
Pheochromocytomas and Paragangliomas—Current Management


Simple Summary
Abstract
1. Introduction
2. The Strategy of the Literature Research
3. Pheochromocytomas and Paragangliomas According to World Health Organization and American Joint Committee on Cancer Classifications
3.1. WHO Classification
3.2. AJCC Classification
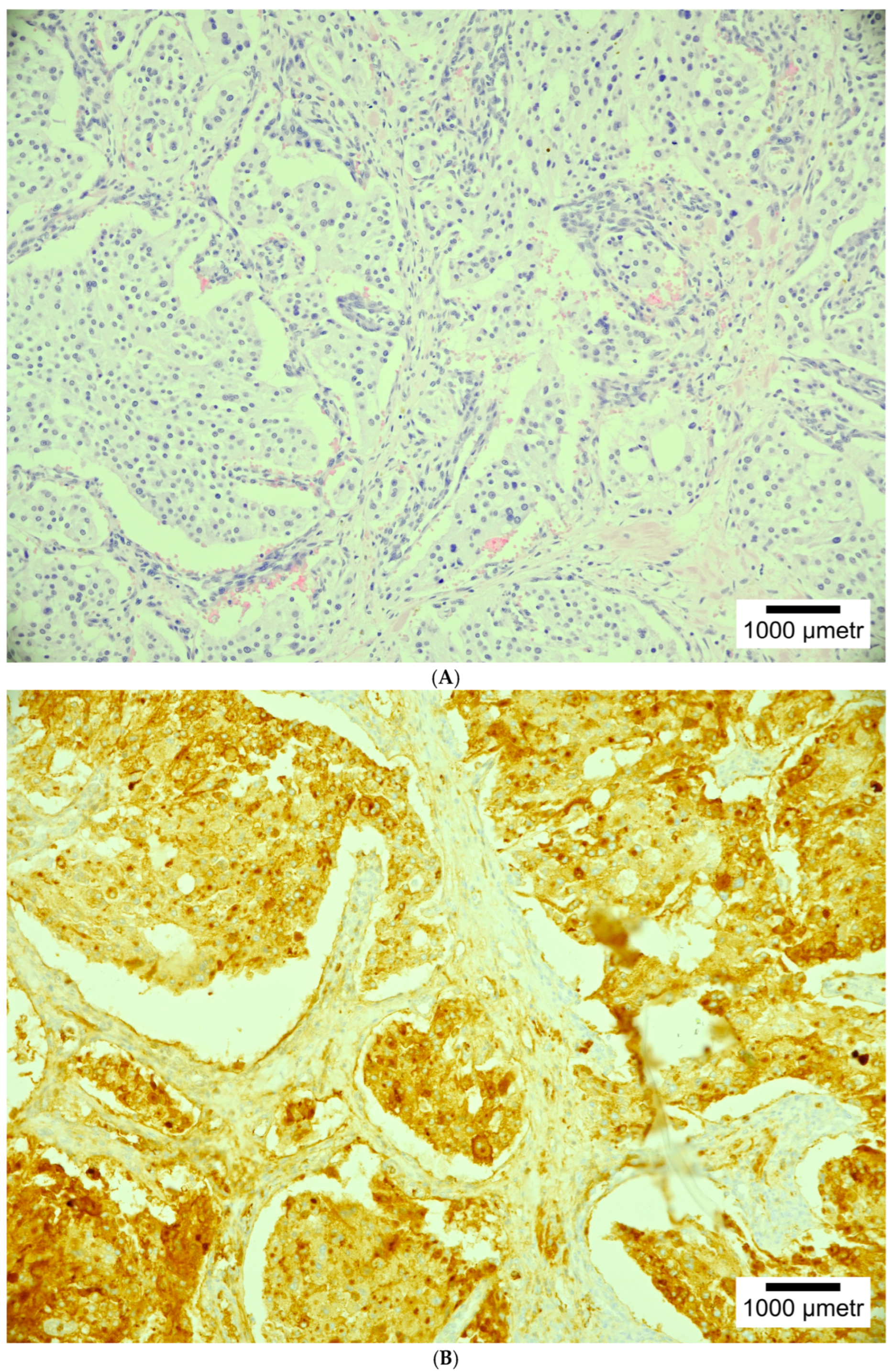
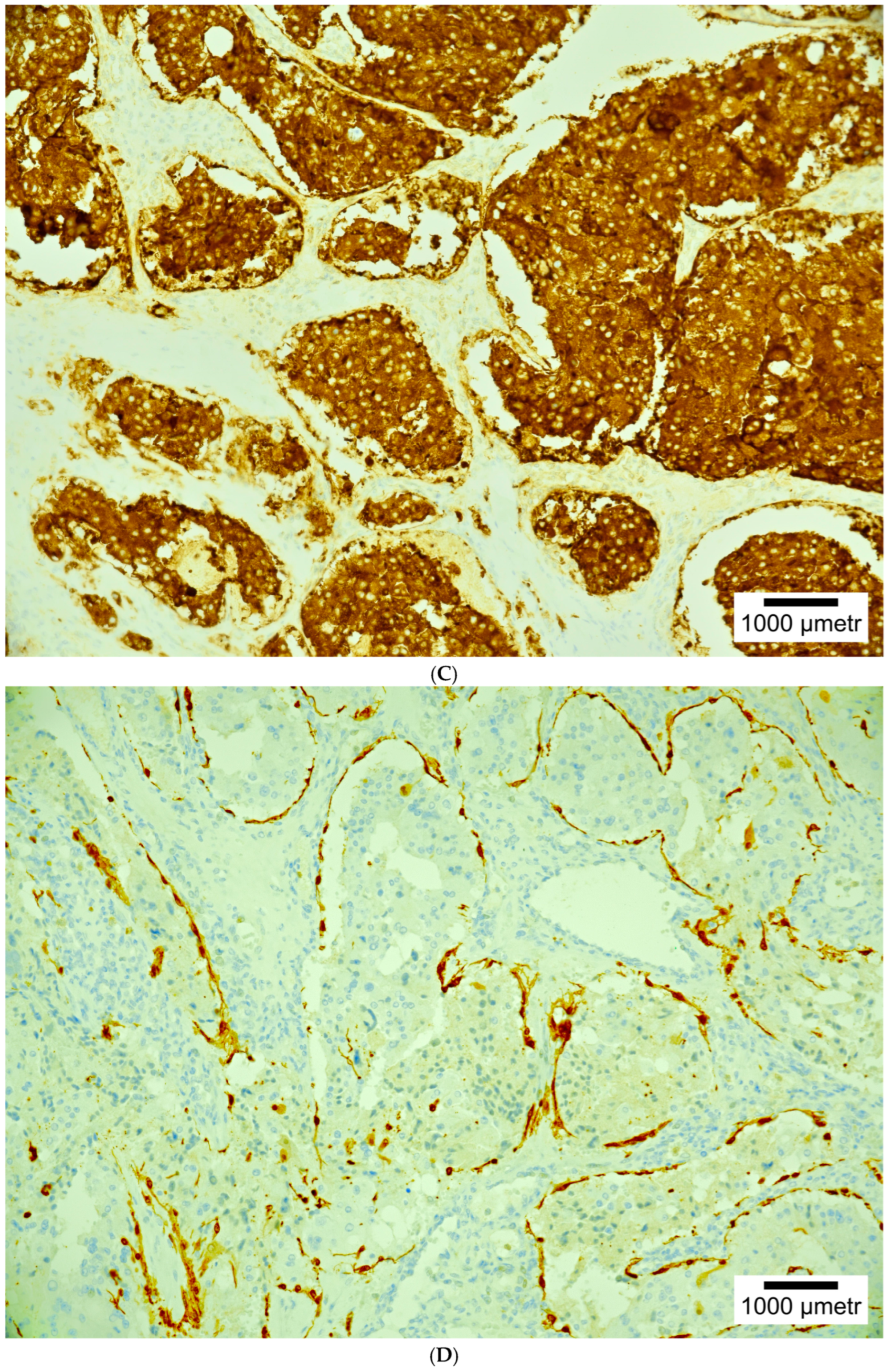
4. Histopathology
5. Epidemiology, Clinical Presentation, Secretory Function, Genetics and Molecular Clusters
5.1. Epidemiology
5.2. Clinical Presentation
5.3. Secretory Function
5.4. Genetics and Molecular Clusters
6. Imaging
6.1. Anatomical Imaging
6.2. Functional Imaging
7. Current Management
7.1. Surgery
7.2. Abdominal EAPGLs and Pheochromocytomas
7.3. Genitourinary Sympathetic EAPGLs
7.4. Thoracic Sympathetic EAPGLs
7.5. Head and Neck PGLs (HNPGLs)
7.5.1. Carotid Body PGL (CB PGL)
7.5.2. Tympano-Jugular PGL (TJ PGL)
7.5.3. Vagal Nerve Paraganglioma (VN PGL)
7.5.4. Laryngeal Paraganglioma (VN PGL)
7.5.5. Multiple Head and Neck Paragangliomas
7.6. Postoperative Surveillance
7.7. Metastatic Pheochromocytomas/Paragangliomas
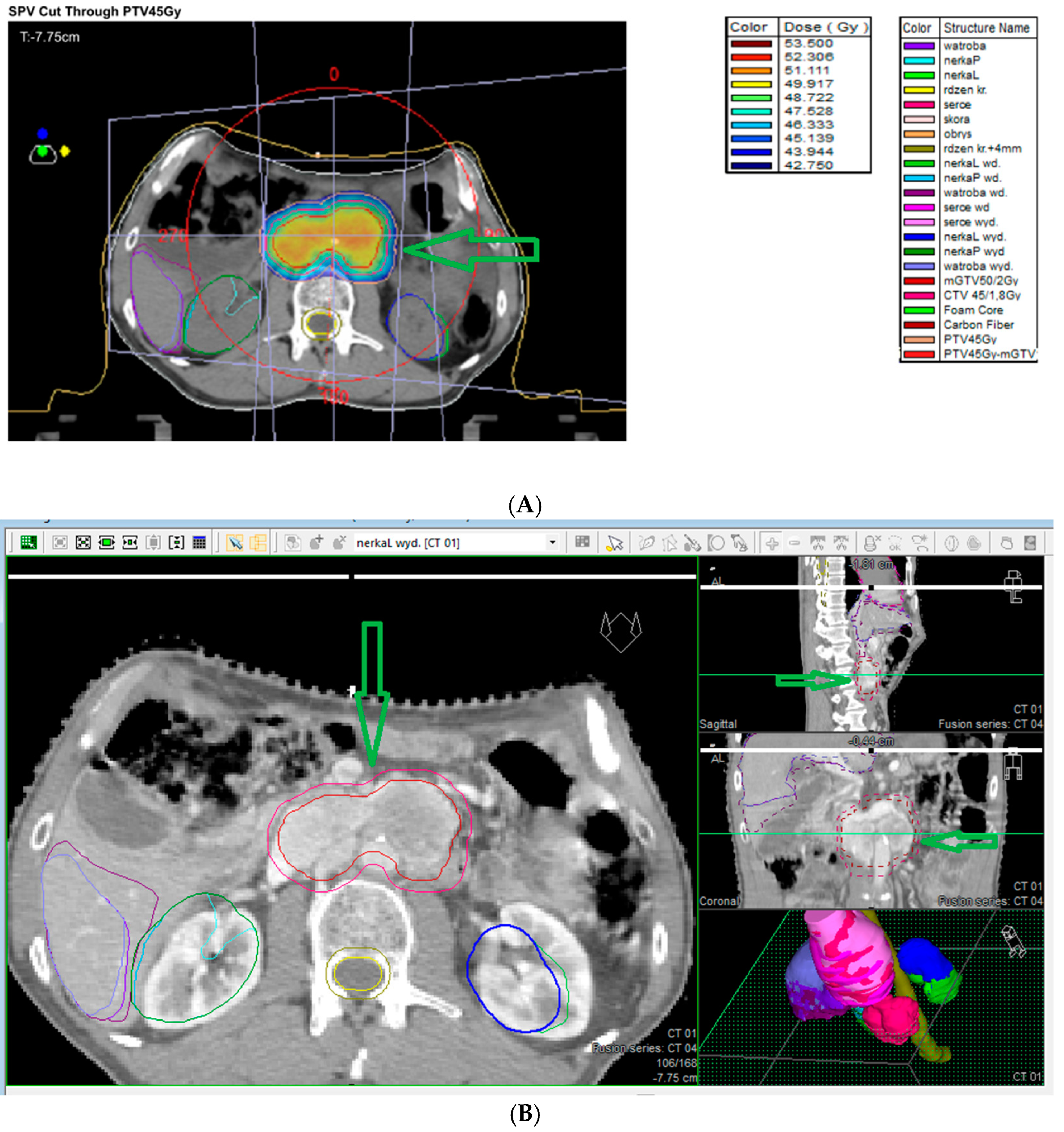
7.8. Stereotactic Radiosurgery (SRS)
7.9. External Beam Radiotherapy (ERBT) and Hypofractionated Stereotactic Radiotherapy (hSRT)
7.10. Systemic Therapy
7.10.1. Chemotherapy
7.10.2. Targeted Therapy
Tyrosine Kinase Inhibitors [TKIs]
mTORC1 Inhibitor Everolimus
Immunotherapy
Cold Somatostatin Analogs (Biotherapy)
Radioligand Therapy [RLT]
7.11. Ablation Therapy
8. Summary and Conclusions
9. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Moore, M.G.; Netterville, J.L.; Mendenhall, W.M.; Isaacson, B.; Nussenbaum, B. Head and Neck Paragangliomas: An Update on Evaluation and Management. Otolaryngol. Head Neck Surg. 2016, 154, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, P.G.; Savargaonkar, P. Paragangliomas: Classification, pathology, and differ ential diagnosis. Otolaryngol. Clin. N. Am. 2001, 34, 845–862. [Google Scholar] [CrossRef] [PubMed]
- Vimawala, S.N.; Graboyes, A.Z.; Bennett, B.; Bonanni, M.; Abbasi, A.; Oliphant, T.; Alonso-Basanta, M.; Rassekh, C.; Cohen, D.; Brant, J.A.; et al. Head and Neck Paragangliomas: Overview of Institutional Experience. Cancers 2024, 16, 1523. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Taïeb, D.; Kaliski, A.; Boedeker, C.C.; Martucci, V.; Fojo, T.; Adler, J.R., Jr.; Pacak, K. Current approaches and recent developments in the management of head and neck paragangliomas. Endocr. Rev. 2014, 35, 795–819. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Erickson, D.; Kudva, Y.C.; Ebersold, M.J.; Thompson, G.B.; Grant, C.S.; van Heerden, J.A.; Young, W.F., Jr. Benign paragangliomas: Clinical presentation and treatment outcomes in 236 patients. J. Clin. Endocrinol. Metab. 2001, 86, 5210–5216. [Google Scholar] [CrossRef] [PubMed]
- Baysal, B.E. Hereditary paraganglioma targets diverse paraganglia. J. Med. Genet. 2002, 39, 617–622. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fishbein, L.; Nathanson, K.L. Pheochromocytoma and paraganglioma: Understanding the complexities of the genetic background. Cancer Genet. 2012, 205, 1–11. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lamas, C.; Febrero, B.; Casteràs, A.; Romero-Lluch, A.; Recio-Córdova, J.M.; Ros, I.; Iglesias, P.; Hanzu, F.A.; Araujo-Castro, M.; Guerrero-Pérez, F.; et al. Current Management of Head and Neck Paragangliomas: A Multicenter Series with Long-Term Follow-Up. Otolaryngol. Head Neck Surg. 2025, 172, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Dahia, P.L. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat. Rev. Cancer 2014, 14, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L. Pheochromocytoma and Paraganglioma: Genetics, Diagnosis, and Treatment. Hematol. Oncol. Clin. N. Am. 2016, 30, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Castro-Vega, L.J.; Letouzé, E.; Burnichon, N.; Buffet, A.; Disderot, P.H.; Khalifa, E.; Loriot, C.; Elarouci, N.; Morin, A.; Menara, M.; et al. Multi-omics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. Nat. Commun. 2015, 6, 6044. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Carlson, M.L.; Sweeney, A.D.; Pelosi, S.; Wanna, G.B.; Glasscock, M.E., 3rd; Haynes, D.S. Glomus tympanicum: A review of 115 cases over 4 decades. Otolaryngol. Head Neck Surg. 2015, 152, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Sandow, L.; Thawani, R.; Kim, M.S.; Heinrich, M.C. Paraganglioma of the Head and Neck: A Review. Endocr. Pract. 2023, 29, 141–147, Erratum in Endocr. Pract. 2024, 30, 790. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Suárez, C.; Sevilla, M.A.; Llorente, J.L. Temporal paragangliomas. Eur. Arch. Otorhinolaryngol. 2007, 264, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, A.D.; Carlson, M.L.; Wanna, G.B.; Bennett, M.L. Glomus tympanicum tumors. Otolaryngol. Clin. N. Am. 2015, 48, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Wanna, G.B.; Sweeney, A.D.; Haynes, D.S.; Carlson, M.L. Contemporary management of jugular paragangliomas. Otolaryngol. Clin. N. Am. 2015, 48, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.P.; Chin, B.B.; Fishbein, L.; Moritani, T.; Montoya, S.P.; Ellika, S.; Newlands, S. Head and Neck Paragangliomas: An Update on the Molecular Classification, State-of-the-Art Imaging, and Management Recommendations. Radiol. Imaging Cancer 2022, 4, e210088. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mete, O.; Asa, S.L.; Gill, A.J.; Kimura, N.; de Krijger, R.R.; Tischler, A. Overview of the 2022 WHO Classification of Paragangliomas and Pheochromocytomas. Endocr. Pathol. 2022, 33, 90–114. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.B.; Greene, F.L.; Edge, S.B.; Compton, C.C.; Gershenwald, J.E.; Brookland, R.K.; Meyer, L.; Gress, D.M.; Byrd, D.R.; Winchester, D.P. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA A Cancer J. Clin. 2017, 67, 93–99. [Google Scholar] [CrossRef]
- Haller, F.; Moskalev, E.A.; Faucz, F.R.; Barthelmeß, S.; Wiemann, S.; Bieg, M.; Assie, G.; Bertherat, J.; Schaefer, I.M.; Otto, C.; et al. Aberrant DNA hypermethylation of SDHC: A novel mechanism of tumor development in Carney triad. Endocr. Relat. Cancer 2014, 21, 567–577. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Palade, D.O.; Hainarosie, R.; Zamfir, A.; Vrinceanu, D.; Pertea, M.; Tusaliu, M.; Mocanu, F.; Voiosu, C. Paragangliomas of the Head and Neck: A Review of the Latest Diagnostic and Treatment Methods. Medicina 2024, 60, 914. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mete, O.; Wenig, B.M. Update from the 5th Edition of the World Health Organization Classification of Head and Neck Tumors: Overview of the 2022 WHO Classification of Head and Neck Neuroendocrine Neoplasms. Head Neck Pathol. 2022, 16, 123–142. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cleere, E.F.; Martin-Grace, J.; Gendre, A.; Sherlock, M.; O’Neill, J.P. Contemporary management of paragangliomas of the head and neck. Laryngoscope Investig. Otolaryngol. 2021, 7, 93–107. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kimura, N.; Shiga, K.; Kaneko, K.; Sugisawa, C.; Katabami, T.; Naruse, M. The Diagnostic Dilemma of GATA3 Immunohistochemistry in Pheochromocytoma and Paraganglioma. Endocr. Pathol. 2020, 31, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.D. Paragangliomas of the head and neck: An overview from diagnosis to genetics. Head Neck Pathol. 2017, 11, 278–287. [Google Scholar] [CrossRef]
