
Design of a Phase I Drug Combination Study with Adaptive Allocation Based on Dose-Limiting Toxicity Attribution


Simple Summary
Abstract
1. Introduction
2. Methods
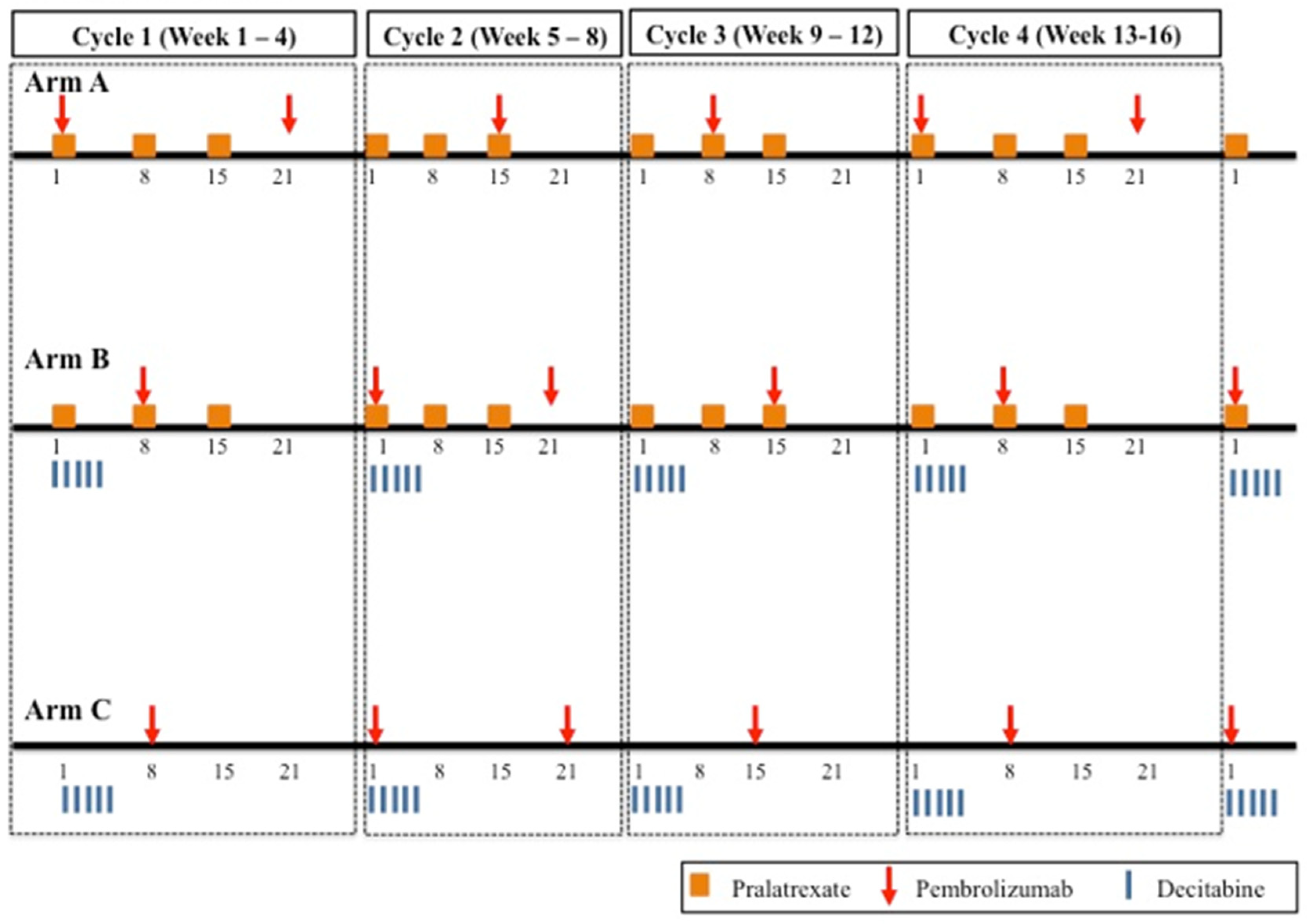
2.1. Design Considerations
2.2. Estimation
2.3. Allocation
- If the most recent participant accrued to the study does not experience a DLT, then the recommended combination for the next participant will be the combination indicated by the model to have an estimated DLT rate closest to 25%. Escalation is restricted to adjacent dose combinations that differ from the current combination by one dose level of one drug.
