
Predicting the Progression from Asymptomatic to Symptomatic Multiple Myeloma and Stage Classification Using Gene Expression Data


Simple Summary
Abstract
1. Introduction
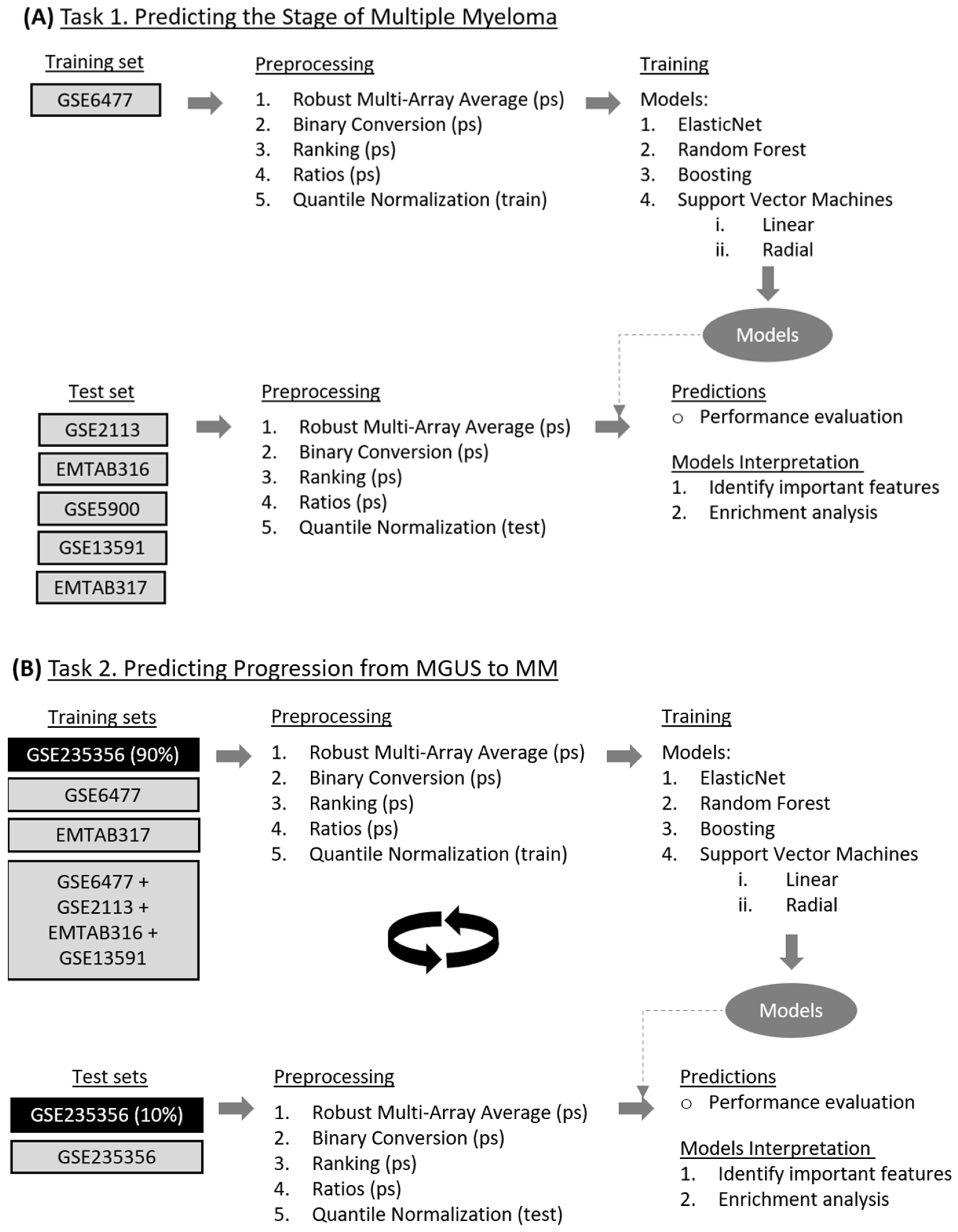
2. Materials and Methods
2.1. Source of Microarray Datasets and Description of Data Variables and Features
2.2. Data Cleaning and Preprocessing Techniques
2.3. Overview of Machine Learning (ML) Algorithms
2.4. Model Training and Interpretation
3. Results
3.1. Task 1—Predicting the Stage of Multiple Myeloma
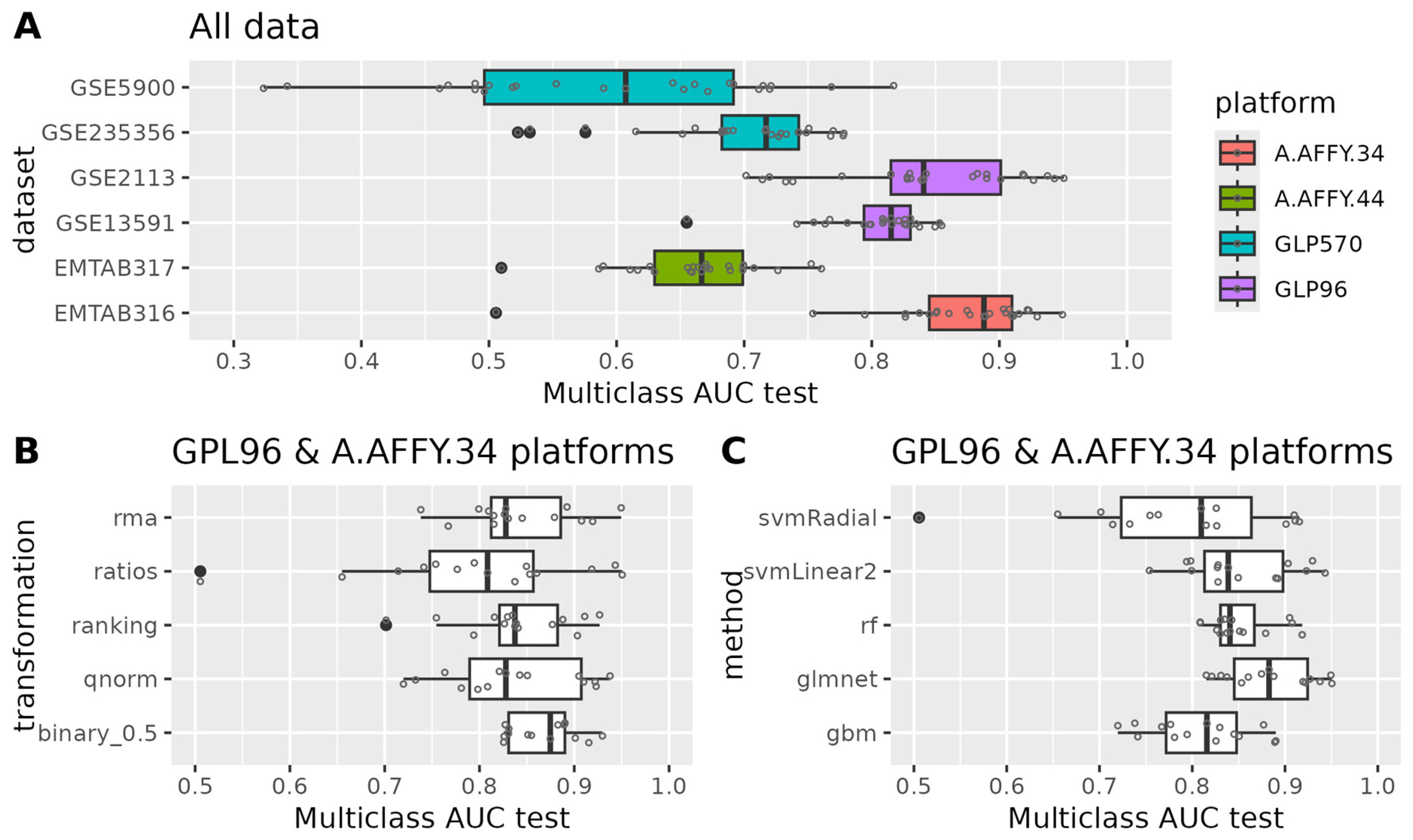
3.1.1. Model Development for Disease Staging
3.1.2. Evaluation of Model Performance
3.1.3. Model Interpretation
3.2. Task 2—Predicting Progression from MGUS to MM
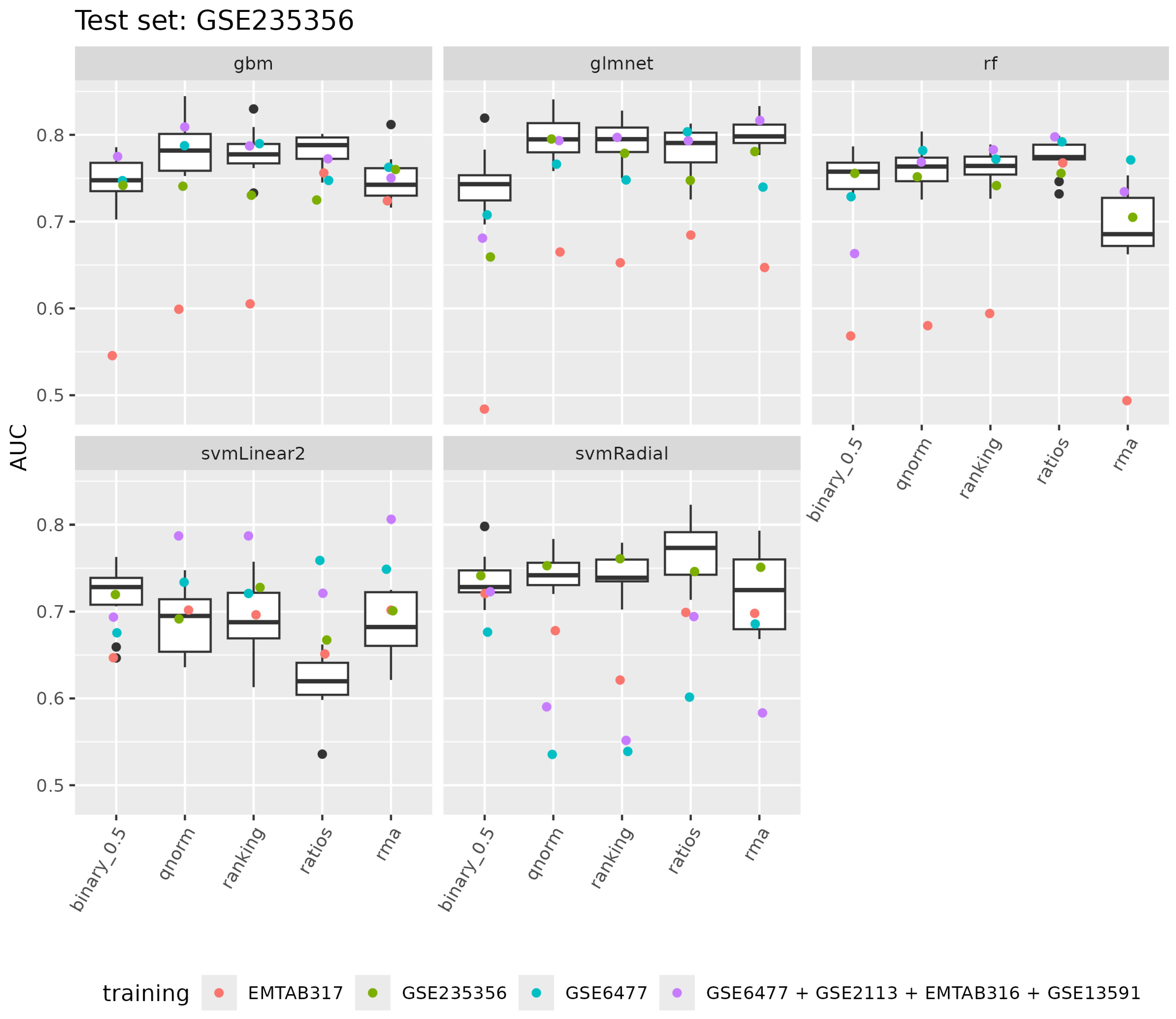
3.2.1. Model Development for Disease Progression Prediction
3.2.2. Evaluation of Model Performance
3.2.3. Model Interpretation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.-V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group Updated Criteria for the Diagnosis of Multiple Myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Kumar, S. Multiple Myeloma Current Treatment Algorithms. Blood Cancer J. 2020, 10, 94. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Larson, D.R.; Plevak, M.F.; Melton, L.J. Incidence of Multiple Myeloma in Olmsted County, Minnesota. Cancer 2004, 101, 2667–2674. [Google Scholar] [CrossRef] [PubMed]
- Landgren, O.; Weiss, B.M. Patterns of Monoclonal Gammopathy of Undetermined Significance and Multiple Myeloma in Various Ethnic/Racial Groups: Support for Genetic Factors in Pathogenesis. Leukemia 2009, 23, 1691–1697. [Google Scholar] [CrossRef]
- Kyle, R.A.; Gertz, M.A.; Witzig, T.E.; Lust, J.A.; Lacy, M.Q.; Dispenzieri, A.; Fonseca, R.; Rajkumar, S.V.; Offord, J.R.; Larson, D.R.; et al. Review of 1027 Patients with Newly Diagnosed Multiple Myeloma. Mayo Clin. Proc. 2003, 78, 21–33. [Google Scholar] [CrossRef]