- Favier, J.; Amar, L.; Gimenez-Roqueplo, A.P. Paraganglioma and phaeochromocytoma: From genetics to personalized medicine. Nat. Rev. Endocrinol. 2015, 11, 101–111. [Google Scholar] [CrossRef]
- Antonio, K.; Valdez, M.M.N.; Mercado-Asis, L.; Taïeb, D.; Pacak, K. Pheochromocytoma/paraganglioma: Recent updates in genetics, biochemistry, immunohistochemistry, metabolomics, imaging and therapeutic options. Gland Surg. 2020, 9, 105–123. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rana, H.Q.; Koeller, D.R.; Walker, M.; Unal, B.; Levine, A.S.; Chittenden, A.; Isidro, R.A.; Hayes, C.P.; Manam, M.D.; Buehler, R.M.; et al. Int2grate Oncology Consortium. Advancing Precision Oncology in Hereditary Paraganglioma-Pheochromocytoma Syndromes: Integrated Interpretation and Data Sharing of the Germline and Tumor Genomes. Cancers 2024, 16, 947. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Turin, C.G.; Crenshaw, M.M.; Fishbein, L. Pheochromocytoma and paraganglioma: Germline genetics and hereditary syndromes. Endocr. Oncol. 2022, 2, R65–R77. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Neumann, H.P.; Bausch, B.; McWhinney, S.R.; Bender, B.U.; Gimm, O.; Franke, G.; Schipper, J.; Klisch, J.; Altehoefer, C.; Zerres, K.; et al. Germ-line mutations in nonsyndromic pheochromocytoma. N. Engl. J. Med. 2002, 346, 1459–1466. [Google Scholar] [CrossRef]
- Bausch, B.; Wellner, U.; Bausch, D.; Schiavi, F.; Barontini, M.; Sanso, G.; Walz, M.K.; Peczkowska, M.; Weryha, G.; Dall’Igna, P.; et al. Long-term prognosis of patients with pediatric pheochromocytoma. Endocr. Relat. Cancer 2013, 21, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Mannelli, M.; Castellano, M.; Schiavi, F.; Filetti, S.; Giacchè, M.; Mori, L.; Pignataro, V.; Bernini, G.; Giachè, V.; Bacca, A.; et al. Clinically guided genetic screening in a large cohort of italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J. Clin. Endocrinol. Metab. 2009, 94, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.D.; Harvey, R.N.; Darr, O.A.; Prince, M.E.; Bradford, C.R.; Wolf, G.T.; Else, T.; Basura, G.J. Head and neck paragangliomas: A two-decade institutional experience and algorithm for management. Laryngoscope Investig. Otolaryngol. 2017, 2, 380–389. [Google Scholar] [CrossRef]
- Robertson, V.; Poli, F.; Hobson, B.; Saratzis, A.; Ross, N.A. A systematic review and meta-analysis of the presentation and surgical management of patients with carotid body tumours. Eur. J. Vasc. Endovasc. Surg. 2019, 57, 477–486. [Google Scholar] [CrossRef]
- Kakamad, F.H.; Mustafa, M.N.; Yasin, S.W.; Xalid, S.S.; Mohammed, A.A.; Othman, S.; Hiwa, D.S.; Abdullah, H.O.; Abdalla, B.A.; Nasralla, H.A.; et al. Carotid body tumor: Characteristics and surgical outcome. J. Cardiothorac. Surg. 2024, 19, 473. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Boedeker, C.C.; Ridder, G.J.; Schipper, J. Paragangliomas of the head and neck: Diagnosis and treatment. Fam. Cancer 2005, 4, 55–59. [Google Scholar] [CrossRef]
- Patetsios, P.; Gable, D.R.; Garrett, W.V.; Lamont, J.P.; Kuhn, J.A.; Shutze, W.P.; Kourlis, H.; Grimsley, B.; Pearl, G.J.; Smith, B.L.; et al. Management of carotid body paragangliomas and review of a 30-year experience. Ann. Vasc. Surg. 2002, 16, 331–338. [Google Scholar] [CrossRef]
- Fisch, U. Infratemporal fossa approach for glomus tumors of the temporal bone. Ann. Otol. Rhinol. Laryngol. 1982, 91 Pt 1, 474–479. [Google Scholar] [CrossRef]
- Mazzoni, A.; Zanoletti, E. Observation and partial targeted surgery in the management of tympano-jugular paraganglioma: A contribution to the multioptional treatment. Eur. Arch. Otorhinolaryngol. 2016, 273, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Fayad, J.N.; Keles, B.; Brackmann, D.E. Jugular foramen tumors: Clinical characteristics and treatment outcomes. Otol. Neurotol. 2010, 31, 299–305. [Google Scholar] [CrossRef]
- Sanna, M.; Al-Khateeb, M.; Yilala, M.H.; Almashhadani, M.; Fancello, G. Gruppo Otologico’s Experience in Managing the So-Called Inoperable Tympanojugular Paraganglioma. Brain Sci. 2024, 14, 745. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sanna, M.; Jain, Y.; De Donato, G.; Rohit, L.L.; Taibah, A. Management of jugular paragangliomas: The Gruppo Otologico experience. Otol. Neurotol. 2004, 25, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.G. Glomus tympanicum and glomus jugulare tumors. Otolaryngol. Clin. N. Am. 2001, 34, 941–970. [Google Scholar] [CrossRef] [PubMed]
- Netterville, J.L.; Jackson, C.G.; Miller, F.R.; Wanamaker, J.R.; Glasscock, M.E. Vagal paraganglioma: A review of 46 patients treated during a 20-year period. Arch. Otolaryngol. Head Neck Surg. 1998, 124, 1133–1140. [Google Scholar] [CrossRef]
- Miller, R.B.; Boon, M.S.; Atkins, J.P.; Lowry, L.D. Vagal paraganglioma: The Jefferson experience. Otolaryngol. Head Neck Surg. 2000, 122, 482–487. [Google Scholar]
- Lenders, J.W.; Duh, Q.Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.-P.; Grebe, S.K.G.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Huynh, T.T.; Hiroi, M.; Pacak, K. Understanding catecholamine metabolism as a guide to the biochemical diagnosis of pheochromocytoma. Rev. Endocr. Metab. Disord. 2001, 2, 297–311. [Google Scholar] [CrossRef]
- Osinga, T.E.; Korpershoek, E.; de Krijger, R.R.; Kerstens, M.N.; Dullaart, R.P.; Kema, I.P.; van der Laan, B.F.A.M.; van der Horst-Schrivers, A.N.A.; Links, T.P. Catecholamines synthesizing enzymes are expressed in parasympathetic head and neck paraganglioma tissue. Neuroendocrinology 2015, 101, 289–295. [Google Scholar] [CrossRef]
- Corssmit, E.P.; Romijn, J.A. Clinical management of paragangliomas. Eur. J. Endocrinol. 2014, 171, R231–R243. [Google Scholar] [CrossRef]
- Colen, T.Y.; Mihm, F.G.; Mason, T.P.; Roberson, J.B. Catecholamine-Secreting Paragangliomas: Recent Progress in Diagnosis and Perioperative Management. Skull Base 2009, 19, 377–385. [Google Scholar] [CrossRef]
- Smith, J.D.; Ellsperman, S.E.; Basura, G.J.; Else, T. Re-evaluating the prevalence and factors characteristic of catecholamine secreting head and neck paragangliomas. Endocrinol. Diabetes Metab. 2021, 4, e00256. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guilmette, J.; Sadow, P.M. A Guide to Pheochromocytomas and Paragangliomas. Surg. Pathol. Clin. 2019, 12, 951–965. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Boedeker, C.C.; Hensen, E.F.; Neumann, H.P.; Maier, W.; van Nederveen, F.H.; Suárez, C.; Kunst, H.P.; Rodrigo, J.P.; Takes, R.P.; Pellitteri, P.K.; et al. Genetics of hereditary head and neck paragangliomas. Head Neck 2014, 36, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.; Pacak, K.; Kunst, H.P.M.; Januszewicz, A.; Nölting, S.; Remde, H.; Robledo, M.; Eisenhofer, G.; Timmers, H.J.L.M.; Pamporaki, C. Management and follow-up strategies for patients with head and neck paraganglioma. Eur. J. Endocrinol. 2024, 191, 389–398. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chase, W.H. Familial and bilateral tumours of the carotid body. J. Pathol. Bacteriol. 1933, 36, 1–2. [Google Scholar] [CrossRef]