- If the most recent participant accrued to the study does experience a DLT, then attribution by study agent occurs such that
- If the DLT is mucositis or thrombocytopenia or mucositis plus neutropenia and/or thrombocytopenia, then the recommended combination for the next participant will be restricted to either the current combination administered to the most recent participant or to de-escalation by one dose level of pralatrexate, based on which combination has an estimated DLT rate closest to 25%. We designate the occurrence of this event as a “Type 1” DLT.
- If the DLT is neutropenia or neutropenia and thrombocytopenia, then the recommended combination for the next participant will be restricted to either the current combination administered to the most recent participant or to de-escalation by one dose level of decitabine, based on which combination has an estimated DLT rate closest to 25%. We designate the occurrence of this event as a “Type 2” DLT.
- If the DLT is neither a Type 1 nor Type 2 event, we designate it as a “Type 3” DLT event. If Type 1 and Type 2 events occur simultaneously, then the attribution cannot be ascertained and the event is considered to be a Type 3 event.
2.4. Stopping the Trial
- Accrual to Arm B will be stopped for safety if the observed DLT rate at the lowest combination (Combination 1) is ≥ the number of DLTs out of the number of participants treated at the lowest combination as displayed in Table 4. The stopping guidelines in Table 4 are based on whether the lower limit of an Agresti–Coull [15] binomial confidence interval (with 80% confidence) for the lowest combination exceeds the target DLT rate. The bounds were generated using the web application at http://uvatrapps.shinyapps.io/pocrm/, accessed on 20 October 2020.
- 2.
- If the recommendation is to assign the next participant to a combination that already has 10 participants (including the four Columbia participants) treated on the combination, accrual to Arm B will be stopped and the recommended combination is declared the MTDC.
- 3.
- Otherwise, the MTDC is defined as the combination that is recommended after the maximum sample size of 30 participants are accrued to the study.
3. Results
3.1. Simulation Studies to Evaluate the Design
3.1.1. Single Simulated Trial Illustration
3.1.2. Operating Characteristics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yin, G.; Yuan, Y. A latent contingency table approach to dose finding for combinations of two agents. Biometrics 2009, 65, 866–875. [Google Scholar] [PubMed]
- Murtaugh, P.A.; Fisher, L.D. Bivariate binary models of efficacy and toxicity in dose-ranging trials. Commun. Stat. Part A-Theory Methods 1990, 19, 2003–2020. [Google Scholar]
- Wheeler, G.M.; Sweeting, M.J.; Mander, A.P.; Lee, S.M.; Cheung, Y.K.K. Modelling semi-attributable toxicity in dual-agent phase I trials with non-concurrent drug administration. Stat. Med. 2017, 36, 225–241. [Google Scholar]
- Iasonos, A.; O’Quigley, J. Phase I designs that allow for uncertainty in the attribution of adverse events. J. R. Stat. Soc. C Appl. Stat. 2017, 66, 1015–1030. [Google Scholar]
- Lee, B.L.; Fan, S.K. A two-dimensional search algorithm for dose-finding trials of two agents. J. Biopharm. Stat. 2012, 22, 802–818. [Google Scholar] [PubMed]