- Landgren, O.; Kyle, R.A.; Pfeiffer, R.M.; Katzmann, J.A.; Caporaso, N.E.; Hayes, R.B.; Dispenzieri, A.; Kumar, S.; Clark, R.J.; Baris, D.; et al. Monoclonal Gammopathy of Undetermined Significance (MGUS) Consistently Precedes Multiple Myeloma: A Prospective Study. Blood 2009, 113, 5412–5417. [Google Scholar] [CrossRef]
- Weiss, B.M.; Abadie, J.; Verma, P.; Howard, R.S.; Kuehl, W.M. A Monoclonal Gammopathy Precedes Multiple Myeloma in Most Patients. Blood 2009, 113, 5418–5422. [Google Scholar] [CrossRef]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Larson, D.R.; Plevak, M.F.; Offord, J.R.; Dispenzieri, A.; Katzmann, J.A.; Melton, L.J. Prevalence of Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2006, 354, 1362–1369. [Google Scholar] [CrossRef]
- Dispenzieri, A.; Katzmann, J.A.; Kyle, R.A.; Larson, D.R.; Melton, L.J.; Colby, C.L.; Therneau, T.M.; Clark, R.; Kumar, S.K.; Bradwell, A.; et al. Prevalence and Risk of Progression of Light-Chain Monoclonal Gammopathy of Undetermined Significance: A Retrospective Population-Based Cohort Study. Lancet 2010, 375, 1721–1728. [Google Scholar] [CrossRef]
- Murray, D.; Kumar, S.K.; Kyle, R.A.; Dispenzieri, A.; Dasari, S.; Larson, D.R.; Vachon, C.; Cerhan, J.R.; Rajkumar, S.V. Detection and Prevalence of Monoclonal Gammopathy of Undetermined Significance: A Study Utilizing Mass Spectrometry-Based Monoclonal Immunoglobulin Rapid Accurate Mass Measurement. Blood Cancer J. 2019, 9, 102. [Google Scholar] [CrossRef] [PubMed]
- Landgren, O.; Graubard, B.I.; Kumar, S.; Kyle, R.A.; Katzmann, J.A.; Murata, K.; Costello, R.; Dispenzieri, A.; Caporaso, N.; Mailankody, S.; et al. Prevalence of Myeloma Precursor State Monoclonal Gammopathy of Undetermined Significance in 12 372 Individuals 10–49 Years Old: A Population-Based Study from the National Health and Nutrition Examination Survey. Blood Cancer J. 2017, 7, e618. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Offord, J.R.; Larson, D.R.; Plevak, M.F.; Melton, L.J. A Long-Term Study of Prognosis in Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2002, 346, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Larson, D.R.; Therneau, T.M.; Dispenzieri, A.; Kumar, S.; Cerhan, J.R.; Rajkumar, S.V. Long-Term Follow-up of Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2018, 378, 241–249. [Google Scholar] [CrossRef]
