
Interplay and Dynamics of Chromatin Architecture and DNA Damage Response: An Overview


 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
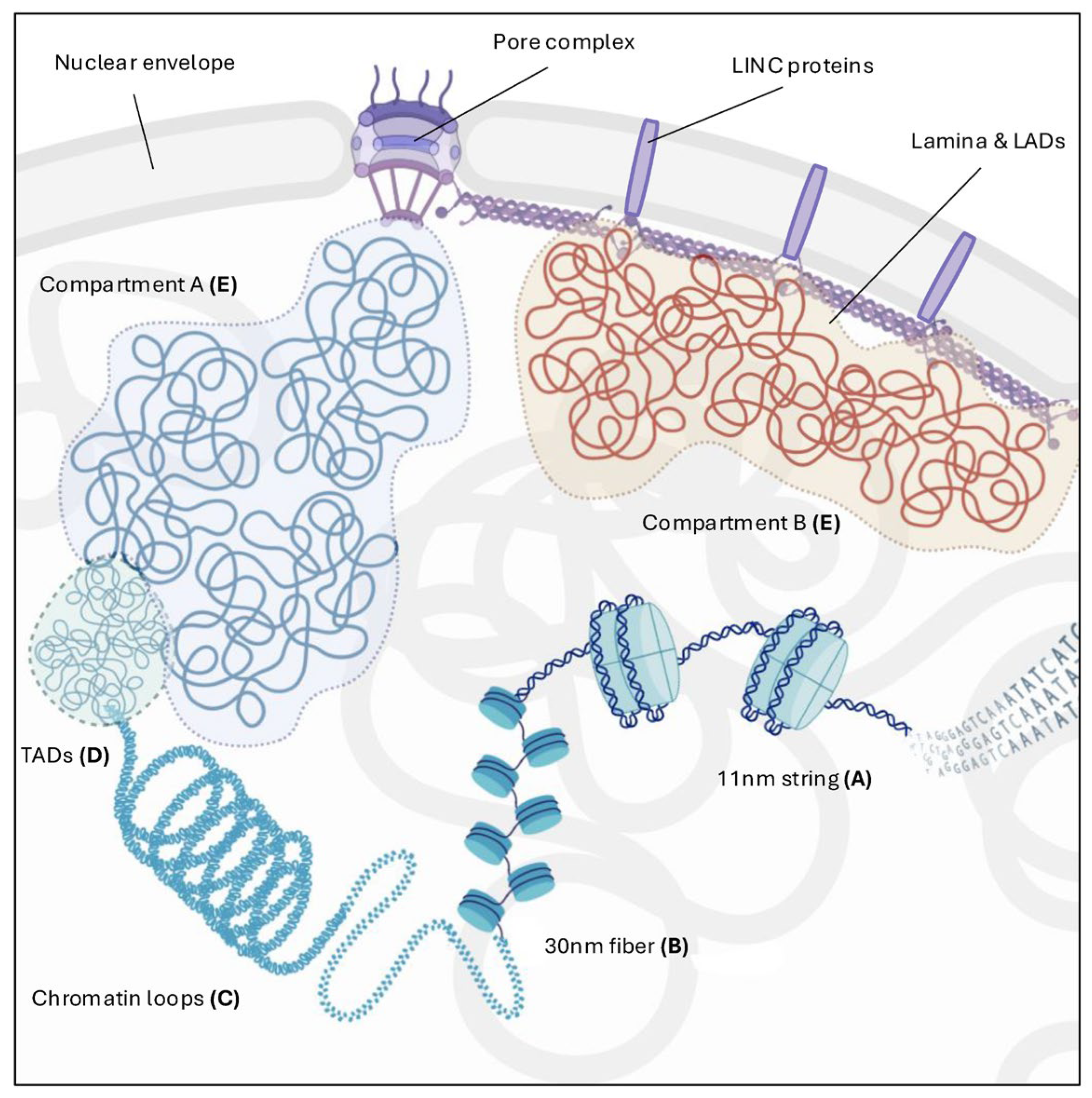
2. Chromatin Architecture: From Nucleosome to Higher Order Organization
3. Chromatin Architecture and DNA Damage Response
3.1. Local Chromatin Reorganization upon DSBs
3.1.1. DSBs Histone Code
3.1.2. Transcription Regulation at DSBs
3.2. Large-Scale Chromatin Reorganization upon DSBs
3.2.1. TADs Lead DDR Chromatin Domains’ Establishment
3.2.2. DNA Repair Is Dependent on Preexisting Chromatin Features
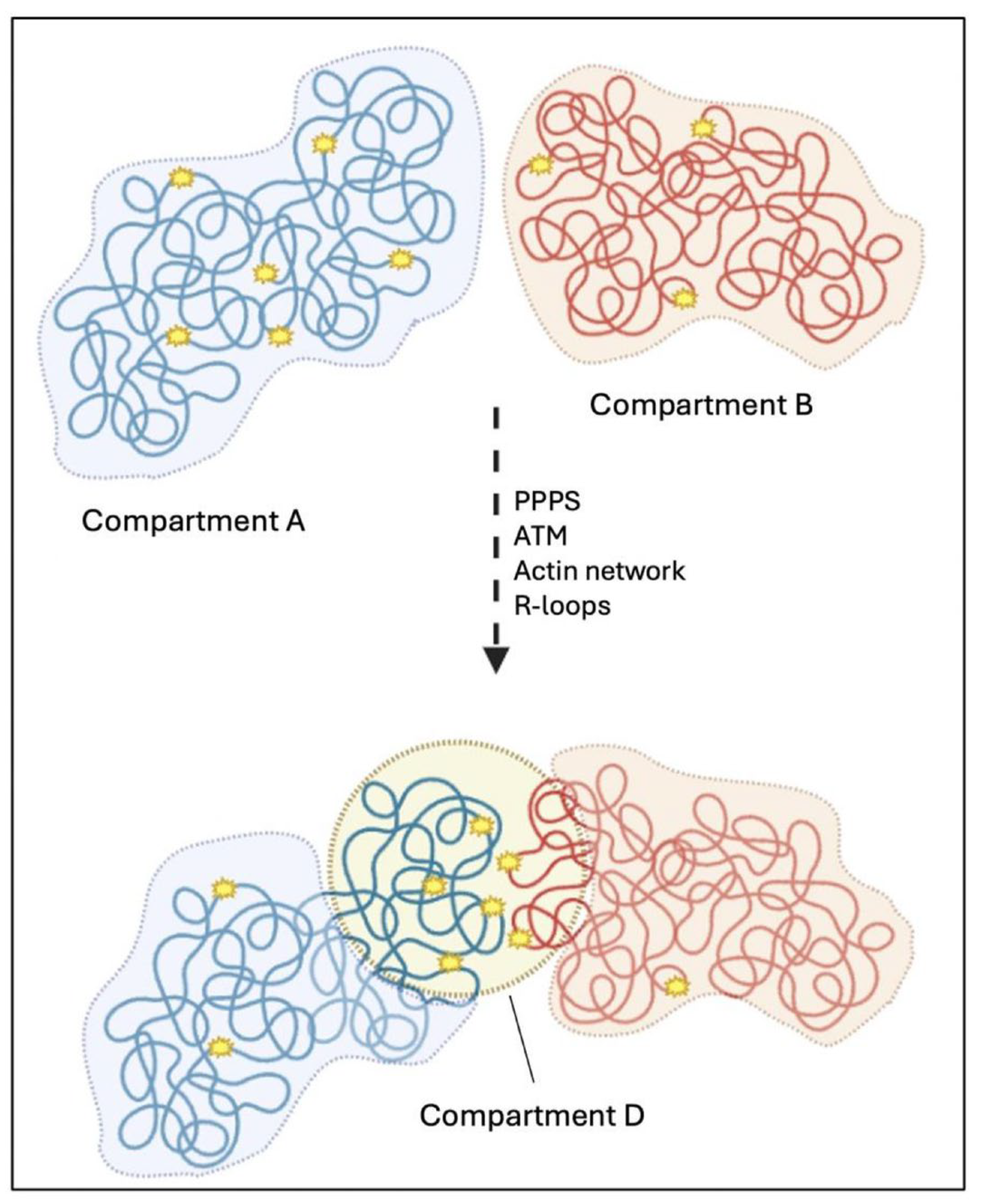
3.2.3. Nuclear Compartmentalization Following DSBs’ Induction
3.2.4. DSBs’ Clustering Depends on the Crosstalk Between Nuclear Structures and DNA Repair
4. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Lai, W.K.M.; Pugh, B.F. Understanding Nucleosome Dynamics and Their Links to Gene Expression and DNA Replication. Nat. Rev. Mol. Cell Biol. 2017, 18, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Millán-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone Post-Translational Modifications—Cause and Consequence of Genome Function. Nat. Rev. Genet. 2022, 23, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Boopathi, R.; Dimitrov, S.; Hamiche, A.; Petosa, C.; Bednar, J. Cryo-Electron Microscopy of the Chromatin Fiber. Curr. Opin. Struct. Biol. 2020, 64, 97–103. [Google Scholar] [CrossRef]
- Ricci, M.A.; Manzo, C.; García-Parajo, M.F.; Lakadamyali, M.; Cosma, M.P. Chromatin Fibers Are Formed by Heterogeneous Groups of Nucleosomes In Vivo. Cell 2015, 160, 1145–1158. [Google Scholar] [CrossRef]
- Dekker, J.; Misteli, T. Long-Range Chromatin Interactions. Cold Spring Harb. Perspect. Biol. 2015, 7, a019356. [Google Scholar] [CrossRef]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Dehingia, B.; Milewska, M.; Janowski, M.; Pękowska, A. CTCF Shapes Chromatin Structure and Gene Expression in Health and Disease. EMBO Rep. 2022, 23, e55146. [Google Scholar] [CrossRef]
- Parelho, V.; Hadjur, S.; Spivakov, M.; Leleu, M.; Sauer, S.; Gregson, H.C.; Jarmuz, A.; Canzonetta, C.; Webster, Z.; Nesterova, T.; et al. Cohesins Functionally Associate with CTCF on Mammalian Chromosome Arms. Cell 2008, 132, 422–433. [Google Scholar] [CrossRef]
- Zhang, H.; Shi, Z.; Banigan, E.J.; Kim, Y.; Yu, H.; Bai, X.; Finkelstein, I.J. CTCF and R-Loops Are Boundaries of Cohesin-Mediated DNA Looping. Mol. Cell 2023, 83, 2856–2871.e8. [Google Scholar] [CrossRef]
- Peters, J.-M.; Tedeschi, A.; Schmitz, J. The Cohesin Complex and Its Roles in Chromosome Biology. Genes Dev. 2008, 22, 3089–3114. [Google Scholar] [CrossRef]
- Davidson, I.F.; Peters, J.-M. Genome Folding through Loop Extrusion by SMC Complexes. Nat. Rev. Mol. Cell Biol. 2021, 22, 445–464. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological Domains in Mammalian Genomes Identified by Analysis of Chromatin Interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.-H.; Ghosh, S.; Noordermeer, D. TADs and Their Borders: Free Movement or Building a Wall? J. Mol. Biol. 2020, 432, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Dekker, J.; Mirny, L.A. The Chromosome Folding Problem and How Cells Solve It. Cell 2024, 187, 6424–6450. [Google Scholar] [CrossRef]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016, 15, 2038–2049. [Google Scholar] [CrossRef]
- Bintu, B.; Mateo, L.J.; Su, J.-H.; Sinnott-Armstrong, N.A.; Parker, M.; Kinrot, S.; Yamaya, K.; Boettiger, A.N.; Zhuang, X. Super-Resolution Chromatin Tracing Reveals Domains and Cooperative Interactions in Single Cells. Science 2018, 362, eaau1783. [Google Scholar] [CrossRef]
- Marnef, A.; Legube, G. Organizing DNA Repair in the Nucleus: DSBs Hit the Road. Curr. Opin. Cell Biol. 2017, 46, 1–8. [Google Scholar] [CrossRef]
- Briand, N.; Collas, P. Lamina-Associated Domains: Peripheral Matters and Internal Affairs. Genome Biol. 2020, 21, 85. [Google Scholar] [CrossRef]