- Wages, N.A.; Conaway, M.R.; O’Quigley, J. Continual reassessment method for partial ordering. Biometrics 2011, 67, 1555–1563. [Google Scholar] [CrossRef] [PubMed]
- Wages, N.A.; Dillon, P.M.; Portell, C.A.; Slingluff, C.L., Jr.; Petroni, G.R. Applications of the partial-order continual reassessment method in the early development of treatment combinations. Clin. Trials 2024, 21, 331–339. [Google Scholar] [CrossRef]
- Patel, A.; Brock, K.; Slade, D.; Gaunt, C.; Kong, A.; Mehanna, H.; Billingham, L.; Gaunt, P. Implementing the time-to-event continual reassessment method in the presence of partial orders in a phase I head and neck cancer trial. BMC Med. Res. Methodol. 2024, 24, 11. [Google Scholar]
- Mozgunov, P.; Jaki, T.; Gounaris, I.; Goddemeier, T.; Victor, A.; Grinberg, M. Practical implementation of the partial ordering continual reassessment method in a Phase I combination-schedule dose-finding trial. Stat. Med. 2022, 41, 5789–5809. [Google Scholar]
- Yap, T.A.; Plummer, E.R.; Tolcher, A.W.; Szucs, Z.; Gounaris, I.; Locatelli, G.; Enderlin, M.; Hicking, C.; Mukker, J.K.; De Bono, J.S. A phase I study of highly potent oral ATR inhibitor (ATRi) tuvusertib plus oral PARP inhibitor (PARPi) niraparib in patients with solid tumors. J. Clin. Oncol. 2024, 42 (Suppl. 16), 3018. [Google Scholar]
- Walker, L.E.; FitzGerald, R.; Saunders, G.; Lyon, R.; Fisher, M.; Martin, K.; Eberhart, I.; Woods, C.; Ewings, S.; Hale, C.; et al. An open label, adaptive, phase 1 trial of high-dose oral nitazoxanide in healthy volunteers: An antiviral candidate for SARS-CoV-2. Clin. Pharmacol. Ther. 2022, 111, 585–594. [Google Scholar] [PubMed]
- O’Quigley, J.; Pepe, M.; Fisher, L. Continual reassessment method: A practical design for Phase I clinical trials in cancer. Biometrics 1990, 46, 33–48. [Google Scholar]
- Paoletti, X.; Kramar, A. A comparison of model choices for the continual reassessment method in phase I clinical trials. Stat. Med. 2009, 28, 3012–3028. [Google Scholar]
- Lee, S.M.; Cheung, Y.K. Model calibration in the continual reassessment method. Clin. Trials 2009, 6, 227–238. [Google Scholar]
- Agresti, A.; Coull, B.A. Approximate is better than ‘exact’ for interval estimation of binomial proportions. Am. Stat. 1998, 52, 119–126. [Google Scholar]
- Love, S.B.; Brown, S.; Weir, C.J.; Harbron, C.; Yap, C.; Gaschler-Markefski, B.; Matcham, J.; Caffrey, L.; McKevitt, C.; Clive, S.; et al. Embracing model-based designs for dose-finding trials. Br. J. Cancer 2017, 117, 332–339. [Google Scholar] [PubMed]
- Iasonos, A.; O’Quigley, J. Adaptive dose-finding studies: A review of model-guided phase I clinical trials. J. Clin. Oncol. 2014, 32, 2505–2511. [Google Scholar]
- Riviere, M.K.; Le Tourneau, C.; Paoletti, X.; Dubois, F.; Zohar, S. Designs of drug-combination phase I trials in oncology: A systematic review of the literature. Ann. Oncol. 2015, 26, 669–674. [Google Scholar]
- Wages, N.A.; Conaway, M.R.; Slingluff, C.L., Jr.; Williams, M.E.; Portell, C.A.; Hwu, P.; Petroni, G.R. Recent developments in the implementation of novel designs for early-phase combination studies. Ann. Oncol. 2015, 26, 1036–1037. [Google Scholar]
- Paoletti, X.; Ezzalfani, M.; Le Tourneau, C. Statistical controversies in clinical research: Requiem for the 3+3 design for phase I trials. Ann. Oncol. 2015, 26, 1808–1812. [Google Scholar]