- Therneau, T.M.; Kyle, R.A.; Melton, L.J.; Larson, D.R.; Benson, J.T.; Colby, C.L.; Dispenzieri, A.; Kumar, S.; Katzmann, J.A.; Cerhan, J.R.; et al. Incidence of Monoclonal Gammopathy of Undetermined Significance and Estimation of Duration before First Clinical Recognition. Mayo Clin. Proc. 2012, 87, 1071–1079. [Google Scholar] [CrossRef]
- Kyle, R.A.; Remstein, E.D.; Therneau, T.M.; Dispenzieri, A.; Kurtin, P.J.; Hodnefield, J.M.; Larson, D.R.; Plevak, M.F.; Jelinek, D.F.; Fonseca, R.; et al. Clinical Course and Prognosis of Smoldering (Asymptomatic) Multiple Myeloma. N. Engl. J. Med. 2007, 356, 2582–2590. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Gupta, V.; Fonseca, R.; Dispenzieri, A.; Gonsalves, W.I.; Larson, D.; Ketterling, R.P.; Lust, J.A.; Kyle, R.A.; Kumar, S.K. Impact of Primary Molecular Cytogenetic Abnormalities and Risk of Progression in Smoldering Multiple Myeloma. Leukemia 2013, 27, 1738–1744. [Google Scholar] [CrossRef]
- Neben, K.; Jauch, A.; Hielscher, T.; Hillengass, J.; Lehners, N.; Seckinger, A.; Granzow, M.; Raab, M.S.; Ho, A.D.; Goldschmidt, H.; et al. Progression in Smoldering Myeloma Is Independently Determined by the Chromosomal Abnormalities Del(17p), t(4;14), Gain 1q, Hyperdiploidy, and Tumor Load. J. Clin. Oncol. 2013, 31, 4325–4332. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple Myeloma: 2022 Update on Diagnosis, Risk Stratification, and Management. Am. J. Hematol. 2022, 97, 1086–1107. [Google Scholar] [CrossRef]
- Ho, M.; Patel, A.; Goh, C.Y.; Moscvin, M.; Zhang, L.; Bianchi, G. Changing Paradigms in Diagnosis and Treatment of Monoclonal Gammopathy of Undetermined Significance (MGUS) and Smoldering Multiple Myeloma (SMM). Leukemia 2020, 34, 3111–3125. [Google Scholar] [CrossRef]