- Markiewicz, E.; Dechat, T.; Foisner, R.; Quinlan, R.A.; Hutchison, C.J. Lamin A/C Binding Protein LAP2α Is Required for Nuclear Anchorage of Retinoblastoma Protein. Mol. Biol. Cell 2002, 13, 4401–4413. [Google Scholar] [CrossRef]
- Moir, R.D.; Montag-Lowy, M.; Goldman, R.D. Dynamic Properties of Nuclear Lamins: Lamin B Is Associated with Sites of DNA Replication. J. Cell Biol. 1994, 125, 1201–1212. [Google Scholar] [CrossRef]
- Ivorra, C.; Kubicek, M.; González, J.M.; Sanz-González, S.M.; Álvarez-Barrientos, A.; O’Connor, J.-E.; Burke, B.; Andrés, V. A Mechanism of AP-1 Suppression through Interaction of c-Fos with Lamin A/C. Genes Dev. 2006, 20, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Sun, J.; Kono, K.; Horikoshi, Y.; Ikura, T.; Tong, X.; Haraguchi, T.; Tashiro, S. Regulation of Homologous Recombinational Repair by Lamin B1 in Radiation-induced DNA Damage. FASEB J. 2015, 29, 2514–2525. [Google Scholar] [CrossRef] [PubMed]
- Redwood, A.B.; Perkins, S.M.; Vanderwaal, R.P.; Feng, Z.; Biehl, K.J.; Gonzalez-Suarez, I.; Morgado-Palacin, L.; Shi, W.; Sage, J.; Roti-Roti, J.L.; et al. A Dual Role for A-Type Lamins in DNA Double-Strand Break Repair. Cell Cycle 2011, 10, 2549–2560. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Park, S.H.; Myung, K.; Lee, K. Lamin A/C Facilitates DNA Damage Response by Modulating ATM Signaling and Homologous Recombination Pathways. Anim. Cells Syst. 2024, 28, 401–416. [Google Scholar] [CrossRef]
- Gibbs-Seymour, I.; Markiewicz, E.; Bekker-Jensen, S.; Mailand, N.; Hutchison, C.J. Lamin A/C-Dependent Interaction with 53BP1 Promotes Cellular Responses to DNA Damage. Aging Cell 2015, 14, 162–169. [Google Scholar] [CrossRef]
- Janetzko, J.; Oeck, S.; Schramm, A. The Multifaceted Roles of Lamins in Lung Cancer and DNA Damage Response. Cancers 2023, 15, 5501. [Google Scholar] [CrossRef]
- Stroud, M.J. Linker of Nucleoskeleton and Cytoskeleton Complex Proteins in Cardiomyopathy. Biophys. Rev. 2018, 10, 1033–1051. [Google Scholar] [CrossRef]
- King, M.C. Dynamic Regulation of LINC Complex Composition and Function across Tissues and Contexts. FEBS Lett. 2023, 597, 2823–2832. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Groelly, F.J.; Fawkes, M.; Dagg, R.A.; Blackford, A.N.; Tarsounas, M. Targeting DNA Damage Response Pathways in Cancer. Nat. Rev. Cancer 2023, 23, 78–94. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA Double-Strand Break Repair-Pathway Choice in Somatic Mammalian Cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Lottersberger, F.; Karssemeijer, R.A.; Dimitrova, N.; de Lange, T. 53BP1 and the LINC Complex Promote Microtubule-Dependent DSB Mobility and DNA Repair. Cell 2015, 163, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Ochs, F.; Somyajit, K.; Altmeyer, M.; Rask, M.-B.; Lukas, J.; Lukas, C. 53BP1 Fosters Fidelity of Homology-Directed DNA Repair. Nat. Struct. Mol. Biol. 2016, 23, 714–721. [Google Scholar] [CrossRef]
- West, S.C.; Blanco, M.G.; Chan, Y.W.; Matos, J.; Sarbajna, S.; Wyatt, H.D.M. Resolution of Recombination Intermediates: Mechanisms and Regulation. Cold Spring Harb. Symp. Quant. Biol. 2015, 80, 103–109. [Google Scholar] [CrossRef]
- Tarsounas, M.; Sung, P. The Antitumorigenic Roles of BRCA1–BARD1 in DNA Repair and Replication. Nat. Rev. Mol. Cell Biol. 2020, 21, 284–299. [Google Scholar] [CrossRef]
- Llorens-Agost, M.; Ensminger, M.; Le, H.P.; Gawai, A.; Liu, J.; Cruz-García, A.; Bhetawal, S.; Wood, R.D.; Heyer, W.-D.; Löbrich, M. POLθ-Mediated End Joining Is Restricted by RAD52 and BRCA2 until the Onset of Mitosis. Nat. Cell Biol. 2021, 23, 1095–1104. [Google Scholar] [CrossRef]
- Uziel, T. Requirement of the MRN Complex for ATM Activation by DNA Damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef]
- So, S.; Davis, A.J.; Chen, D.J. Autophosphorylation at Serine 1981 Stabilizes ATM at DNA Damage Sites. J. Cell Biol. 2009, 187, 977–990. [Google Scholar] [CrossRef]