- Goekoop, V.C. Fibro-Haemangiom des Felsenbeines und des Mittelohres bei drei Schwestern. Acta Oto-Laryngol. 1933, 18, 153–162. [Google Scholar] [CrossRef]
- Linn, H.J.; Proctor, B. Tumor of the ganglion nodosum of the vagus nerve. Laryngoscope 1956, 66, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; van der Mey, A.; Taschner, P.E.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Niemann, S.; Müller, U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat. Genet. 2000, 26, 268–270. [Google Scholar] [CrossRef] [PubMed]
- Astuti, D.; Latif, F.; Dallol, A.; Dahia, P.L.; Douglas, F.; George, E.; Sköldberg, F.; Husebye, E.S.; Eng, C.; Maher, E.R. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 2001, 69, 49–54, Erratum in Am. J. Hum. Genet. 2002, 70, 565. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hao, H.X.; Khalimonchuk, O.; Schraders, M.; Dephoure, N.; Bayley, J.P.; Kunst, H.; Devilee, P.; Cremers, C.W.; Schiffman, J.D.; Bentz, B.G.; et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 2009, 325, 1139–1142. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pillai, S.; Gopalan, V.; Smith, R.A.; Lam, A.K. Updates on the genetics and the clinical impacts on phaeochromocytoma and paraganglioma in the new era. Crit. Rev. Oncol. Hematol. 2016, 100, 190–208. [Google Scholar] [CrossRef] [PubMed]
- Taïeb, D.; Timmers, H.J.; Hindié, E.; Guillet, B.A.; Neumann, H.P.; Walz, M.K.; Opocher, G.; de Herder, W.W.; Boedeker, C.C.; de Krijger, R.R.; et al. EANM 2012 guidelines for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 1977–1995. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Neumann, H.P.; Pawlu, C.; Peczkowska, M.; Bausch, B.; McWhinney, S.R.; Muresan, M.; Buchta, M.; Franke, G.; Klisch, J.; Bley, T.A.; et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 2004, 292, 943–951, Erratum in JAMA 2004, 292, 1686. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L.; Del Rivero, J.; Else, T.; Howe, J.R.; Asa, S.L.; Cohen, D.L.; Dahia, P.L.M.; Fraker, D.L.; Goodman, K.A.; Hope, T.A.; et al. The North American Neuroendocrine Tumor Society Consensus Guidelines for Surveillance and Management of Metastatic and/or Unresectable Pheochromocytoma and Paraganglioma. Pancreas 2021, 50, 469–493. [Google Scholar] [CrossRef] [PubMed]
- Bayley, J.P.; Kunst, H.P.; Cascon, A.; Sampietro, M.L.; Gaal, J.; Korpershoek, E.; Hinojar-Gutierrez, A.; Timmers, H.J.; Hoefsloot, L.H.; Hermsen, M.A.; et al. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol. 2010, 11, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Taïeb, D.; Hicks, R.J.; Hindié, E.; Guillet, B.A.; Avram, A.; Ghedini, P.; Timmers, H.J.; Scott, A.T.; Elojeimy, S.; Rubello, D.; et al. European Association of Nuclear Medicine Practice Guideline/Society of Nuclear Medicine and Molecular Imaging Procedure Standard 2019 for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2112–2137. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guichard, J.P.; Fakhry, N.; Franc, J.; Herman, P.; Righini, C.A.; Taieb, D. Morphological and functional imaging of neck paragangliomas. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2017, 134, 243–248. [Google Scholar] [CrossRef]
- Chen, H.; Sippel, R.S.; O’Dorisio, M.S.; Vinik, A.I.; Lloyd, R.V.; Pacak, K.; North American Neuroendocrine Tumor Society (NANETS). The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: Pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas 2010, 39, 775–783. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Taïeb, D.; Nölting, S.; Perrier, N.D.; Fassnacht, M.; Carrasquillo, J.A.; Grossman, A.B.; Clifton-Bligh, R.; Wanna, G.B.; Schwam, Z.G.; Amar, L.; et al. Management of phaeochromocytoma and paraganglioma in patients with germline SDHB pathogenic variants: An international expert Consensus statement. Nat. Rev. Endocrinol. 2024, 20, 168–184. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; El-Tamer, M.; Schifftner, T.; Turrentine, F.E.; Henderson, W.G.; Khuri, S.; Hanks, J.B.; Inabnet, W.B., 3rd. Open and laparoscopic adrenalectomy: Analysis of the National Surgical Quality Improvement Program. J. Am. Coll. Surg. 2008, 206, 953–959; discussion 959–961. [Google Scholar] [CrossRef] [PubMed]
- Assadipour, Y.; Sadowski, S.M.; Alimchandani, M.; Quezado, M.; Steinberg, S.M.; Nilubol, N.; Patel, D.; Prodanov, T.; Pacak, K.; Kebebew, E. SDHB mutation status and tumor size but not tumor grade are important predictors of clinical outcome in pheochromocytoma and abdominal paraganglioma. Surgery 2017, 161, 230–239. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schovanek, J.; Martucci, V.; Wesley, R.; Fojo, T.; Del Rivero, J.; Huynh, T.; Adams, K.; Kebebew, E.; Frysak, Z.; Stratakis, C.A.; et al. The size of the primary tumor and age at initial diagnosis are independent predictors of the metastatic behavior and survival of patients with SDHB-related pheochromocytoma and paraganglioma: A retrospective cohort study. BMC Cancer 2014, 14, 523. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zelinka, T.; Musil, Z.; Dušková, J.; Burton, D.; Merino, M.J.; Milosevic, D.; Widimský, J., Jr.; Pacak, K. Metastatic pheochromocytoma: Does the size and age matter? Eur. J. Clin. Investig. 2011, 41, 1121–1128. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Timmers, H.J.; Chen, C.C.; Carrasquillo, J.A.; Whatley, M.; Ling, A.; Eisenhofer, G.; King, K.S.; Rao, J.U.; Wesley, R.A.; Adams, K.T.; et al. Staging and functional characterization of pheochromocytoma and paraganglioma by 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography. J. Natl. Cancer Inst. 2012, 104, 700–708. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Turkova, H.; Prodanov, T.; Maly, M.; Martucci, V.; Adams, K.; Widimsky, J., Jr.; Chen, C.C.; Ling, A.; Kebebew, E.; Stratakis, C.A.; et al. Characteristics and Outcomes of Metastatic SDHB and Sporadic Pheochromocytoma/Paraganglioma: An National Institutes of Health Study. Endocr. Pract. 2016, 22, 302–314. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gimenez-Roqueplo, A.P.; Favier, J.; Rustin, P.; Rieubland, C.; Kerlan, V.; Plouin, P.F.; Rötig, A.; Jeunemaitre, X. Functional consequences of a SDHB gene mutation in an apparently sporadic pheochromocytoma. J. Clin. Endocrinol. Metab. 2002, 87, 4771–4774. [Google Scholar] [CrossRef] [PubMed]
- Gimenez-Roqueplo, A.P.; Favier, J.; Rustin, P.; Rieubland, C.; Crespin, M.; Nau, V.; Khau Van Kien, P.; Corvol, P.; Plouin, P.F.; Jeunemaitre, X.; et al. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003, 63, 5615–5621. [Google Scholar] [PubMed]
- Brouwers, F.M.; Eisenhofer, G.; Tao, J.J.; Kant, J.A.; Adams, K.T.; Linehan, W.M.; Pacak, K. High frequency of SDHB germline mutations in patients with malignant catecholamine-producing paragangliomas: Implications for genetic testing. J. Clin. Endocrinol. Metab. 2006, 91, 4505–4509. [Google Scholar] [CrossRef] [PubMed]