- Mateos, M.-V.; Hernández, M.-T.; Giraldo, P.; de la Rubia, J.; de Arriba, F.; Corral, L.L.; Rosiñol, L.; Paiva, B.; Palomera, L.; Bargay, J.; et al. Lenalidomide plus Dexamethasone for High-Risk Smoldering Multiple Myeloma. N. Engl. J. Med. 2013, 369, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Jacobus, S.; Fonseca, R.; Weiss, M.; Kumar, S.; Orlowski, R.Z.; Kaufman, J.L.; Yacoub, A.M.; Buadi, F.K.; O’Brien, T.; et al. Randomized Trial of Lenalidomide Versus Observation in Smoldering Multiple Myeloma. J. Clin. Oncol. 2020, 38, 1126–1137. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Kyle, R.A.; Therneau, T.M.; Melton, L.J.; Bradwell, A.R.; Clark, R.J.; Larson, D.R.; Plevak, M.F.; Dispenzieri, A.; Katzmann, J.A. Serum Free Light Chain Ratio Is an Independent Risk Factor for Progression in Monoclonal Gammopathy of Undetermined Significance. Blood 2005, 106, 812–817. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Persona, E.; Vidriales, M.-B.; Mateo, G.; García-Sanz, R.; Mateos, M.-V.; de Coca, A.G.; Galende, J.; Martín-Nuñez, G.; Alonso, J.M.; de las Heras, N.; et al. New Criteria to Identify Risk of Progression in Monoclonal Gammopathy of Uncertain Significance and Smoldering Multiple Myeloma Based on Multiparameter Flow Cytometry Analysis of Bone Marrow Plasma Cells. Blood 2007, 110, 2586–2592. [Google Scholar] [CrossRef] [PubMed]
- Dispenzieri, A.; Rajkumar, S.V.; Gertz, M.A.; Lacy, M.Q.; Kyle, R.A.; Greipp, P.R.; Witzig, T.E.; Lust, J.A.; Russell, S.J.; Hayman, S.R.; et al. Treatment of Newly Diagnosed Multiple Myeloma Based on Mayo Stratification of Myeloma and Risk-Adapted Therapy (MSMART): Consensus Statement. Mayo Clin. Proc. 2007, 82, 323–341. [Google Scholar] [CrossRef]
- Allegra, A.; Tonacci, A.; Sciaccotta, R.; Genovese, S.; Musolino, C.; Pioggia, G.; Gangemi, S. Machine Learning and Deep Learning Applications in Multiple Myeloma Diagnosis, Prognosis, and Treatment Selection. Cancers 2022, 14, 606. [Google Scholar] [CrossRef]