- Caron, P.; Choudjaye, J.; Clouaire, T.; Bugler, B.; Daburon, V.; Aguirrebengoa, M.; Mangeat, T.; Iacovoni, J.S.; Álvarez-Quilón, A.; Cortés-Ledesma, F.; et al. Non-Redundant Functions of ATM and DNA-PKcs in Response to DNA Double-Strand Breaks. Cell Rep. 2015, 13, 1598–1609. [Google Scholar] [CrossRef]
- Lou, Z.; Minter-Dykhouse, K.; Franco, S.; Gostissa, M.; Rivera, M.A.; Celeste, A.; Manis, J.P.; Van Deursen, J.; Nussenzweig, A.; Paull, T.T.; et al. MDC1 Maintains Genomic Stability by Participating in the Amplification of ATM-Dependent DNA Damage Signals. Mol. Cell 2006, 21, 187–200. [Google Scholar] [CrossRef]
- Mattiroli, F.; Vissers, J.H.A.; van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 Ubiquitinates K13-15 on H2A/H2AX to Drive DNA Damage Signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.-L.; Bie, S.-Y.; Deng, Z.-H.; Bai, S.-M.; Shi, J.; Qin, C.-L.; Liu, H.-L.; Li, J.-X.; Chen, W.-Y.; Zhou, J.-Y.; et al. Ubiquitin-Induced RNF168 Condensation Promotes DNA Double-Strand Break Repair. Proc. Natl. Acad. Sci. USA 2024, 121, e2322972121. [Google Scholar] [CrossRef] [PubMed]
- Le, J.; Perez, E.; Nemzow, L.; Gong, F. Role of Deubiquitinases in DNA Damage Response. DNA Repair 2019, 76, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.G.; So, S.; Gupta, A.; Kumar, R.; Cayrou, C.; Avvakumov, N.; Bhadra, U.; Pandita, R.K.; Porteus, M.H.; Chen, D.J.; et al. MOF and Histone H4 Acetylation at Lysine 16 Are Critical for DNA Damage Response and Double-Strand Break Repair. Mol. Cell. Biol. 2010, 30, 3582–3595. [Google Scholar] [CrossRef]
- Lashgari, A.; Kougnassoukou Tchara, P.-E.; Lambert, J.-P.; Côté, J. New Insights into the DNA Repair Pathway Choice with NuA4/TIP60. DNA Repair 2022, 113, 103315. [Google Scholar] [CrossRef]
- Hsiao, K.-Y.; Mizzen, C.A. Histone H4 Deacetylation Facilitates 53BP1 DNA Damage Signaling and Double-Strand Break Repair. J. Mol. Cell Biol. 2013, 5, 157–165. [Google Scholar] [CrossRef]
- Tang, J.; Cho, N.W.; Cui, G.; Manion, E.M.; Shanbhag, N.M.; Botuyan, M.V.; Mer, G.; Greenberg, R.A. Acetylation Limits 53BP1 Association with Damaged Chromatin to Promote Homologous Recombination. Nat. Struct. Mol. Biol. 2013, 20, 317–325. [Google Scholar] [CrossRef]
- Horikoshi, N.; Sharma, D.; Leonard, F.; Pandita, R.K.; Charaka, V.K.; Hambarde, S.; Horikoshi, N.T.; Gaur Khaitan, P.; Chakraborty, S.; Cote, J.; et al. Pre-Existing H4K16ac Levels in Euchromatin Drive DNA Repair by Homologous Recombination in S-Phase. Commun. Biol. 2019, 2, 253. [Google Scholar] [CrossRef]
- Costelloe, T.; Lowndes, N.F. Chromatin Assembly and Signalling the End of DNA Repair Requires Acetylation of Histone H3 on Lysine 56. In Genome Stability and Human Diseases; Nasheuer, H.-P., Ed.; Subcellular Biochemistry; Springer: Dordrecht, The Netherlands, 2010; Volume 50, pp. 43–54. ISBN 978-90-481-3470-0. [Google Scholar]
- Chen, J.; Wang, Z.; Guo, X.; Li, F.; Wei, Q.; Chen, X.; Gong, D.; Xu, Y.; Chen, W.; Liu, Y.; et al. TRIM66 Reads Unmodified H3R2K4 and H3K56ac to Respond to DNA Damage in Embryonic Stem Cells. Nat. Commun. 2019, 10, 4273. [Google Scholar] [CrossRef]
- Hajji, N.; Wallenborg, K.; Vlachos, P.; Füllgrabe, J.; Hermanson, O.; Joseph, B. Opposing Effects of hMOF and SIRT1 on H4K16 Acetylation and the Sensitivity to the Topoisomerase II Inhibitor Etoposide. Oncogene 2010, 29, 2192–2204. [Google Scholar] [CrossRef]
- Pfister, S.X.; Ahrabi, S.; Zalmas, L.-P.; Sarkar, S.; Aymard, F.; Bachrati, C.Z.; Helleday, T.; Legube, G.; La Thangue, N.B.; Porter, A.C.G.; et al. SETD2-Dependent Histone H3K36 Trimethylation Is Required for Homologous Recombination Repair and Genome Stability. Cell Rep. 2014, 7, 2006–2018. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Nishikawa, H.; Fukuda, T.; Vittal, V.; Asano, M.; Miyoshi, Y.; Klevit, R.E.; Ohta, T. Interaction of BARD1 and HP1 Is Required for BRCA1 Retention at Sites of DNA Damage. Cancer Res. 2015, 75, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Saredi, G.; Becker, J.R.; Foster, B.M.; Nguyen, N.V.; Beyer, T.E.; Cesa, L.C.; Faull, P.A.; Lukauskas, S.; Frimurer, T.; et al. H4K20me0 Recognition by BRCA1–BARD1 Directs Homologous Recombination to Sister Chromatids. Nat. Cell Biol. 2019, 21, 311–318. [Google Scholar] [CrossRef]