- Nie, L.; Rubin, E.H.; Mehrotra, N.; Pinheiro, J.; Fernandes, L.L.; Roy, A.; Bailey, S.; de Alwis, D.P. Rendering the 3+3 design to rest: More efficient approaches to targeted oncology dose-finding trials. Clin. Cancer Res. 2016, 22, 2623–2629. [Google Scholar] [PubMed]
- Kurzrock, R.; Lin, C.C.; Wu, T.C.; Hobbs, B.P.; Pestana, R.C.; Hong, D.S. Moving beyond 3+3: The future of clinical trial design. Am. Soc. Clin. Oncol. Educ. Book 2021, 41, e133–e144. [Google Scholar] [PubMed]
- Silva, R.B.; Yap, C.; Carvajal, R.; Lee, S.M. Would the recommended dose have been different using novel dose-finding designs? Comparing dose-finding designs in published trials. JCO Precis. Oncol. 2021, 5, 1024–1034. [Google Scholar]
- Clertant, M. Early-phase oncology trials: Why so many designs? J. Clin. Oncol. 2022, 40, 3529–3536. [Google Scholar]
- Chiuzan, C.; Dehbi, H.M. The 3+3 design in dose-finding studies with small sample sizes: Pitfalls and possible remedies. Clin. Trials 2024, 21, 350–357. [Google Scholar]
- Ratain, M.J. Designing dose-finding phase I clinical trials: Top questions that should be discussed with your clinical pharmacologist. JCO Precis. Oncol. 2021, 5, 935–936. [Google Scholar]
- Petroni, G.R.; Wages, N.A.; Paux, G.; Dubois, F. Implementation of adaptive methods in early-phase clinical trials. Stat. Med. 2017, 36, 215–224. [Google Scholar] [PubMed]
- Iasonos, A.; Gönen, M.; Bosl, G.J. Scientific review of phase I protocols with novel dose-escalation designs: How much information is needed? J. Clin. Oncol. 2015, 33, 2221–2225. [Google Scholar]
- Wei, L.; Pan, X.; Fernandez, S. Practical considerations for the implementation of adaptive designs for oncology Phase I dose-finding trials. Future Drug Discov. 2019, 1, FDD18. [Google Scholar]
- Yap, C.; Solovyeva, O.; De Bono, J.; Rekowski, J.; Patel, D.; Jaki, T.; Mander, A.; Evans TR, J.; Peck, R.; Hayward, K.S.; et al. Enhancing quality and impact of early phase dose-finding clinical trials: CONSORT-DEFINE & SPIRIT-DEFINE guidance. BMJ 2023, 383, e076387. [Google Scholar]
- Conaway, M.R.; Petroni, G.R. The impact of early-phase trial design in the drug development process. Clin. Cancer Res. 2019, 25, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Ratain, M.J. The role of early-phase design. Clin. Cancer Res. 2019, 25, 3190. [Google Scholar] [CrossRef] [PubMed]
- Jaki, T.; Burdon, A.; Chen, X.; Mozgunov, P.; Zheng, H.; Baird, R. Early phase clinical trials in oncology: Realising the potential of seamless designs. Eur. J. Cancer 2023, 189, 112916. [Google Scholar] [CrossRef] [PubMed]
- Op‘t Hoog, C.J.; Mehra, N.; Maliepaard, M.; Bol, K.; Gelderblom, H.; Sonke, G.S.; de Langen, A.J.; van de Donk, N.W.; Janssen, J.J.; Minnema, M.C.; et al. Dose selection of novel anticancer drugs: Exposing the gap between selected and required doses. Lancet Oncol. 2024, 25, e340–e351. [Google Scholar] [CrossRef]

{kind=link}
| Pralatrexate | 30 mg days 1, 8, and 15 | Combination 12 | Combination 14 | Combination 15 |
| 30 mg days 1 and 15 | Combination 9 | Combination 11 | Combination 13 | |
| 20 mg days 1, 8, and 15 | Combination 6 | Combination 8 | Combination 10 | |
| 20 mg days 1 and 15 | Combination 3 | Combination 5 | Combination 7 | |