- Pamporaki, C.; Berends, A.M.A.; Filippatos, A.; Prodanov, T.; Meuter, L.; Prejbisz, A.; Beuschlein, F.; Fassnacht, M.; Timmers, H.J.L.M.; Nölting, S.; et al. Prediction of metastatic pheochromocytoma and paraganglioma: A machine learning modelling study using data from a cross-sectional cohort. Lancet Digit. Health 2023, 5, e551–e559. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rijken, J.A.; van Hulsteijn, L.T.; Dekkers, O.M.; Niemeijer, N.D.; Leemans, C.R.; Eijkelenkamp, K.; van der Horst-Schrivers, A.N.A.; Kerstens, M.N.; van Berkel, A.; Timmers, H.J.L.M.; et al. Increased Mortality in SDHB but Not in SDHD Pathogenic Variant Carriers. Cancers 2019, 11, 103. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Friedman, L.R.; Ramamoorthy, B.; Nilubol, N. Progress in surgical approaches and outcomes of patients with pheochromocytoma and paraganglioma. Best Pract. Res. Clin. Endocrinol. Metab. 2024, 39, 101954. [Google Scholar] [CrossRef] [PubMed]
- Fang, A.M.; Rosen, J.; Saidian, A.; Bae, S.; Tanno, F.Y.; Chambo, J.L.; Bloom, J.; Gordetsky, J.; Srougi, V.; Phillips, J.; et al. Perioperative outcomes of laparoscopic, robotic, and open approaches to pheochromocytoma. J. Robot. Surg. 2020, 14, 849–854. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gan, L.; Peng, L.; Li, J.; Meng, C.; Li, K.; Wu, J.; Zhang, Z.; Li, Y. Comparison of the effectiveness and safety of robotic-assisted and laparoscopic in adrenalectomy: A systematic review and meta-analysis. Int. J. Surg. 2022, 105, 106853. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Peng, L.; Meng, C.; Zheng, L.; Zeng, Z.; Ge, S.; Wang, Z.; Li, K.; Li, Y. The role of laparoscopic adrenalectomy in the treatment of large pheochromocytomas (>6 cm): A meta-analysis and systematic review. Int. J. Surg. 2023, 109, 1459–1469. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Garcia-Carbonero, R.; Matute Teresa, F.; Mercader-Cidoncha, E.; Mitjavila-Casanovas, M.; Robledo, M.; Tena, I.; Alvarez-Escola, C.; Arístegui, M.; Bella-Cueto, M.R.; Ferrer-Albiach, C.; et al. Multidisciplinary practice guidelines for the diagnosis, genetic counseling and treatment of pheochromocytomas and paragangliomas. Clin. Transl. Oncol. 2021, 23, 1995–2019. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wald, O.; Shapira, O.M.; Murar, A.; Izhar, U. Paraganglioma of the mediastinum: Challenges in diagnosis and surgical management. J. Cardiothorac. Surg. 2010, 5, 19. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Al-Jehani, Y.; Saleh, W.; Al Halees, Z. Successful resection of a huge paraganglioma utilizing cardiopulmonary bypass. Asian Cardiovasc. Thorac. Ann. 2012, 20, 482–485. [Google Scholar] [CrossRef]
- Wherley, E.M.; Gross, D.J.; Nguyen, D.M. Extracorporeal membrane oxygenation in the surgical management of large mediastinal masses: A narrative review. J. Thorac. Dis. 2023, 15, 5248–5255. [Google Scholar] [CrossRef]
- Iacobone, M.; Belluzzi, A.; Torresan, F. Surgical approaches and results of treatment for hereditary paragangliomas. Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101298. [Google Scholar] [CrossRef]
- Shamblin, W.R.; ReMine, W.H.; Sheps, S.G.; Harrison, E.G., Jr. Carotid body tumor (chemodectoma). Clinicopathologic analysis of ninety cases. Am. J. Surg. 1971, 122, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Luna-Ortiz, K.; Rascon-Ortiz, M.; Villavicencio-Valencia, V.; Herrera-Gomez, A. Does Shamblin’s classification predict postoperative morbidity in carotid body tumors? A proposal to modify Shamblin’s classification. Eur. Arch. Otorhinolaryngol. 2006, 263, 171–175, Erratum in Eur. Arch. Otorhinolaryngol. 2006, 263, 1161. [Google Scholar] [CrossRef] [PubMed]
- Ng, E.; Duncan, G.; Choong, A.M.; Francis, L.; Foster, W.; Kruger, A. Sclerosing paragangliomas of the carotid body: A series of a rare variant and review of the literature. Ann. Vasc. Surg. 2015, 29, 1454.e5–1454.e12. [Google Scholar] [CrossRef]
- Abu-Ghanem, S.; Yehuda, M.; Carmel, N.N.; Abergel, A.; Fliss, D.M. Impact of preoperative embolization on the outcomes of carotid body tumor surgery: A meta-analysis and review of the literature. Head Neck 2016, 38, E2386e94. [Google Scholar] [CrossRef]
- Wang, Y.H.; Yang, J.; Zhong, H.; Wu, J.J.; Wu, K.; Hu, A.; Wu, J.Y.; Zhu, J.H. Prevalence, characteristics, evaluation, and management of carotid body tumors: Systematic analysis based on available evidence. J. Vasc. Surg. 2024, 80, 574–585.e4. [Google Scholar] [CrossRef] [PubMed]
- Jansen, T.T.G.; Timmers, H.J.L.M.; Marres, H.A.M.; Kunst, H.P.M. Feasibility of a wait-and-scan period as initial management strategy forhead and neck paraganglioma. Head Neck 2017, 39, 2088e94. [Google Scholar] [CrossRef]
- Graham, N.J.; Smith, J.D.; Else, T.; Basura, G.J. Paragangliomas of the head and neck: A contemporary review. Endocr. Oncol. 2022, 2, R153–R162. [Google Scholar] [CrossRef]
- Fancello, G.; Fancello, V.; Ehsani, D.; Porpiglia, V.; Piras, G.; Caruso, A.; Sanna, M. Tumor progression in tympanojugular paragangliomas: The role of radiotherapy and wait and scan. Eur. Arch. Otorhinolaryngol. 2024, 281, 2779–2789, Erratum in Eur. Arch. Otorhinolaryngol. 2024, 281, 2791. [Google Scholar] [CrossRef] [PubMed]
- Jansen, T.T.G.; Timmers, H.J.L.M.; Marres, H.A.M.; Kaanders, J.H.A.M.; Kunst, H.P.M. Results of a systematic literature review of treatment modalities for jugulotympanic paraganglioma, stratified per Fisch class. Clin. Otolaryngol. 2018, 43, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Fisch, U.; Mattox, D.E. Microsurgery of the Skull Base; Thieme-Verlag: Stuttgart, Germany; New York, NY, USA, 1988; pp. 149–152. [Google Scholar]
- Moe, K.S.; Li, D.; Linder, T.E.; Schmid, S.; Fisch, U. An Update on the Surgical Treatment of Temporal Bone Paraganglioma. Skull Base Surg. 1999, 9, 185–194. [Google Scholar] [CrossRef]
- Jackson, C.G.; Glasscock, M.E., 3rd; Harris, P.F. Glomus Tumors: Diagnosis, classification, and management of large lesions. Arch. Otolaryngol. 1982, 108, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Zanoletti, E.; Mazzoni, A. Vagal paraganglioma. Skull Base 2006, 16, 161–167. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Singh, P.; Singh, S.P. Vagal Paraganglioma of Neck Mimicking Carotid Body Tumor: Clues to Differentiate on Imaging. Indian J. Otolaryngol. Head Neck Surg. 2022, 74 (Suppl. S3), 6258–6261. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- González-Orús Álvarez-Morujo, R.; Arístegui Ruiz, M.; Martin Oviedo, C.; Álvarez Palacios, I.; Scola Yurrita, B. Management of vagal paragangliomas: Review of 17 patients. Eur. Arch. Oto-Rhino-Laryngol. 2015, 272, 2403–2414. [Google Scholar] [CrossRef]
- Mendenhall, W.M.; Amdur, R.J.; Vaysberg, M.; Mendenhall, C.M.; Werning, J.W. Head and neck paragangliomas. Head Neck 2011, 33, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Papagiannopoulos, P.; Raman, A.; Miller, C.; Jhaveri, M.; Ghai, R.; Husain, I. Laryngeal paraganglioma with chronic cough: A case report. Turk. Arch. Otorhinolaryngol. 2018, 56, 233. [Google Scholar] [CrossRef]