- Simonetta, M.; De Krijger, I.; Serrat, J.; Moatti, N.; Fortunato, D.; Hoekman, L.; Bleijerveld, O.B.; Altelaar, A.F.M.; Jacobs, J.J.L. H4K20me2 Distinguishes Pre-Replicative from Post-Replicative Chromatin to Appropriately Direct DNA Repair Pathway Choice by 53BP1-RIF1-MAD2L2. Cell Cycle 2018, 17, 124–136. [Google Scholar] [CrossRef]
- Acs, K.; Luijsterburg, M.S.; Ackermann, L.; Salomons, F.A.; Hoppe, T.; Dantuma, N.P. The AAA-ATPase VCP/P97 Promotes 53BP1 Recruitment by Removing L3MBTL1 from DNA Double-Strand Breaks. Nat. Struct. Mol. Biol. 2011, 18, 1345–1350. [Google Scholar] [CrossRef]
- Mallette, F.A.; Mattiroli, F.; Cui, G.; Young, L.C.; Hendzel, M.J.; Mer, G.; Sixma, T.K.; Richard, S. RNF8- and RNF168-Dependent Degradation of KDM4A/JMJD2A Triggers 53BP1 Recruitment to DNA Damage Sites: RNF8- and RNF168-Dependent Degradation of KDM4A/JMJD2A. EMBO J. 2012, 31, 1865–1878. [Google Scholar] [CrossRef]
- Karl, L.A.; Peritore, M.; Galanti, L.; Pfander, B. DNA Double Strand Break Repair and Its Control by Nucleosome Remodeling. Front. Genet. 2022, 12, 821543. [Google Scholar] [CrossRef]
- Peritore, M.; Reusswig, K.-U.; Bantele, S.C.S.; Straub, T.; Pfander, B. Strand-Specific ChIP-Seq at DNA Breaks Distinguishes ssDNA versus dsDNA Binding and Refutes Single-Stranded Nucleosomes. Mol. Cell 2021, 81, 1841–1853.e4. [Google Scholar] [CrossRef]
- Gospodinov, A.; Vaissiere, T.; Krastev, D.B.; Legube, G.; Anachkova, B.; Herceg, Z. Mammalian Ino80 Mediates Double-Strand Break Repair through Its Role in DNA End Strand Resection. Mol. Cell. Biol. 2011, 31, 4735–4745. [Google Scholar] [CrossRef]
- He, L.; Moon, J.; Cai, C.; Hao, Y.; Lee, H.; Kim, W.; Zhao, F.; Lou, Z. The Interplay between Chromatin Remodeling and DNA Double-Strand Break Repair: Implications for Cancer Biology and Therapeutics. DNA Repair 2025, 146, 103811. [Google Scholar] [CrossRef]
- Min, S.; Ji, J.-H.; Heo, Y.; Cho, H. Transcriptional Regulation and Chromatin Dynamics at DNA Double-Strand Breaks. Exp. Mol. Med. 2022, 54, 1705–1712. [Google Scholar] [CrossRef] [PubMed]
- Ginjala, V.; Nacerddine, K.; Kulkarni, A.; Oza, J.; Hill, S.J.; Yao, M.; Citterio, E.; Van Lohuizen, M.; Ganesan, S. BMI1 Is Recruited to DNA Breaks and Contributes to DNA Damage-Induced H2A Ubiquitination and Repair. Mol. Cell. Biol. 2011, 31, 1972–1982. [Google Scholar] [CrossRef] [PubMed]
- Ui, A.; Nagaura, Y.; Yasui, A. Transcriptional Elongation Factor ENL Phosphorylated by ATM Recruits Polycomb and Switches Off Transcription for DSB Repair. Mol. Cell 2015, 58, 468–482. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Liu, R.; Jia, Z.; Li, R.; Zhu, F.; Zhu, W.; Shao, Y.; Jin, Y.; Xue, Y.; Huang, J.; et al. Poly(ADP-Ribosylation) of P-TEFb by PARP1 Disrupts Phase Separation to Inhibit Global Transcription after DNA Damage. Nat. Cell Biol. 2022, 24, 513–525. [Google Scholar] [CrossRef]
- Awwad, S.W.; Abu-Zhayia, E.R.; Guttmann-Raviv, N.; Ayoub, N. NELF-E Is Recruited to DNA Double-strand Break Sites to Promote Transcriptional Repression and Repair. EMBO Rep. 2017, 18, 745–764. [Google Scholar] [CrossRef]
- Michelini, F.; Pitchiaya, S.; Vitelli, V.; Sharma, S.; Gioia, U.; Pessina, F.; Cabrini, M.; Wang, Y.; Capozzo, I.; Iannelli, F.; et al. Damage-Induced lncRNAs Control the DNA Damage Response through Interaction with DDRNAs at Individual Double-Strand Breaks. Nat. Cell Biol. 2017, 19, 1400–1411. [Google Scholar] [CrossRef]
- Ouyang, J.; Yadav, T.; Zhang, J.-M.; Yang, H.; Rheinbay, E.; Guo, H.; Haber, D.A.; Lan, L.; Zou, L. RNA Transcripts Stimulate Homologous Recombination by Forming DR-Loops. Nature 2021, 594, 283–288. [Google Scholar] [CrossRef]
- Bonath, F.; Domingo-Prim, J.; Tarbier, M.; Friedländer, M.R.; Visa, N. Next-Generation Sequencing Reveals Two Populations of Damage-Induced Small RNAs at Endogenous DNA Double-Strand Breaks. Nucleic Acids Res. 2018, 46, 11869–11882. [Google Scholar] [CrossRef]
