.; Dong, K.; Sung, D.; Dao, A.; Gosa, L.; Nathanson, D.; Bhat, K.; Duhachek-Muggy, S.; et al. PK-M2-Mediated Metabolic Changes in Breast Cancer Cells Induced by Ionizing Radiation. Breast Cancer Res. Treat 2019, 178, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.; Tsolou, A.; Pouliliou, S.; Lamprou, I.; Papadopoulou, M.; Ilemosoglou, M.; Kostoglou, G.; Ananiadou, D.; Sivridis, E.; Giatromanolaki, A. Blocking LDHA Glycolytic Pathway Sensitizes Glioblastoma Cells to Radiation and Temozolomide. Biochem. Biophys. Res. Commun. 2017, 491, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lu, Y.; Hang, J.; Zhang, J.; Zhang, T.; Huo, Y.; Liu, J.; Lai, S.; Luo, D.; Wang, L.; et al. Lactate-Modulated Immunosuppression of Myeloid-Derived Suppressor Cells Contributes to the Radioresistance of Pancreatic Cancer. Cancer Immunol. Res. 2020, 8, 1440–1451. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A Mitochondria-K+ Channel Axis Is Suppressed in Cancer and Its Normalization Promotes Apoptosis and Inhibits Cancer Growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef]
- Dong, G.; Chen, Q.; Jiang, F.; Yu, D.; Mao, Q.; Xia, W.; Shi, R.; Wang, J.; Xu, L. Diisopropylamine Dichloroacetate Enhances Radiosensitization in Esophageal Squamous Cell Carcinoma by Increasing Mitochondria-Derived Reactive Oxygen Species Levels. Oncotarget 2016, 7, 68170–68178. [Google Scholar] [CrossRef]
- Yu, Y.; Yu, J.; Ge, S.; Su, Y.; Fan, X. Novel Insight into Metabolic Reprogrammming in Cancer Radioresistance: A Promising Therapeutic Target in Radiotherapy. Int. J. Biol. Sci. 2023, 19, 811–828. [Google Scholar] [CrossRef]
- Khan, A.; Andrews, D.; Blackburn, A.C. Long-Term Stabilization of Stage 4 Colon Cancer Using Sodium Dichloroacetate Therapy. World J. Clin. Cases 2016, 4, 336–343. [Google Scholar] [CrossRef]
- Powell, S.F.; Mazurczak, M.; Dib, E.G.; Bleeker, J.S.; Geeraerts, L.H.; Tinguely, M.; Lohr, M.M.; McGraw, S.C.; Jensen, A.W.; Ellison, C.A.; et al. Phase II Study of Dichloroacetate, an Inhibitor of Pyruvate Dehydrogenase, in Combination with Chemoradiotherapy for Unresected, Locally Advanced Head and Neck Squamous Cell Carcinoma. Investig. New Drugs 2022, 40, 622–633. [Google Scholar] [CrossRef]
- Bonora, M.; Missiroli, S.; Perrone, M.; Fiorica, F.; Pinton, P.; Giorgi, C. Mitochondrial Control of Genomic Instability in Cancer. Cancers 2021, 13, 1914. [Google Scholar] [CrossRef]
- Fu, Y.; Zou, T.; Shen, X.; Nelson, P.J.; Li, J.; Wu, C.; Yang, J.; Zheng, Y.; Bruns, C.; Zhao, Y.; et al. Lipid Metabolism in Cancer Progression and Therapeutic Strategies. MedComm 2021, 2, 27–59. [Google Scholar] [CrossRef]
- Vanauberg, D.; Schulz, C.; Lefebvre, T. Involvement of the Pro-Oncogenic Enzyme Fatty Acid Synthase in the Hallmarks of Cancer: A Promising Target in Anti-Cancer Therapies. Oncogenesis 2023, 12, 16. [Google Scholar] [CrossRef] [PubMed]
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef] [PubMed]