- Lenders, J.W.M.; Kerstens, M.N.; Amar, L.; Prejbisz, A.; Robledo, M.; Taieb, D.; Pacak, K.; Crona, J.; Zelinka, T.; Mannelli, M.; et al. Genetics, diagnosis, management and future directions of research of phaeochromocytoma and paraganglioma: A position statement and consensus of the Working Group on Endocrine Hypertension of the European Society of Hypertension. J. Hypertens. 2020, 38, 1443–1456. [Google Scholar] [CrossRef]
- Plouin, P.F.; Amar, L.; Lepoutre, C. Phaeochromocytomas and functional paragangliomas, clinical management. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 933–941. [Google Scholar] [CrossRef]
- Passman, J.E.; Wachtel, H. Management of pheochromocytomas and paragangliomas. Surg. Clin. N. Am. 2024, 104, 863–881. [Google Scholar] [CrossRef]
- Patel, D.; Phay, J.E.; Yen, T.W.F.; Dickson, P.V.; Wang, T.S.; Garcia, R.; Yang, A.D.; Kim, L.T.; Solórzano, C.C. Update on pheochromocytoma and paraganglioma from the SSO Endocrine and Head and Neck Disease Site Working Group, part 2 of 2: Perioperative management and outcomes of pheochromocytoma and paraganglioma. Ann. Surg. Oncol. 2020, 27, 1338–1347. [Google Scholar] [CrossRef]
- Shah, M.H.; Goldner, W.S.; Benson, A.B.; Bergsland, E.; Blaszkowsky, L.S.; Brock, P.; Chan, J.; Das, S.; Dickson, P.V.; Fanta, P.; et al. Neuroendocrine and adrenal tumors, version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 839–868. [Google Scholar] [CrossRef] [PubMed]
- Fassnacht, M.; Assie, G.; Baudin, E.; Eisenhofer, G.; de la Fouchardiere, C.; Haak, H.; de Krijger, R.; Porpiglia, F.; Terzolo, M.; Berruti, A. Adrenocortical carcinomas and malignant phaeochromocytomas: ESMO-EURACAN clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 1476–1490. [Google Scholar] [CrossRef] [PubMed]
- Torresan, F.; Beber, A.; Schiavone, D.; Zovato, S.; Galuppini, F.; Crimì, F.; Ceccato, F.; Iacobone, M. Long-Term Outcomes after Surgery for Pheochromocytoma and Sympathetic Paraganglioma. Cancers 2023, 15, 2890. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jimenez, C.; Ma, J.; Roman Gonzalez, A.; Varghese, J.; Zhang, M.; Perrier, N.; Habra, M.A.; Graham, P.; Waguespack, S.G. TNM Staging and Overall Survival in Patients With Pheochromocytoma and Sympathetic Paraganglioma. J. Clin. Endocrinol. Metab. 2023, 108, 1132–1142. [Google Scholar] [CrossRef] [PubMed]
- Ong, V.; Bourcier, A.J.; Florence, T.J.; Mozaffari, K.; Mekonnen, M.; Sheppard, J.P.; Duong, C.; Ding, K.; Yang, I. Stereotactic Radiosurgery for Glomus Jugulare Tumors: Systematic Review and Meta-Analysis. World Neurosurg. 2022, 162, e49–e57. [Google Scholar] [CrossRef] [PubMed]
- Dharnipragada, R.; Butterfield, J.T.; Dhawan, S.; Adams, M.E.; Venteicher, A.S. Modern Management of Complex Tympanojugular Paragangliomas: Systematic Review and Meta-Analysis. World Neurosurg. 2023, 170, 149–156.e3. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.E.; Sughrue, M.E.; Clark, A.J.; Kane, A.J.; Aranda, D.; Barani, I.J.; Parsa, A.T. A meta-analysis of tumor control rates and treatment-related morbidity for patients with glomus jugulare tumors. J. Neurosurg. 2011, 114, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Garg, K.; Singh, M. Role of Stereotactic Radiosurgery in Skull Base Paragangliomas—A Narrative Review. Neurol. India 2023, 71 (Suppl. S1), S153–S160. [Google Scholar] [CrossRef]
- Fatima, N.; Pollom, E.; Soltys, S.; Chang, S.D.; Meola, A. Stereotactic radiosurgery for head and neck paragangliomas: A systematic review and meta-analysis. Neurosurg. Rev. 2021, 44, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Gigliotti, M.J.; Hasan, S.; Liang, Y.; Chen, D.; Fuhrer, R.; Wegner, R.E. A 10-year experience of linear accelerator-based stereotactic radiosurgery/radiotherapy (SRS/SRT) for paraganglioma: A single institution experience and review of the literature. J. Radiosurg. SBRT 2018, 5, 183–190. [Google Scholar] [PubMed] [PubMed Central]
- Bracigliano, A.; Marretta, A.L.; Guerrera, L.P.; Simioli, R.; Clemente, O.; Granata, V.; Minopoli, A.; Della Vittoria Scarpati, G.; Picozzi, F.; Cannella, L.; et al. The Management of Phaeochromocytomas and Paragangliomas in the Era of Precision Medicine: Where Are We Now? Evidence-Based Systemic Treatment Options and Future Cluster Oriented Perspectives. Pharmaceuticals 2024, 17, 354. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Keiser, H.R.; Goldstein, D.S.; Wade, J.L.; Douglas, F.L.; Averbuch, S.D. Treatment of malignant pheochromocytoma with combination chemotherapy. Hypertension 1985, 7 Pt 2, I18–I24. [Google Scholar] [CrossRef] [PubMed]
- Kobayakawa, M.; Shiga, T.; Takahashi, K.; Sugawara, S.; Nomura, K.; Hanada, K.; Ishizuka, N.; Ito, H. Evaluation of pharmacokinetics, safety, and efficacy of [211At] meta-astatobenzylguanidine ([211At] MABG) in patients with pheochromocytoma or paraganglioma (PPGL): A study protocol. PLoS ONE 2024, 19, e0303623. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Niemeijer, N.D.; Alblas, G.; van Hulsteijn, L.T.; Dekkers, O.M.; Corssmit, E.P. Chemotherapy with cyclophosphamide, vincristine and dacarbazine for malignant paraganglioma and pheochromocytoma: Systematic review and meta-analysis. Clin. Endocrinol. 2014, 81, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Jawed, I.; Velarde, M.; Därr, R.; Wolf, K.I.; Adams, K.; Venkatesan, A.M.; Balasubramaniam, S.; Poruchynsky, M.S.; Reynolds, J.C.; Pacak, K.; et al. Continued Tumor Reduction of Metastatic Pheochromocytoma/Paraganglioma Harboring Succinate Dehydrogenase Subunit B Mutations with Cyclical Chemotherapy. Cell. Mol. Neurobiol. 2018, 38, 1099–1106. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nölting, S.; Bechmann, N.; Taieb, D.; Beuschlein, F.; Fassnacht, M.; Kroiss, M.; Eisenhofer, G.; Grossman, A.; Pacak, K. Personalized Management of Pheochromocytoma and Paraganglioma. Endocr. Rev. 2022, 43, 199–239, Erratum in Endocr. Rev. 2022, 43, 440; Erratum in Endocr. Rev. 2022, 43, 437–439. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhou, Y.; Cui, Y.; Zhang, D.; Tong, A. Efficacy and Safety of Tyrosine Kinase Inhibitors in Patients with Metastatic Pheochromocytomas/Paragangliomas. J. Clin. Endocrinol. Metab. 2023, 108, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, C.; Subbiah, V.; Stephen, B.; Ma, J.; Milton, D.; Xu, M.; Zarifa, A.; Akhmedzhanov, F.O.; Tsimberidou, A.; Habra, M.A.; et al. Phase II Clinical Trial of Pembrolizumab in Patients with Progressive Metastatic Pheochromocytomas and Paragangliomas. Cancers 2020, 12, 2307. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Naing, A.; Meric-Bernstam, F.; Stephen, B.; Karp, D.D.; Hajjar, J.; Rodon Ahnert, J.; Piha-Paul, S.A.; Colen, R.R.; Jimenez, C.; Raghav, K.P.; et al. Phase 2 study of pembrolizumab in patients with advanced rare cancers. J. Immunother. Cancer 2020, 8, e000347, Erratum in J. Immunother. Cancer 2020, 8, e000347corr1. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fischer, A.; Kloos, S.; Maccio, U.; Friemel, J.; Remde, H.; Fassnacht, M.; Pamporaki, C.; Eisenhofer, G.; Timmers, H.J.L.M.; Robledo, M.; et al. Metastatic Pheochromocytoma and Paraganglioma: Somatostatin Receptor 2 Expression, Genetics, and Therapeutic Responses. J. Clin. Endocrinol. Metab. 2023, 108, 2676–2685. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Castellani, M.R.; Aktolun, C.; Buzzoni, R.; Seregni, E.; Chiesa, C.; Maccauro, M.; Aliberti, G.L.; Vellani, C.; Lorenzoni, A.; Bombardieri, E. Iodine-131 metaiodobenzylguanidine (I-131 MIBG) diagnosis and therapy of pheochromocytoma and paraganglioma: Current problems, critical issues and presentation of a sample case. Q. J. Nucl. Med. Mol. Imaging 2013, 57, 146–152. [Google Scholar] [PubMed]