- D’Alessandro, G.; d’Adda Di Fagagna, F. Transcription and DNA Damage: Holding Hands or Crossing Swords? J. Mol. Biol. 2017, 429, 3215–3229. [Google Scholar] [CrossRef]
- Francia, S.; Michelini, F.; Saxena, A.; Tang, D.; De Hoon, M.; Anelli, V.; Mione, M.; Carninci, P.; d’Adda Di Fagagna, F. Site-Specific DICER and DROSHA RNA Products Control the DNA-Damage Response. Nature 2012, 488, 231–235. [Google Scholar] [CrossRef]
- Pessina, F.; Giavazzi, F.; Yin, Y.; Gioia, U.; Vitelli, V.; Galbiati, A.; Barozzi, S.; Garre, M.; Oldani, A.; Flaus, A.; et al. Functional Transcription Promoters at DNA Double-Strand Breaks Mediate RNA-Driven Phase Separation of Damage-Response Factors. Nat. Cell Biol. 2019, 21, 1286–1299. [Google Scholar] [CrossRef] [PubMed]
- Altmeyer, M.; Neelsen, K.J.; Teloni, F.; Pozdnyakova, I.; Pellegrino, S.; Grøfte, M.; Rask, M.-B.D.; Streicher, W.; Jungmichel, S.; Nielsen, M.L.; et al. Liquid Demixing of Intrinsically Disordered Proteins Is Seeded by Poly(ADP-Ribose). Nat. Commun. 2015, 6, 8088. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-L.; Zhao, W.-W.; Shi, J.; Wan, X.-B.; Zheng, J.; Fan, X.-J. Liquid-Liquid Phase Separation in DNA Double-Strand Breaks Repair. Cell Death Dis. 2023, 14, 746. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, D.; Furst, A.; Meaburn, K.; Lezaja, A.; Wen, Y.; Altmeyer, M.; Reina-San-Martin, B.; Soutoglou, E. Activation of Homologous Recombination in G1 Preserves Centromeric Integrity. Nature 2021, 600, 748–753. [Google Scholar] [CrossRef]
- Wei, L.; Nakajima, S.; Böhm, S.; Bernstein, K.A.; Shen, Z.; Tsang, M.; Levine, A.S.; Lan, L. DNA Damage during the G0/G1 Phase Triggers RNA-Templated, Cockayne Syndrome B-Dependent Homologous Recombination. Proc. Natl. Acad. Sci. USA 2015, 112, E3495–E3504. [Google Scholar] [CrossRef]
- Podhorecka, M.; Skladanowski, A.; Bozko, P. H2AX Phosphorylation: Its Role in DNA Damage Response and Cancer Therapy. J. Nucleic Acids 2010, 2010, 920161. [Google Scholar] [CrossRef]
- Iacovoni, J.S.; Caron, P.; Lassadi, I.; Nicolas, E.; Massip, L.; Trouche, D.; Legube, G. High-Resolution Profiling of γH2AX around DNA Double Strand Breaks in the Mammalian Genome. EMBO J. 2010, 29, 1446–1457. [Google Scholar] [CrossRef]
- Natale, F.; Rapp, A.; Yu, W.; Maiser, A.; Harz, H.; Scholl, A.; Grulich, S.; Anton, T.; Hörl, D.; Chen, W.; et al. Identification of the Elementary Structural Units of the DNA Damage Response. Nat. Commun. 2017, 8, 15760. [Google Scholar] [CrossRef]
- Varga, D.; Majoros, H.; Ujfaludi, Z.; Erdélyi, M.; Pankotai, T. Quantification of DNA Damage Induced Repair Focus Formation via Super-Resolution dSTORM Localization Microscopy. Nanoscale 2019, 11, 14226–14236. [Google Scholar] [CrossRef]
- Collins, P.L.; Purman, C.; Porter, S.I.; Nganga, V.; Saini, A.; Hayer, K.E.; Gurewitz, G.L.; Sleckman, B.P.; Bednarski, J.J.; Bassing, C.H.; et al. DNA Double-Strand Breaks Induce H2Ax Phosphorylation Domains in a Contact-Dependent Manner. Nat. Commun. 2020, 11, 3158. [Google Scholar] [CrossRef]
- Sanders, J.T.; Freeman, T.F.; Xu, Y.; Golloshi, R.; Stallard, M.A.; Hill, A.M.; San Martin, R.; Balajee, A.S.; McCord, R.P. Radiation-Induced DNA Damage and Repair Effects on 3D Genome Organization. Nat. Commun. 2020, 11, 6178. [Google Scholar] [CrossRef] [PubMed]
- Arnould, C.; Rocher, V.; Finoux, A.-L.; Clouaire, T.; Li, K.; Zhou, F.; Caron, P.; Mangeot, P.E.; Ricci, E.P.; Mourad, R.; et al. Loop Extrusion as a Mechanism for Formation of DNA Damage Repair Foci. Nature 2021, 590, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Fedkenheuer, M.; Shang, Y.; Jung, S.; Fedkenheuer, K.; Park, S.; Mazza, D.; Sebastian, R.; Nagashima, H.; Zong, D.; Tan, H.; et al. A Dual Role of Cohesin in DNA DSB Repair. Nat. Commun. 2025, 16, 843. [Google Scholar] [CrossRef]
- Allshire, R.C.; Madhani, H.D. Ten Principles of Heterochromatin Formation and Function. Nat. Rev. Mol. Cell Biol. 2018, 19, 229–244. [Google Scholar] [CrossRef]