- De Martino, M.; Daviaud, C.; Minns, H.E.; Lazarian, A.; Wacker, A.; Costa, A.P.; Attarwala, N.; Chen, Q.; Choi, S.-W.; Rabadàn, R.; et al. Radiation Therapy Promotes Unsaturated Fatty Acids to Maintain Survival of Glioblastoma. Cancer Lett. 2023, 570, 216329. [Google Scholar] [CrossRef] [PubMed]
- Rae, C.; Fragkoulis, G.I.; Chalmers, A.J. Cytotoxicity and Radiosensitizing Activity of the Fatty Acid Synthase Inhibitor C75 Is Enhanced by Blocking Fatty Acid Uptake in Prostate Cancer Cells. Adv. Radiat. Oncol. 2020, 5, 994–1005. [Google Scholar] [CrossRef]
- Zhan, N.; Li, B.; Xu, X.; Xu, J.; Hu, S. Inhibition of FASN Expression Enhances Radiosensitivity in Human Non-Small Cell Lung Cancer. Oncol. Lett. 2018, 15, 4578–4584. [Google Scholar] [CrossRef]
- Rae, C.; Haberkorn, U.; Babich, J.W.; Mairs, R.J. Inhibition of Fatty Acid Synthase Sensitizes Prostate Cancer Cells to Radiotherapy. Radiat Res. 2015, 184, 482–493. [Google Scholar] [CrossRef]
- Falchook, G.; Infante, J.; Arkenau, H.-T.; Patel, M.R.; Dean, E.; Borazanci, E.; Brenner, A.; Cook, N.; Lopez, J.; Pant, S.; et al. First-in-Human Study of the Safety, Pharmacokinetics, and Pharmacodynamics of First-in-Class Fatty Acid Synthase Inhibitor TVB-2640 Alone and with a Taxane in Advanced Tumors. eClinicalMedicine 2021, 34, 100797. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of Fatty Acid Metabolism in Cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef]
- Dheeraj, A.; Agarwal, C.; Schlaepfer, I.R.; Raben, D.; Singh, R.; Agarwal, R.; Deep, G. A Novel Approach to Target Hypoxic Cancer Cells via Combining β-Oxidation Inhibitor Etomoxir with Radiation. Hypoxia 2018, 6, 23–33. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Jin, Y.; Tan, Y.; Wu, J.; Ren, Z. Lipid Droplets: A Cellular Organelle Vital in Cancer Cells. Cell Death Discov. 2023, 9, 254. [Google Scholar] [CrossRef] [PubMed]
- Tirinato, L.; Marafioti, M.G.; Pagliari, F.; Jansen, J.; Aversa, I.; Hanley, R.; Nisticò, C.; Garcia-Calderón, D.; Genard, G.; Guerreiro, J.F.; et al. Lipid Droplets and Ferritin Heavy Chain: A Devilish Liaison in Human Cancer Cell Radioresistance. eLife 2021, 10, e72943. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Ganji, S.; Pass, I.; Ardecky, R.; Peddibhotla, M.; Loribelle, M.; Heynen-Genel, S.; Sauer, M.; Pass, I.; Vasile, S.; et al. Potent Inhibitors of Lipid Droplet Formation. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Nisticò, C.; Pagliari, F.; Chiarella, E.; Fernandes Guerreiro, J.; Marafioti, M.G.; Aversa, I.; Genard, G.; Hanley, R.; Garcia-Calderón, D.; Bond, H.M.; et al. Lipid Droplet Biosynthesis Impairment through DGAT2 Inhibition Sensitizes MCF7 Breast Cancer Cells to Radiation. Int. J. Mol. Sci. 2021, 22, 10102. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine Reliance in Cell Metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- Liu, X.; Ren, B.; Ren, J.; Gu, M.; You, L.; Zhao, Y. The Significant Role of Amino Acid Metabolic Reprogramming in Cancer. Cell Commun. Signal. 2024, 22, 380. [Google Scholar] [CrossRef]
- Glutamine Reliance in Cell Metabolism | Experimental & Molecular Medicine. Available online: https://www.nature.com/articles/s12276-020-00504-8 (accessed on 27 October 2024).