- Marretta, A.L.; Ottaiano, A.; Iervolino, D.; Bracigliano, A.; Clemente, O.; Di Gennaro, F.; Tafuto, R.; Santorsola, M.; Lastoria, S.; Tafuto, S. Response to Peptide Receptor Radionuclide Therapy in Pheocromocytomas and Paragangliomas: A Systematic Review and Meta-Analysis. J. Clin. Med. 2023, 12, 1494. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kohlenberg, J.; Welch, B.; Hamidi, O.; Callstrom, M.; Morris, J.; Sprung, J.; Bancos, I.; Young, W., Jr. Efficacy and Safety of Ablative Therapy in the Treatment of Patients with Metastatic Pheochromocytoma and Paraganglioma. Cancers 2019, 11, 195. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wachtel, H.; Nathanson, K.L. Molecular Genetics of Pheochromocytoma/Paraganglioma. Curr. Opin. Endocr. Metab. Res. 2024, 36, 100527. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, L.; Zeng, W.; Wu, Y.; Gong, Z. Comparison of clinical efficacy and safety between robotic-assisted and laparoscopic adrenalectomy for pheochromocytoma: A systematic review and meta-analysis. J. Robot. Surg. 2024, 18, 115. [Google Scholar] [CrossRef] [PubMed]
- Schiavone, D.; Ballo, M.; Filardo, M.; Dughiero, S.; Torresan, F.; Rossi, G.P.; Iacobone, M. Total adrenalectomy versus subtotal adrenalectomy for bilateral pheochromocytoma: Meta-analysis. BJS Open 2023, 7, zrad109. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, J.; Liu, Q.; Jiang, S.; Zhang, J.; He, J.; Li, Y.; Wang, D. Preoperative α-blockade versus no blockade for pheochromocytoma-paraganglioma patients undergoing surgery: A systematic review and updated meta-analysis. Int. J. Surg. 2023, 109, 1470–1480. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zawadzka, K.; Tylec, P.; Małczak, P.; Major, P.; Pędziwiatr, M.; Pisarska-Adamczyk, M. Total versus partial adrenalectomy in bilateral pheochromocytoma—A systematic review and meta-analysis. Front. Endocrinol. 2023, 14, 1127676. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gan, L.; Meng, C.; Li, K.; Lei Peng Li, J.; Wu, J.; Li, Y. Safety and effectiveness of minimally invasive adrenalectomy versus open adrenalectomy in patients with large adrenal tumors (≥5 cm): A meta-analysis and systematic review. Int. J. Surg. 2022, 104, 106779. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.Q.; Wang, S.Y.; Chen, Q.; Liu, Y.T.; Li, Z.L.; Sun, T. Laparoscopic versus open surgery for pheochromocytoma: A meta-analysis. BMC Surg. 2020, 20, 167. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, J.; Wang, Y.; Chang, X.; Han, Z. Laparoscopic adrenalectomy (LA) vs open adrenalectomy (OA) for pheochromocytoma (PHEO): A systematic review and meta-analysis. Eur. J. Surg. Oncol. 2020, 46, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.L.; Qian, L.J.; Li, Z.; Wang, K.E.; Zhou, X.L.; Zhou, J.; Ye, C.H. Comparison of the retroperitoneal versus Transperitoneal laparoscopic Adrenalectomy perioperative outcomes and safety for Pheochromocytoma: A meta-analysis. BMC Surg. 2020, 20, 12. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schimmack, S.; Kaiser, J.; Probst, P.; Kalkum, E.; Diener, M.K.; Strobel, O. Meta-analysis of α-blockade versus no blockade before adrenalectomy for phaeochromocytoma. Br. J. Surg. 2020, 107, e102–e108. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, O.; Young, W.F., Jr.; Gruber, L.; Smestad, J.; Yan, Q.; Ponce, O.J.; Prokop, L.; Murad, M.H.; Bancos, I. Outcomes of patients with metastatic phaeochromocytoma and paraganglioma: A systematic review and meta-analysis. Clin. Endocrinol. 2017, 87, 440–450. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Texakalidis, P.; Charisis, N.; Giannopoulos, S.; Xenos, D.; Rangel-Castilla, L.; Tassiopoulos, A.K.; Jabbour, P.; Grossberg, J.A.; Machinis, T. Role of Preoperative Embolization in Carotid Body Tumor Surgery: A Systematic Review and Meta-Analysis. World Neurosurg. 2019, 129, 503–513.e2. [Google Scholar] [CrossRef] [PubMed]
- Koh, E.S.; Dabsha, A.; Rahouma, M.; Zappi, K.; Srinivasan, Y.; Hickner, A.; Kutler, D.I. Succinate dehydrogenase mutations in head and neck paragangliomas: A systematic review and meta-analysis of individual patients’ data. Head Neck 2024, 46, 1795–1808. [Google Scholar] [CrossRef] [PubMed]
- Napoli, G.; Tritto, R.; Moscarelli, M.; Forleo, C.; La Marca, M.G.C.; Yang, L.; Biondi-Zoccai, G.; Giordano, A.; Tshomba, Y.; Pepe, M. Role of pre-operative embolization in carotid body tumor surgery according to Shamblin classification: A systematic review and meta-analysis. Head Neck 2023, 45, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.S.; Myhill, J.A.; Padhya, T.A.; McCaffrey, J.C.; McCaffrey, T.V.; Mhaskar, R.S. The Effects of Preoperative Embolization on Carotid Body Paraganglioma Surgery: A Systematic Review and Meta-analysis. Otolaryngol. Head Neck Surg. 2015, 153, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Ghanaati, H.; Zarei, D.; Issaiy, M.; Ghavami, N.; Shakiba, M.; Zebardast, J.; Abbastabar, H.; Jalali, A.H.; Firouznia, K. Efficacy and Safety of Preoperative Embolization in Glomus Jugulare Tumors: A Systematic Review and Meta-analysis of Clinical Outcomes and Complications. Cardiovasc. Intervent. Radiol. 2024, 47, 416–431. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, P.; Tatsui, C.; Jessop, A.; Thosani, S.; Jimenez, C. Treatment for Malignant Pheochromocytomas and Paragangliomas: 5 Years of Progress. Curr. Oncol. Rep. 2017, 19, 83. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PASS (Points) | GAPP (Points) |
|---|---|
Histologic pattern
| Histologic pattern
|
| Cellularity
|
|
|
|
|
|
|
Other features
|
|
| Maximum Score = 20 | Maximum Score = 10 |
| Clinical Manifestation in Patients with Pheochromocytomas and Paragangliomas |
|---|
|
| Gene | Syndrome Name | Head and Neck | Thoracic | Abdominal Adrenal (PCC) | Abdominal Extraadrenal | Malignancy Risk |
|---|---|---|---|---|---|---|
| SDHA | PGL5 | 30–<60% | <10% | 30–<60% | <10% | |
| SDHB | PGL4 | 10–<30% | 10–<30% | 10–<30% | 30–<60% | 30–<60% |
| SDHC | PGL3 | 90–100% | <10% | <10% | <10% | <10% |
| SDHD | PGL1 | 30–<60% | 10–<30% | 30–<60% | 30–<60% | <10% |
| SDHAF2 (SDH5) | PGL2 | 90–100% | Never reported | Never reported | Never reported | Never reported |
| RET | MEN2 | <10% | Never reported | 90–100% | <10% | <10% |
| VHL | VHL | <10% | <10% | 90–100% | 10–<30% | <10% |
| NF1 | NF1 | Never reported | Never reported | 90–100% | <10% | 10–<30% |
| TMEM127 | - | 10–<30% | Never reported | 90–100% | 10–<30% | Never reported |