- Seman, M.; Levashkevich, A.; Larkin, A.; Huang, F.; Ragunathan, K. Uncoupling the Distinct Functions of HP1 Proteins during Heterochromatin Establishment and Maintenance. Cell Rep. 2023, 42, 113428. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Noon, A.T.; Deckbar, D.; Ziv, Y.; Shiloh, Y.; Löbrich, M.; Jeggo, P.A. ATM Signaling Facilitates Repair of DNA Double-Strand Breaks Associated with Heterochromatin. Mol. Cell 2008, 31, 167–177. [Google Scholar] [CrossRef]
- Bolderson, E.; Savage, K.I.; Mahen, R.; Pisupati, V.; Graham, M.E.; Richard, D.J.; Robinson, P.J.; Venkitaraman, A.R.; Khanna, K.K. Krüppel-Associated Box (KRAB)-Associated Co-Repressor (KAP-1) Ser-473 Phosphorylation Regulates Heterochromatin Protein 1β (HP1-β) Mobilization and DNA Repair in Heterochromatin. J. Biol. Chem. 2012, 287, 28122–28131. [Google Scholar] [CrossRef]
- Chiolo, I.; Minoda, A.; Colmenares, S.U.; Polyzos, A.; Costes, S.V.; Karpen, G.H. Double-Strand Breaks in Heterochromatin Move Outside of a Dynamic HP1a Domain to Complete Recombinational Repair. Cell 2011, 144, 732–744. [Google Scholar] [CrossRef]
- Jakob, B.; Splinter, J.; Conrad, S.; Voss, K.-O.; Zink, D.; Durante, M.; Löbrich, M.; Taucher-Scholz, G. DNA Double-Strand Breaks in Heterochromatin Elicit Fast Repair Protein Recruitment, Histone H2AX Phosphorylation and Relocation to Euchromatin. Nucleic Acids Res. 2011, 39, 6489–6499. [Google Scholar] [CrossRef]
- Janssen, A.; Breuer, G.A.; Brinkman, E.K.; Van Der Meulen, A.I.; Borden, S.V.; Van Steensel, B.; Bindra, R.S.; LaRocque, J.R.; Karpen, G.H. A Single Double-Strand Break System Reveals Repair Dynamics and Mechanisms in Heterochromatin and Euchromatin. Genes Dev. 2016, 30, 1645–1657. [Google Scholar] [CrossRef]
- Tsouroula, K.; Furst, A.; Rogier, M.; Heyer, V.; Maglott-Roth, A.; Ferrand, A.; Reina-San-Martin, B.; Soutoglou, E. Temporal and Spatial Uncoupling of DNA Double Strand Break Repair Pathways within Mammalian Heterochromatin. Mol. Cell 2016, 63, 293–305. [Google Scholar] [CrossRef]
- Stephan, A.K.; Kliszczak, M.; Morrison, C.G. The Nse2/Mms21 SUMO Ligase of the Smc5/6 Complex in the Maintenance of Genome Stability. FEBS Lett. 2011, 585, 2907–2913. [Google Scholar] [CrossRef] [PubMed]
- Ryu, T.; Bonner, M.R.; Chiolo, I. Cervantes and Quijote Protect Heterochromatin from Aberrant Recombination and Lead the Way to the Nuclear Periphery. Nucleus 2016, 7, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Lukasova, E.; Gabrielova, B.; Ondrej, V.; Kozubek, S. Chromatin Dynamics during DSB Repair. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2007, 1773, 1534–1545. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Lukasova, E.; Kozubek, S. Higher-Order Chromatin Structure in DSB Induction, Repair and Misrepair. Mutat. Res. Rev. Mutat. Res. 2010, 704, 88–100. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Kurka, T.; Jeggo, P.A. KAP-1 Phosphorylation Regulates CHD3 Nucleosome Remodeling during the DNA Double-Strand Break Response. Nat. Struct. Mol. Biol. 2011, 18, 831–839. [Google Scholar] [CrossRef]
- Fortuny, A.; Chansard, A.; Caron, P.; Chevallier, O.; Leroy, O.; Renaud, O.; Polo, S.E. Imaging the Response to DNA Damage in Heterochromatin Domains Reveals Core Principles of Heterochromatin Maintenance. Nat. Commun. 2021, 12, 2428. [Google Scholar] [CrossRef]
- Fortuny, A.; Polo, S.E. The Response to DNA Damage in Heterochromatin Domains. Chromosoma 2018, 127, 291–300. [Google Scholar] [CrossRef]
- Falk, M.; Hausmann, M. A Paradigm Revolution or Just Better Resolution—Will Newly Emerging Superresolution Techniques Identify Chromatin Architecture as a Key Factor in Radiation-Induced DNA Damage and Repair Regulation? Cancers 2020, 13, 18. [Google Scholar] [CrossRef]
- Doksani, Y.; De Lange, T. Telomere-Internal Double-Strand Breaks Are Repaired by Homologous Recombination and PARP1/Lig3-Dependent End-Joining. Cell Rep. 2016, 17, 1646–1656. [Google Scholar] [CrossRef]