| 15 mg days 1 and 15 | Combination 1 | Combination 2 | Combination 4 | |
| 10 mg days 1–3 | 10 mg days 1–5 | 20 mg days 1–3 | ||
| Decitabine | ||||
|
|
|
|
|
|
| Combination Labels | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Order | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 |
| 1 | 0.001 | 0.004 | 0.010 | 0.03 | 0.06 | 0.11 | 0.17 | 0.25 | 0.33 | 0.42 | 0.50 | 0.58 | 0.65 | 0.71 | 0.76 |
| 2 | 0.001 | 0.004 | 0.010 | 0.11 | 0.06 | 0.03 | 0.17 | 0.25 | 0.33 | 0.58 | 0.50 | 0.42 | 0.65 | 0.71 | 0.76 |
| 3 | 0.001 | 0.110 | 0.004 | 0.50 | 0.17 | 0.01 | 0.58 | 0.25 | 0.03 | 0.65 | 0.33 | 0.06 | 0.71 | 0.42 | 0.76 |
| 4 | 0.001 | 0.004 | 0.030 | 0.01 | 0.06 | 0.17 | 0.11 | 0.25 | 0.42 | 0.33 | 0.50 | 0.65 | 0.58 | 0.71 | 0.76 |
| 5 | 0.001 | 0.010 | 0.004 | 0.11 | 0.06 | 0.03 | 0.33 | 0.25 | 0.17 | 0.58 | 0.50 | 0.42 | 0.71 | 0.65 | 0.76 |
| 6 | 0.001 | 0.010 | 0.004 | 0.03 | 0.06 | 0.11 | 0.33 | 0.25 | 0.17 | 0.42 | 0.50 | 0.58 | 0.71 | 0.65 | 0.76 |
| Number of Participants | Boundary |
|---|---|
| 2–3 | ≥2 |
| 4–6 | ≥3 |
| 7–9 | ≥4 |
| 10 | ≥5 |
| Participant ID | Dose Pair Assigned | True DLT Rate of Assigned Dose Pair | DLT Outcome | DLT Type |
|---|---|---|---|---|
| 1 | 8 | 0.47 | 0 | - |
| 2 | 11 | 0.56 | 1 | 1 |
| 3 | 8 | 0.47 | 1 | 2 |
| 4 | 8 | 0.47 | 1 | 3 |
| 5 | 5 | 0.25 | 0 | - |
| 6 | 4 | 0.25 | 0 | - |
| 7 | 7 | 0.47 | 1 | - |
| 8 | 4 | 0.25 | 1 | - |
| 9 | 4 | 0.25 | 0 | - |
| 10 | 5 | 0.25 | 1 | 2 |
| 11 | 5 | 0.25 | 1 | 1 |
| 12 | 2 | 0.18 | 0 | - |
| 13 | 2 | 0.18 | 0 | - |
| 14 | 4 | 0.25 | 0 | - |
| 15 | 4 | 0.25 | 1 | 1 |
| 16 | 4 | 0.25 | 0 | - |
| 17 | 4 | 0.25 | 1 | 1 |
| 18 | 4 | 0.25 | 0 | - |
| 19 | 5 | 0.25 | 1 | 2 |
| 20 | 3 | 0.18 | 0 | - |
| 21 | 2 | 0.18 | 0 | - |
| 22 | 2 | 0.18 | 0 | - |
| 23 | 4 | 0.25 | 0 | - |
| 24 | 4 | 0.25 | 0 | - |
| Scenario 1 | Scenario 2 | Scenario 3 | Scenario 4 | Scenario 5 | |
|---|---|---|---|---|---|
| PCS | 38.9 | 67.9 | 38.9 | 42.7 | 48.7 |
| # of observed DLTs | 5.0 | 6.1 | 4.0 | 8.0 | 9.3 |
| # of average sample size | 22.8 | 21.6 | 22.9 | 23.9 | 24.8 |
| # of participants treated at MTDC | 6.8 | 11.3 | 4.8 | 6.9 | 7.0 |
| % of early stopping for safety | 0.0 | 0.0 | 0.0 | 0.1 | 0.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wages, N.A.; Horton, B.J.; Liu, L.; Marchi, E.; Petroni, G.R. Design of a Phase I Drug Combination Study with Adaptive Allocation Based on Dose-Limiting Toxicity Attribution. Cancers 2025, 17, 1038. https://doi.org/10.3390/cancers17061038
Wages NA, Horton BJ, Liu L, Marchi E, Petroni GR. Design of a Phase I Drug Combination Study with Adaptive Allocation Based on Dose-Limiting Toxicity Attribution. Cancers. 2025; 17(6):1038. https://doi.org/10.3390/cancers17061038
Chicago/Turabian StyleWages, Nolan A., Bethany J. Horton, Li Liu, Enrica Marchi, and Gina R. Petroni. 2025. "Design of a Phase I Drug Combination Study with Adaptive Allocation Based on Dose-Limiting Toxicity Attribution" Cancers 17, no. 6: 1038. https://doi.org/10.3390/cancers17061038
APA StyleWages, N. A., Horton, B. J., Liu, L., Marchi, E., & Petroni, G. R. (2025). Design of a Phase I Drug Combination Study with Adaptive Allocation Based on Dose-Limiting Toxicity Attribution. Cancers, 17(6), 1038. https://doi.org/10.3390/cancers17061038