- Sun, F.; Cheng, Y.; Ying, J.; Mery, D.; Al Hadidi, S.; Wanchai, V.; Siegel, E.R.; Xu, H.; Gai, D.; Ashby, T.C.; et al. A Gene Signature Can Predict Risk of MGUS Progressing to Multiple Myeloma. J. Hematol. Oncol. 2023, 16, 70. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models Via Coordiante Descent. J. Stat. Softw. 2010, 33, 1. [Google Scholar] [CrossRef]
- Hastie, T.; Tibshirani, R.; James, G.; Witten, D. An Introduction to Statistical Learning, with Applications in R; Springer: Berlin/Heidelberg, Germany, 2021; Volume 102, ISBN 9780387781884. [Google Scholar]
- Natekin, A.; Knoll, A. Gradient Boosting Machines, a Tutorial. Front. Neurorobot. 2013, 7, 21. [Google Scholar] [CrossRef]
- Meyer, D. Support Vector Machines: The Interface to Libsvm in Package E1071, 1st ed.; Springer: New York, NY, USA, 2014; pp. 1–8. [Google Scholar] [CrossRef]
- Karatzoglou, A.; Smola, A.; Hornik, K.; Zeileis, A. Kernlab—An S4 Package for Kernel Methods in R. J. Stat. Softw. 2004, 11, 389–393. [Google Scholar] [CrossRef]
- Kuhn, M. Building Predictive Models in R Using the Caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Yan, G.R.; He, Q.Y. DOSE: An R/Bioconductor Package for Disease Ontology Semantic and Enrichment Analysis. Bioinformatics 2015, 31, 608–609. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Qu, Y.; Wang, M.; Chu, B.; Chen, W.; Zheng, Y.; Niu, T.; Qian, Z. Pathogenesis and Treatment of Multiple Myeloma. MedComm 2022, 3, e146. [Google Scholar] [CrossRef]
- John, L.; Krauth, M.T.; Podar, K.; Raab, M.S. Pathway-Directed Therapy in Multiple Myeloma. Cancers 2021, 13, 1668. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Kumar, S.; VanWier, S.; Ahmann, G.; Price-Troska, T.; Henderson, K.; Chung, T.H.; Kim, S.; Mulligan, G.; Bryant, B.; et al. Molecular Dissection of Hyperdiploid Multiple Myeloma by Gene Expression Profiling. Cancer Res. 2007, 67, 2982–2989. [Google Scholar] [CrossRef]
- Zhong, H.; Huang, D.; Wu, J.; Chen, X.; Chen, Y.; Huang, C. 18F-FDG PET/CT Based Radiomics Features Improve Prediction of Prognosis: Multiple Machine Learning Algorithms and Multimodality Applications for Multiple Myeloma. BMC Med. Imaging 2023, 23, 87. [Google Scholar] [CrossRef]