- Lemaître, C.; Grabarz, A.; Tsouroula, K.; Andronov, L.; Furst, A.; Pankotai, T.; Heyer, V.; Rogier, M.; Attwood, K.M.; Kessler, P.; et al. Nuclear Position Dictates DNA Repair Pathway Choice. Genes Dev. 2014, 28, 2450–2463. [Google Scholar] [CrossRef] [PubMed]
- Schep, R.; Brinkman, E.K.; Leemans, C.; Vergara, X.; Van Der Weide, R.H.; Morris, B.; Van Schaik, T.; Manzo, S.G.; Peric-Hupkes, D.; Van Den Berg, J.; et al. Impact of Chromatin Context on Cas9-Induced DNA Double-Strand Break Repair Pathway Balance. Mol. Cell 2021, 81, 2216–2230.e10. [Google Scholar] [CrossRef] [PubMed]
- Torres-Rosell, J.; Sunjevaric, I.; De Piccoli, G.; Sacher, M.; Eckert-Boulet, N.; Reid, R.; Jentsch, S.; Rothstein, R.; Aragón, L.; Lisby, M. The Smc5–Smc6 Complex and SUMO Modification of Rad52 Regulates Recombinational Repair at the Ribosomal Gene Locus. Nat. Cell Biol. 2007, 9, 923–931. [Google Scholar] [CrossRef]
- Van Sluis, M.; McStay, B. A Localized Nucleolar DNA Damage Response Facilitates Recruitment of the Homology-Directed Repair Machinery Independent of Cell Cycle Stage. Genes Dev. 2015, 29, 1151–1163. [Google Scholar] [CrossRef]
- Harding, S.M.; Boiarsky, J.A.; Greenberg, R.A. ATM Dependent Silencing Links Nucleolar Chromatin Reorganization to DNA Damage Recognition. Cell Rep. 2015, 13, 251–259. [Google Scholar] [CrossRef]
- Chen, Z.; Tyler, J.K. The Chromatin Landscape Channels DNA Double-Strand Breaks to Distinct Repair Pathways. Front. Cell Dev. Biol. 2022, 10, 909696. [Google Scholar] [CrossRef]
- Aymard, F.; Bugler, B.; Schmidt, C.K.; Guillou, E.; Caron, P.; Briois, S.; Iacovoni, J.S.; Daburon, V.; Miller, K.M.; Jackson, S.P.; et al. Transcriptionally Active Chromatin Recruits Homologous Recombination at DNA Double-Strand Breaks. Nat. Struct. Mol. Biol. 2014, 21, 366–374. [Google Scholar] [CrossRef]
- Aymard, F.; Aguirrebengoa, M.; Guillou, E.; Javierre, B.M.; Bugler, B.; Arnould, C.; Rocher, V.; Iacovoni, J.S.; Biernacka, A.; Skrzypczak, M.; et al. Genome-Wide Mapping of Long-Range Contacts Unveils Clustering of DNA Double-Strand Breaks at Damaged Active Genes. Nat. Struct. Mol. Biol. 2017, 24, 353–361. [Google Scholar] [CrossRef]
- Schrank, B.R.; Aparicio, T.; Li, Y.; Chang, W.; Chait, B.T.; Gundersen, G.G.; Gottesman, M.E.; Gautier, J. Nuclear ARP2/3 Drives DNA Break Clustering for Homology-Directed Repair. Nature 2018, 559, 61–66. [Google Scholar] [CrossRef]
- Hagiwara, Y.; Niimi, A.; Isono, M.; Yamauchi, M.; Yasuhara, T.; Limsirichaikul, S.; Oike, T.; Sato, H.; Held, K.D.; Nakano, T.; et al. 3D-Structured Illumination Microscopy Reveals Clustered DNA Double-Strand Break Formation in Widespread γH2AX Foci after High LET Heavy-Ion Particle Radiation. Oncotarget 2017, 8, 109370–109381. [Google Scholar] [CrossRef]
- Arnould, C.; Rocher, V.; Saur, F.; Bader, A.S.; Muzzopappa, F.; Collins, S.; Lesage, E.; Le Bozec, B.; Puget, N.; Clouaire, T.; et al. Chromatin Compartmentalization Regulates the Response to DNA Damage. Nature 2023, 623, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Zagelbaum, J.; Schooley, A.; Zhao, J.; Schrank, B.R.; Callen, E.; Zha, S.; Gottesman, M.E.; Nussenzweig, A.; Rabadan, R.; Dekker, J.; et al. Multiscale Reorganization of the Genome Following DNA Damage Facilitates Chromosome Translocations via Nuclear Actin Polymerization. Nat. Struct. Mol. Biol. 2023, 30, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.W. The Functional Importance of Lamins, Actin, Myosin, Spectrin and the LINC Complex in DNA Repair. Exp. Biol. Med. 2019, 244, 1382–1406. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, K.S.; Tapley, E.C.; Cruz, V.E.; Li, Q.; Aung, K.; Hart, K.C.; Schwartz, T.U.; Starr, D.A.; Engebrecht, J. LINC Complexes Promote Homologous Recombination in Part through Inhibition of Nonhomologous End Joining. J. Cell Biol. 2016, 215, 801–821. [Google Scholar] [CrossRef]
- Camps, J.; Erdos, M.R.; Ried, T. The Role of Lamin B1 for the Maintenance of Nuclear Structure and Function. Nucleus 2015, 6, 8–14. [Google Scholar] [CrossRef]