- Sappington, D.R.; Siegel, E.R.; Hiatt, G.; Desai, A.; Penney, R.B.; Jamshidi-Parsian, A.; Griffin, R.J.; Boysen, G. Glutamine Drives Glutathione Synthesis and Contributes to Radiation Sensitivity of A549 and H460 Lung Cancer Cell Lines. Biochim. Biophys. Acta 2016, 1860, 836–843. [Google Scholar] [CrossRef]
- Mattaini, K.R.; Sullivan, M.R.; Vander Heiden, M.G. The Importance of Serine Metabolism in Cancer. J. Cell Biol. 2016, 214, 249–257. [Google Scholar] [CrossRef]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and Glycine Metabolism in Cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef]
- Lee, C.M.; Hwang, Y.; Kim, M.; Park, Y.-C.; Kim, H.; Fang, S. PHGDH: A Novel Therapeutic Target in Cancer. Exp. Mol. Med. 2024, 56, 1513–1522. [Google Scholar] [CrossRef]
- Keshet, R.; Erez, A. Arginine and the Metabolic Regulation of Nitric Oxide Synthesis in Cancer. Dis. Model Mech. 2018, 11, dmm033332. [Google Scholar] [CrossRef]
- Martí i Líndez, A.-A.; Reith, W. Arginine-Dependent Immune Responses. Cell. Mol. Life Sci. 2021, 78, 5303–5324. [Google Scholar] [CrossRef] [PubMed]
- Marullo, R.; Castro, M.; Yomtoubian, S.; Calvo-Vidal, M.N.; Revuelta, M.V.; Krumsiek, J.; Cho, A.; Morgado, P.C.; Yang, S.; Medina, V.; et al. The Metabolic Adaptation Evoked by Arginine Enhances the Effect of Radiation in Brain Metastases. Sci. Adv. 2021, 7, eabg1964. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.N.; Capuk, O.; Patel, S.M.; Sun, D. The Role of Metabolic Plasticity of Tumor-Associated Macrophages in Shaping the Tumor Microenvironment Immunity. Cancers 2022, 14, 3331. [Google Scholar] [CrossRef]
- Fernando, V.; Zheng, X.; Sharma, V.; Furuta, S. Reprogramming of Breast Tumor-Associated Macrophages with Modulation of Arginine Metabolism. bioRxiv 2023. [Google Scholar] [CrossRef]
- Chen, C.-L.; Hsu, S.-C.; Ann, D.K.; Yen, Y.; Kung, H.-J. Arginine Signaling and Cancer Metabolism. Cancers 2021, 13, 3541. [Google Scholar] [CrossRef]
- Feun, L.; Savaraj, N. Pegylated Arginine Deiminase: A Novel Anticancer Enzyme Agent. Expert Opin. Investig. Drugs 2006, 15, 815–822. [Google Scholar] [CrossRef]
- Park, H.; Lee, J.-B.; Shim, Y.-J.; Shin, Y.-J.; Jeong, S.-Y.; Oh, J.; Park, G.-H.; Lee, K.-H.; Min, B.-H. Arginine Deiminase Enhances MCF-7 Cell Radiosensitivity by Inducing Changes in the Expression of Cell Cycle-Related Proteins. Mol. Cells 2008, 25, 305–311. [Google Scholar] [CrossRef]
- Singh, P.K.; Deorukhkar, A.A.; Venkatesulu, B.P.; Li, X.; Tailor, R.; Bomalaski, J.S.; Krishnan, S. Exploiting Arginine Auxotrophy with Pegylated Arginine Deiminase (ADI-PEG20) to Sensitize Pancreatic Cancer to Radiotherapy via Metabolic Dysregulation. Mol. Cancer Ther. 2019, 18, 2381–2393. [Google Scholar] [CrossRef]
- Yuan, Q.; Yin, L.; He, J.; Zeng, Q.; Liang, Y.; Shen, Y.; Zu, X. Metabolism of Asparagine in the Physiological State and Cancer. Cell Commun. Signal. 2024, 22, 163. [Google Scholar] [CrossRef]
- Chiu, M.; Taurino, G.; Bianchi, M.G.; Kilberg, M.S.; Bussolati, O. Asparagine Synthetase in Cancer: Beyond Acute Lymphoblastic Leukemia. Front. Oncol. 2020, 9, 1480. [Google Scholar] [CrossRef]
- Wang, X.; Gong, W.; Xiong, X.; Jia, X.; Xu, J. Asparagine: A Key Metabolic Junction in Targeted Tumor Therapy. Pharmacol Res 2024, 206, 107292. [Google Scholar] [CrossRef] [PubMed]