- Franks, J.M.; Cai, G.; Whitfield, M.L. Feature Specific Quantile Normalization Enables Cross-Platform Classification of Molecular Subtypes Using Gene Expression Data. Bioinformatics 2018, 34, 1868–1874. [Google Scholar] [CrossRef]
- Foltz, S.M.; Greene, C.S.; Taroni, J.N. Cross-Platform Normalization Enables Machine Learning Model Training on Microarray and RNA-Seq Data Simultaneously. Commun. Biol. 2023, 6, 222. [Google Scholar] [CrossRef]
- Awada, H.; Thapa, B.; Awada, H.; Dong, J.; Gurnari, C.; Hari, P.; Dhakal, B. A Comprehensive Review of the Genomics of Multiple Myeloma: Evolutionary Trajectories, Gene Expression Profiling, and Emerging Therapeutics. Cells 2021, 10, 1961. [Google Scholar] [CrossRef]
- Lu, Q.; Yang, D.; Li, H.; Niu, T.; Tong, A. Multiple Myeloma: Signaling Pathways and Targeted Therapy; Springer Nature: Singapore, 2024; Volume 5, ISBN 4355602400. [Google Scholar]
- Isa, R.; Horinaka, M.; Tsukamoto, T.; Mizuhara, K.; Fujibayashi, Y.; Taminishi-Katsuragawa, Y.; Okamoto, H.; Yasuda, S.; Kawaji-Kanayama, Y.; Matsumura-Kimoto, Y.; et al. The Rationale for the Dual-Targeting Therapy for RSK2 and AKT in Multiple Myeloma. Int. J. Mol. Sci. 2022, 23, 2919. [Google Scholar] [CrossRef]
- Bahar, M.E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK Pathway for Cancer Therapy: From Mechanism to Clinical Studies. Signal Transduct. Target. Ther. 2023, 8, 455. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Bi, Z.; Liu, Y.; Qin, F.; Wei, Y.; Wei, X. Targeting RAS–RAF–MEK–ERK Signaling Pathway in Human Cancer: Current Status in Clinical Trials. Genes Dis. 2023, 10, 76–88. [Google Scholar] [CrossRef]
- Spaan, I.; Raymakers, R.A.; van de Stolpe, A.; Peperzak, V. Wnt Signaling in Multiple Myeloma: A Central Player in Disease with Therapeutic Potential. J. Hematol. Oncol. 2018, 11, 67. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cui, S.; Lyu, C.; Wang, Z.; Li, Z.; Han, C.; Liu, W.; Wang, Y.; Xu, R. Molecular Precision Medicine: Multi-Omics-Based Stratification Model for Acute Myeloid Leukemia. Heliyon 2024, 10, e36155. [Google Scholar] [CrossRef] [PubMed]