- Dubik, N.; Mai, S. Lamin A/C: Function in Normal and Tumor Cells. Cancers 2020, 12, 3688. [Google Scholar] [CrossRef]
- Lamm, N.; Rogers, S.; Cesare, A.J. Chromatin Mobility and Relocation in DNA Repair. Trends Cell Biol. 2021, 31, 843–855. [Google Scholar] [CrossRef]
- De Luca, K.L.; Rullens, P.M.J.; Karpinska, M.A.; De Vries, S.S.; Gacek-Matthews, A.; Pongor, L.S.; Legube, G.; Jachowicz, J.W.; Oudelaar, A.M.; Kind, J. Genome-Wide Profiling of DNA Repair Proteins in Single Cells. Nat. Commun. 2024, 15, 9918. [Google Scholar] [CrossRef]
- Hausmann, M.; Neitzel, C.; Bobkova, E.; Nagel, D.; Hofmann, A.; Chramko, T.; Smirnova, E.; Kopečná, O.; Pagáčová, E.; Boreyko, A.; et al. Single Molecule Localization Microscopy Analyses of DNA-Repair Foci and Clusters Detected Along Particle Damage Tracks. Front. Phys. 2020, 8, 578662. [Google Scholar] [CrossRef]
- Hayashi, K.; Horisaka, K.; Harada, Y.; Ogawa, Y.; Yamashita, T.; Kitano, T.; Wakita, M.; Fukusumi, T.; Inohara, H.; Hara, E.; et al. Polyploidy Mitigates the Impact of DNA Damage While Simultaneously Bearing Its Burden. Cell Death Discov. 2024, 10, 436. [Google Scholar] [CrossRef]
- Anatskaya, O.V.; Vinogradov, A.E. Polyploidy as a Fundamental Phenomenon in Evolution, Development, Adaptation and Diseases. Int. J. Mol. Sci. 2022, 23, 3542. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Liu, X.; Pauklin, S. 3D Chromatin Architecture and Epigenetic Regulation in Cancer Stem Cells. Protein Cell 2021, 12, 440–454. [Google Scholar] [CrossRef] [PubMed]
- Dozmorov, M.G.; Marshall, M.A.; Rashid, N.S.; Grible, J.M.; Valentine, A.; Olex, A.L.; Murthy, K.; Chakraborty, A.; Reyna, J.; Figueroa, D.S.; et al. Rewiring of the 3D Genome during Acquisition of Carboplatin Resistance in a Triple-Negative Breast Cancer Patient-Derived Xenograft. Sci. Rep. 2023, 13, 5420. [Google Scholar] [CrossRef]
- Drew, Y.; Zenke, F.T.; Curtin, N.J. DNA Damage Response Inhibitors in Cancer Therapy: Lessons from the Past, Current Status and Future Implications. Nat. Rev. Drug Discov. 2025, 24, 19–39. [Google Scholar] [CrossRef]
- Lazo, P.A. Targeting Histone Epigenetic Modifications and DNA Damage Responses in Synthetic Lethality Strategies in Cancer? Cancers 2022, 14, 4050. [Google Scholar] [CrossRef]
- Kantidze, O.L.; Luzhin, A.V.; Nizovtseva, E.V.; Safina, A.; Valieva, M.E.; Golov, A.K.; Velichko, A.K.; Lyubitelev, A.V.; Feofanov, A.V.; Gurova, K.V.; et al. The Anti-Cancer Drugs Curaxins Target Spatial Genome Organization. Nat. Commun. 2019, 10, 1441. [Google Scholar] [CrossRef]
- Achinger-Kawecka, J.; Stirzaker, C.; Portman, N.; Campbell, E.; Chia, K.-M.; Du, Q.; Laven-Law, G.; Nair, S.S.; Yong, A.; Wilkinson, A.; et al. The Potential of Epigenetic Therapy to Target the 3D Epigenome in Endocrine-Resistant Breast Cancer. Nat. Struct. Mol. Biol. 2024, 31, 498–512. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ambrosio, S.; Noviello, A.; Di Fusco, G.; Gorini, F.; Piscone, A.; Amente, S.; Majello, B. Interplay and Dynamics of Chromatin Architecture and DNA Damage Response: An Overview. Cancers 2025, 17, 949. https://doi.org/10.3390/cancers17060949
Ambrosio S, Noviello A, Di Fusco G, Gorini F, Piscone A, Amente S, Majello B. Interplay and Dynamics of Chromatin Architecture and DNA Damage Response: An Overview. Cancers. 2025; 17(6):949. https://doi.org/10.3390/cancers17060949
Chicago/Turabian StyleAmbrosio, Susanna, Anna Noviello, Giovanni Di Fusco, Francesca Gorini, Anna Piscone, Stefano Amente, and Barbara Majello. 2025. "Interplay and Dynamics of Chromatin Architecture and DNA Damage Response: An Overview" Cancers 17, no. 6: 949. https://doi.org/10.3390/cancers17060949
APA StyleAmbrosio, S., Noviello, A., Di Fusco, G., Gorini, F., Piscone, A., Amente, S., & Majello, B. (2025). Interplay and Dynamics of Chromatin Architecture and DNA Damage Response: An Overview. Cancers, 17(6), 949. https://doi.org/10.3390/cancers17060949