| MAX | - | Never reported | Never reported | 90–100% | 10–<30% | 10–<30% |
| Gene Mutation | Molecular Cluster | Biochemical Profile |
|---|---|---|
| SDHx or VHL | Pseudohypoxia | Noradrenergic |
| SDHx (often SDHB) | Pseudohypoxia | Dopaminergic or Methoxytyramine |
| RET or NF1 | Kinase-signaling | Adrenergic |
| Class | Type | Description | Degree of Resection Difficulty | Type of Resection |
|---|---|---|---|---|
| I | Localized | The tumor separates the ICA and ECA, adjacent to the bifurcation | Easy resection | Routine dissection |
| II | Partially wrapped | The tumor is adherent and partially surrounding the carotid arteries | More difficult resection | Sub-adventitial dissection |
| III | Wrapped | The tumor is completely encasing the carotid arteries | Very difficult resection and interrupting cerebral circulation is almost always inevitable | Partial or complete vascular resection |
| Class | Type | Description | Resection |
|---|---|---|---|
| I | Localized | Smaller tumors with minimal vessel involvement or the contact between CBT and ICA ≤ 180 degrees on MRI. | The tumor can be resected with minimal morbidity |
| II | Partially wrapped | Larger tumors with possible ICA vs. ECA involvement (≥180 degrees and ≤270 degrees of contact to ICA). | Resection is still deemed possible with a greater risk of morbidity. |
| III | Wrapped | Large tumors with circumferential involvement of the carotids or ≥270 degrees of contact between CBT and ICA. | Internal carotid artery reconstruction or ligation is predicted. |
| Class | Tumor Size | Description | Resection Difficulty |
|---|---|---|---|
| I | <4 cm | No involvement or invasion the carotid arteries | No difficulty |
| II | >4 cm | Partial involvement or invasion the carotid arteries | Difficult |
| IIIA | >4 cm | Close involvement or invasion of the carotid arteries | Difficult, requires repair, removal or replacement of the carotid artery |
| IIIB | Each size | Class I, II or III according to the original Shamblin classification with invasion of the carotid arteries | It is necessary to confirm the invasion of the vessel wall clinically and/or histopathologically |
| Tumor Class | Tumor Location and Extension |
|---|---|
| A | Tumors that arise along the tympanic plexus on promontory. |
| B | Tumors with invasion of hypotympanum; cortical bone over jugular bulb intact. |
| C1 | Tumors with erosion of carotid foramen. |
| C2 | Tumors with destruction of carotid canal. |
| C3 | Tumors with invasion of carotid canal; foramen lacerum intact. |
| C4 | Tumors with invasion of foramen lacerum and cavernous sinus. |
De
| Tumors with intracranial but extradural extension:
|
Di
| Tumors with intracranial and intradural extension:
|
| Glomus Tympanicum | Glomus Jugular | ||
|---|---|---|---|
| I | Small mass limited to the promontory | I | Small tumor involving jugular bulb, middle ear, and mastoid |
| II | Tumor filling middle ear space | II | Tumor extending under internal auditory canal; may have intracranial canal extension (ICE) |
| III | Tumor filling middle ear and extending into the mastoid | III | Tumor extending into petrous apex; may have ICE |
| IV | Tumor filling middle ear, extending into the mastoid or through tympanic membrane to fill the external auditory canal; may extend anterior to the carotid. | IV | Tumor extending beyond petrous apex into clivus or infratemporal fossa; may have ICE |
| Authors | Year | Number of Studies (Patients) | Aim of Meta-Analysis | Results/Conclusions |
|---|---|---|---|---|
| Gan et al. [83] | 2022 | 26 studies (2985 patients) | Comparison of robotic-assisted and laparoscopic adrenalectomy for adrenal PGL | Robotic technique superior to conventional laparoscopy for blood loss,), hospitalization duration, and conversion to open. Similar duration of operation, complication and readmission rates Longer duration of operation in retroperitoneal robotic-assisted surgery compared to laparoscopic approach. |
| Gan et al. [84] | 2023 | 8 studies (600 patients) | The role of laparoscopic adrenalectomy in treatment of large adrenal PGL (>6 cm) | Similar complication rate in laparoscopic approach in small and large PPC. Longer duration of operation, duration of hospitalization, greater blood loss, hypertension, hypotension, and conversion in large tumors. Transabdominal is superior to retroperitoneal LS. |
| Wang et al. [135] | 2024 | 6 studies (658 patients) | Comparison of robotic-assisted and laparoscopic adrenalectomy for adrenal PGL | No differences in duration of operation, transfusion rate, conversion rate, complication rate, intraoperative max SBP, intraoperative min SBP between RA and LA. Less blood loss, a shorter duration of hospitalization in RA compared to LA. |
| Schiavone et al. [136] | 2024 | 10 studies (1202 patients) | Comparison of total adrenalectomy and subtotal adrenalectomy for bilateral adrenal PGL | Less post-surgical primary adrenal insufficiency after subtotal adrenalectomy compared to total adrenalectomy. Higher postoperative recurrence rate after subtotal adrenalectomy compared to total adrenalectomy. |
| Wangs et al. [137] | 2023 | 15 studies (3542 patients) | Comparison of preoperative α-blockade and no blockade for PPGL patients undergoing surgery | Prolonged hypotension and vasopressor usage following α-blockade compared ton no α-blockade. Similar intensive care unit admission, duration of operation, overall cardiovascular morbidity, and mortality in both groups. |
| Zawadzka et al. [138] | 2023 | 25 studies (1444 patients) | Comparison of partial and total adrenalectomy in bilateral adrenal PGL | A lower risk of postoperative loss of adrenal hormone function and acute adrenal crisis following partial adrenalectomy compared to total adrenalectomy. A higher risk of postoperative PPC recurrence following partial adrenalectomy compared to total adrenalectomy. Similar risk of metastasis and overall mortality in both groups. |
| Gan et al. [139] | 2022 | 10 studies (898 patients) | Comparison of minimally invasive adrenalectomy (MIA) with open adrenalectomy (OA) in patients with large adrenal tumors (≥5 cm) | MIA superior to OA for duration of hospitalization, drainage duration, and fasting duration estimated blood loss and transfusion. Similar duration of operation and complication rate in both groups. |
| Fu et al. [140] | 2020 | 14 studies (626 patients) | Comparison of open surgery and laparoscopic surgery for PPC | Lower rates of intraoperative hemodynamic instability, less intraoperative blood loss, lower blood transfusion rates, earlier ambulation, and food intake, shorter drainage tube indwelling time and postoperative stay and lower overall complication rates in LS compared to OS. Similar duration of operation, postoperative blood pressure control, severe complications rate, postoperative hypotension or cardiovascular disease in both groups. |
| Li et al. [141] | 2020 | 14 studies (743 patients) | Comparison of laparoscopic surgery versus open surgery for adrenal PGL | Smaller tumor size and higher body mass index in LS compared to OS. lower estimated blood loss, lower transfusion rate, lower hemodynamic instability, less postoperative complications, lower Clavien–Dindo score ≥3 complications, shorter return to diet time, and shorter duration of hospitalization in LS compared to OS. |