- Correa-Aguila, R.; Alonso-Pupo, N.; Hernández-Rodríguez, E.W. Multi-Omics Data Integration Approaches for Precision Oncology. Mol. Omics 2022, 18, 469–479. [Google Scholar] [CrossRef]
- Li, Y.; Wu, X.; Fang, D.; Luo, Y. Informing Immunotherapy with Multi-Omics Driven Machine Learning. npj Digit. Med. 2024, 7, 67. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Platform | Dataset | Normal | MGUS | Progressing MGUS | MM | Total Number of Samples |
|---|---|---|---|---|---|---|
| GLP96 | GSE2113 | 7 | 39 | 46 | ||
| A-AFFY-33 A-AFFY-34 | EMTAB316 | 7 | 65 | 72 | ||
| GLP570 | GSE5900 | 22 | 44 | 66 | ||
| GLP96 | GSE6477 | 15 | 22 | 73 | 110 | |
| GLP96 | GSE13591 | 5 | 11 | 133 | 149 | |
| A-AFFY-44 | EMTAB317 | 23 | 226 | 249 | ||
| GLP570 | GSE235356 | 319 | 39 | 358 | ||
| Total | 7 | 42 | 433 | 39 | 536 | 1050 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karathanasis, N.; Spyrou, G.M. Predicting the Progression from Asymptomatic to Symptomatic Multiple Myeloma and Stage Classification Using Gene Expression Data. Cancers 2025, 17, 332. https://doi.org/10.3390/cancers17020332
Karathanasis N, Spyrou GM. Predicting the Progression from Asymptomatic to Symptomatic Multiple Myeloma and Stage Classification Using Gene Expression Data. Cancers. 2025; 17(2):332. https://doi.org/10.3390/cancers17020332
Chicago/Turabian StyleKarathanasis, Nestoras, and George M. Spyrou. 2025. "Predicting the Progression from Asymptomatic to Symptomatic Multiple Myeloma and Stage Classification Using Gene Expression Data" Cancers 17, no. 2: 332. https://doi.org/10.3390/cancers17020332
APA StyleKarathanasis, N., & Spyrou, G. M. (2025). Predicting the Progression from Asymptomatic to Symptomatic Multiple Myeloma and Stage Classification Using Gene Expression Data. Cancers, 17(2), 332. https://doi.org/10.3390/cancers17020332