- Nie, C.; He, T.; Zhang, W.; Zhang, G.; Ma, X. Branched Chain Amino Acids: Beyond Nutrition Metabolism. Int. J. Mol. Sci. 2018, 19, 954. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.K.; Okekunle, A.P.; Lee, J.E.; Sung, M.K.; Lim, Y.J. Role of Branched-Chain Amino Acid Metabolism in Tumor Development and Progression. J. Cancer Prev. 2021, 26, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Xu, E.; Ji, B.; Jin, K.; Chen, Y. Branched-Chain Amino Acids Catabolism and Cancer Progression: Focus on Therapeutic Interventions. Front. Oncol. 2023, 13, 1220638. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Metabolic Pathway | Function in Cancer | Effect of Radiotherapy | Inhibitors |
|---|---|---|---|
| Glycolysis | Rapid energy production via glucose conversion to lactate (Warburg effect) | Increased reliance on glycolysis due to mitochondrial dysfunction | 2-DG, Lonidamine, Gossypol |
| Oxidative Phosphorylation (OXPHOS) | Energy production through mitochondrial activity | Mitochondrial damage impairs ATP generation | Metformin |
| Fatty Acid Synthesis | Lipid production for membranes and energy storage | Disruption of lipid synthesis pathways | Orlistat, TVB-2640 |
| Fatty Acid Oxidation (FAO) | Breakdown of fatty acids for energy, especially in hypoxic conditions | Impaired FAO due to mitochondrial damage | Etomoxir |
| Lactate Metabolism | Lactate accumulation supports tumour microenvironment, immune evasion | Increased acidity in the tumour microenvironment | Galloflavin, DCA |
| Glutamine Metabolism | Supports nucleotide synthesis, redox balance (glutathione production) | Increased oxidative stress depletes glutathione | CB-839 (Glutaminase inhibitor) |
| Serine and Glycine Metabolism | One-carbon metabolism for nucleotide synthesis, redox balance | Increased demand for nucleotide synthesis for DNA repair | PHGDH inhibitors |
| Arginine Metabolism | Polyamine and nitric oxide production, immune modulation | Modifies immune responses and NO production | Arginine Deiminase (ADI) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiorica, F.; Tebano, U.; Napoli, G.; Franceschetto, A.; Muraro, M.; Giorgi, C.; Pinton, P. Metabolic-Modulating Effects of Radiation: Undetectable Yet Deadly—A Review on Radiotherapy. Cancers 2025, 17, 54. https://doi.org/10.3390/cancers17010054
Fiorica F, Tebano U, Napoli G, Franceschetto A, Muraro M, Giorgi C, Pinton P. Metabolic-Modulating Effects of Radiation: Undetectable Yet Deadly—A Review on Radiotherapy. Cancers. 2025; 17(1):54. https://doi.org/10.3390/cancers17010054
Chicago/Turabian StyleFiorica, Francesco, Umberto Tebano, Giuseppe Napoli, Antonella Franceschetto, Marco Muraro, Carlotta Giorgi, and Paolo Pinton. 2025. "Metabolic-Modulating Effects of Radiation: Undetectable Yet Deadly—A Review on Radiotherapy" Cancers 17, no. 1: 54. https://doi.org/10.3390/cancers17010054
APA StyleFiorica, F., Tebano, U., Napoli, G., Franceschetto, A., Muraro, M., Giorgi, C., & Pinton, P. (2025). Metabolic-Modulating Effects of Radiation: Undetectable Yet Deadly—A Review on Radiotherapy. Cancers, 17(1), 54. https://doi.org/10.3390/cancers17010054