| Jiang et al. [142] | 2020 | 4 studies (203 patients) | Comparison of of transperitoneal laparoscopic adrenalectomy with retroperitoneal laparoscopic adrenalectomy for adrenal PGL | Shorter duration of operation, less intraoperative blood loss, shorter duration of hospitalization in retroperitoneal laparoscopic adrenalectomy compared to transperitoneal one. Similar complication rate and incidence of hemodynamic crisis in both groups. |
| Schimmack et al. [143] | 2020 | 4 studies (603 patients) | Comparison of preoperative α-blockade and no blockade for adrenal PGL patients undergoing surgery | Similar mortality, cardiovascular complications, mean maximal intraoperative systolic and diastolic BP, and mean maximal intraoperative heart rate in patients with or without α-blockade. |
| Hamidi et al. [144] | 2017 | 20 studies (1338 patients) | Analysis of predictors for mortality rates in patients with metastatic PPGL | Higher mortality associated with male gender and synchronous metastases |
| Abu-Ghanem et al. [93] | 2016 | 15 studies (470 patients) | Comparison of preoperative embolization and no preoperative embolization in patients undergoing surgery for CBT | Similar estimated blood loss, duration of operation, duration of hospitalization, risks of cranial nerve injury, vascular injury, and stroke in embolization and no-embolization groups. |
| Texakalidis et al. [145] | 2019 | 25 studies (1326 patients) | Comparison of preoperative embolization and no preoperative embolization in patients undergoing surgery for CBT | Shorter duration of operation, lower intraoperative blood loss in embolization group compared to no-embolization group. Similar rates of cranial nerve injuries, stroke, transient ischemic attacks, duration of hospitalization in both groups. |
| Wang et al. [94] | 2024 | 25 studies (1326 patients) | Analysis of characteristics, management and operative complications of CBT | Lower estimated blood loss and shorter duration of operation, higher rate of stroke in embolization group compared to no-embolization group. Higher Shamblin grade tumors associated with more operative complications. More frequent relevant family history and more symptoms in SDHx mutation-positive patients compared to patient without mutation. |
| Robertson et al. [34] | 2019 | 104 studies (4418 patients) | Analysis of characteristics, management and operative complications of CBT | Correlation of Shamblin status with stroke: I CBT associated with a 1.89% stroke rate, 2.71% for Shamblin II CBT and 3.99% for Shamblin III tumors. Correlation of Shamblin status with CNI rates: 3.76% for Shamblin I CBT, 14.14% for Shamblin II, and 17.10% for Shamblin III CBT. Similar drainage loss, rate of neck hematoma, and re-exploration rate due to hematoma in embolization and no-embolization groups. |
| Koh et al. [146] | 2024 | 43 studies (8849 patients) | Analysis of demographics, clinical characteristics, treatment methods, and outcomes of SDH-mutated HNPGLs | Correlation between SDHD mutations and multifocality. Correlation between SDHB mutations and distant metastases. No correlation between SDH-related mutations and gender, age, tumor size, and familial occurrences. |
| Napoli et al. [147] | 2023 | 5 studies (245 patients) | Analysis of effectiveness of a preoperative embolization according to different Shamblin classes | Lower blood loss in embolization group compared to no-embolization group. Similar duration of operation in both groups. |
| Fatima et al. [119] | 2021 | 37 studies (11174 patients) | Analysis of effectiveness of stereotactic radiosurgery HNPGL | Surgery more frequently related to transient or permanent deficits compared to SRS. No difference in local control depending upon the SRS technique. SRS in HNPGLs associated with good clinical and radiological outcome. |
| Jackson et al. [148] | 2015 | 22 studies (578 patients) | Assessment of effects of preoperative embolization on CBT surgery | Less estimated blood loss and shorter duration of operation in embolization group compared to no-embolization group. |
| Ghanaati H et al. [149] | 2024 | 19 studies (328 patients) | Assessment of effects of preoperative embolization on GJT surgery | Less estimated blood loss and shorter duration of operation in embolization group. |
| Ong et al. [115] | 2022 | 23 studies (460 patients) | Assessment of stereotactic radiosurgery for GJTs | 95% tumor control rate 95% and 47% symptomatic improvement following SRS that may be a suitable treatment modality for GJTs. |
| Dharnipragada et al. [116] | 2023 | 19 studies (852 patients) | Comparison of radiosurgery and surgical resection for TJP | Similar in both groups—3.5% tumor growth rate following radiosurgery vs. 3.9% recurrence rate in surgery. Lower (7.6%) complication rate for radiosurgery compared to surgery (29.6%). |
| Ivan et al. [117] | 2011 | 46 studies (869 GJT patients) | Comparison of recurrence and cranial neuropathy after subtotal resection (STR), gross-total resection (GTR), STR with adjuvant postoperative radiosurgery (STR+SRS), and stereotactic radiosurgery alone (SRS) | Complete response, partial response and stable disease of, respectively, 4% (95% CI: 1–15%), 37%(95% CI: 25–51%) and 14% (95% CI: 7–27%). |
| Niemeijer et al. [124] | 2014 | 4 studies (50 patients) | Assessment of chemotherapy with CVD on tumor volume in patients with malignant PPGL | 4% complete response, 37% partial response and 14% stable disease. 14% complete, 40% partial and 20% stable hormonal response. |
| Zhou et al. [127] | 2023 | 7 studies (160 patients) | Assessment of efficacy and safety of TKIs in metastatic PPGLs | 0.320 partial response, 0.520 stable disease, and 0.856 disease control rates. Progression-free survival 8.9 months. |
| Marretta et al. [132] | 2023 | 12 studies (213 patients) | Assessment of peptide receptor radionuclide therapy (PRRT) with 177Lu-DOTATATE and 90Y-DOTATOC in the metastatic PPGLs | 0.83 and 0.76 disease control rate and 0.76 for 177Lu- and 90Y-PRRT, respectively. 0.81 pooled disease control rate for PRRT. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brewczyński, A.; Kolasińska-Ćwikła, A.; Jabłońska, B.; Wyrwicz, L. Pheochromocytomas and Paragangliomas—Current Management. Cancers 2025, 17, 1029. https://doi.org/10.3390/cancers17061029
Brewczyński A, Kolasińska-Ćwikła A, Jabłońska B, Wyrwicz L. Pheochromocytomas and Paragangliomas—Current Management. Cancers. 2025; 17(6):1029. https://doi.org/10.3390/cancers17061029
Chicago/Turabian StyleBrewczyński, Adam, Agnieszka Kolasińska-Ćwikła, Beata Jabłońska, and Lucjan Wyrwicz. 2025. "Pheochromocytomas and Paragangliomas—Current Management" Cancers 17, no. 6: 1029. https://doi.org/10.3390/cancers17061029
APA StyleBrewczyński, A., Kolasińska-Ćwikła, A., Jabłońska, B., & Wyrwicz, L. (2025). Pheochromocytomas and Paragangliomas—Current Management. Cancers, 17(6), 1029. https://doi.org/10.3390/cancers17061029