
Opportunities and Challenges in Antibody–Drug Conjugates for Cancer Therapy: A New Era for Cancer Treatment


Simple Summary
Abstract
1. Introduction
2. The Structure of an ADC
2.1. The Antibody Moiety and Target Antigen
2.2. The Chemical Linker
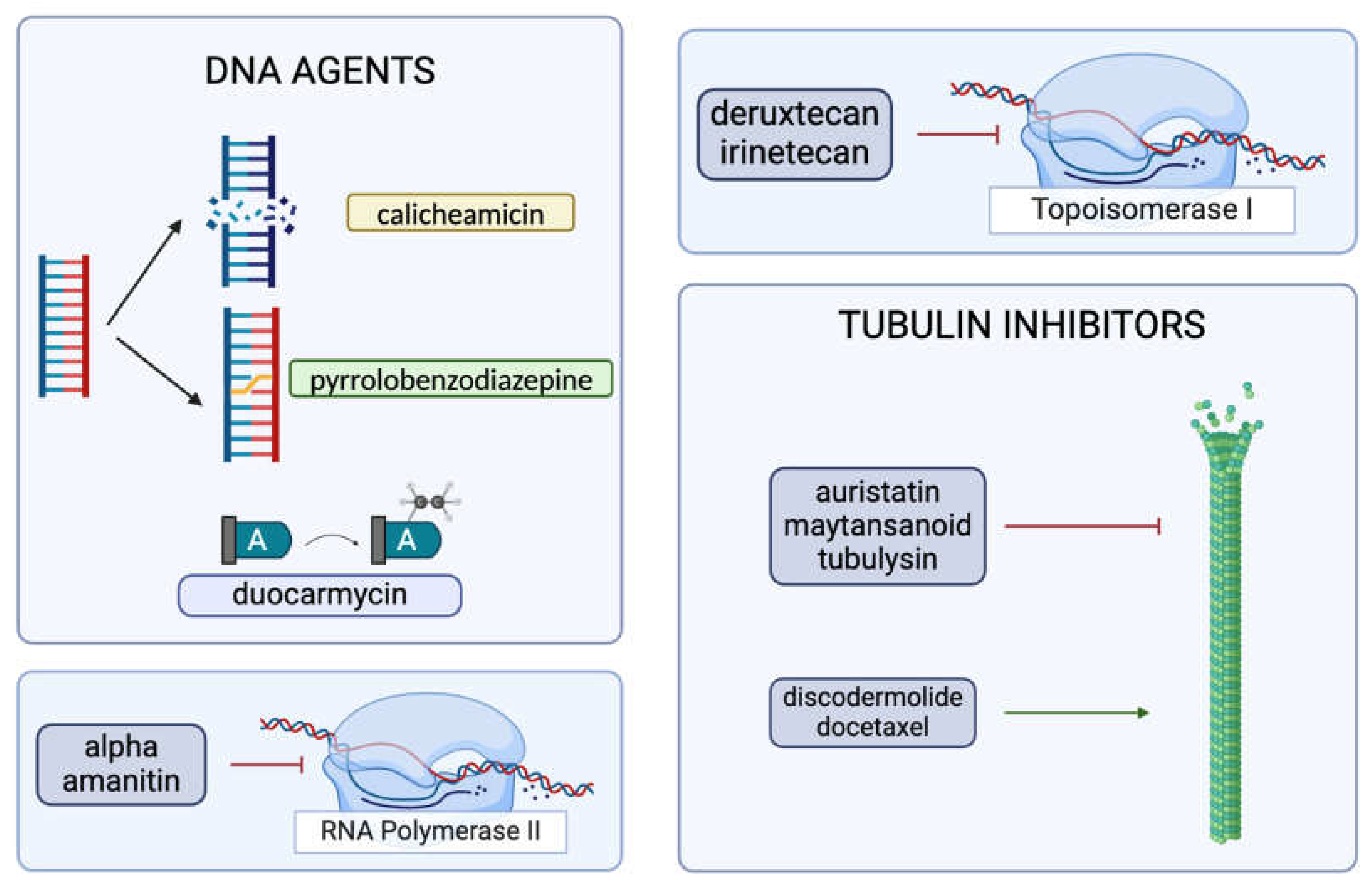
2.3. The Cytotoxic Payload
3. Mechanisms of Action of ADCs
4. Limitations of an ADC
4.1. Toxicities
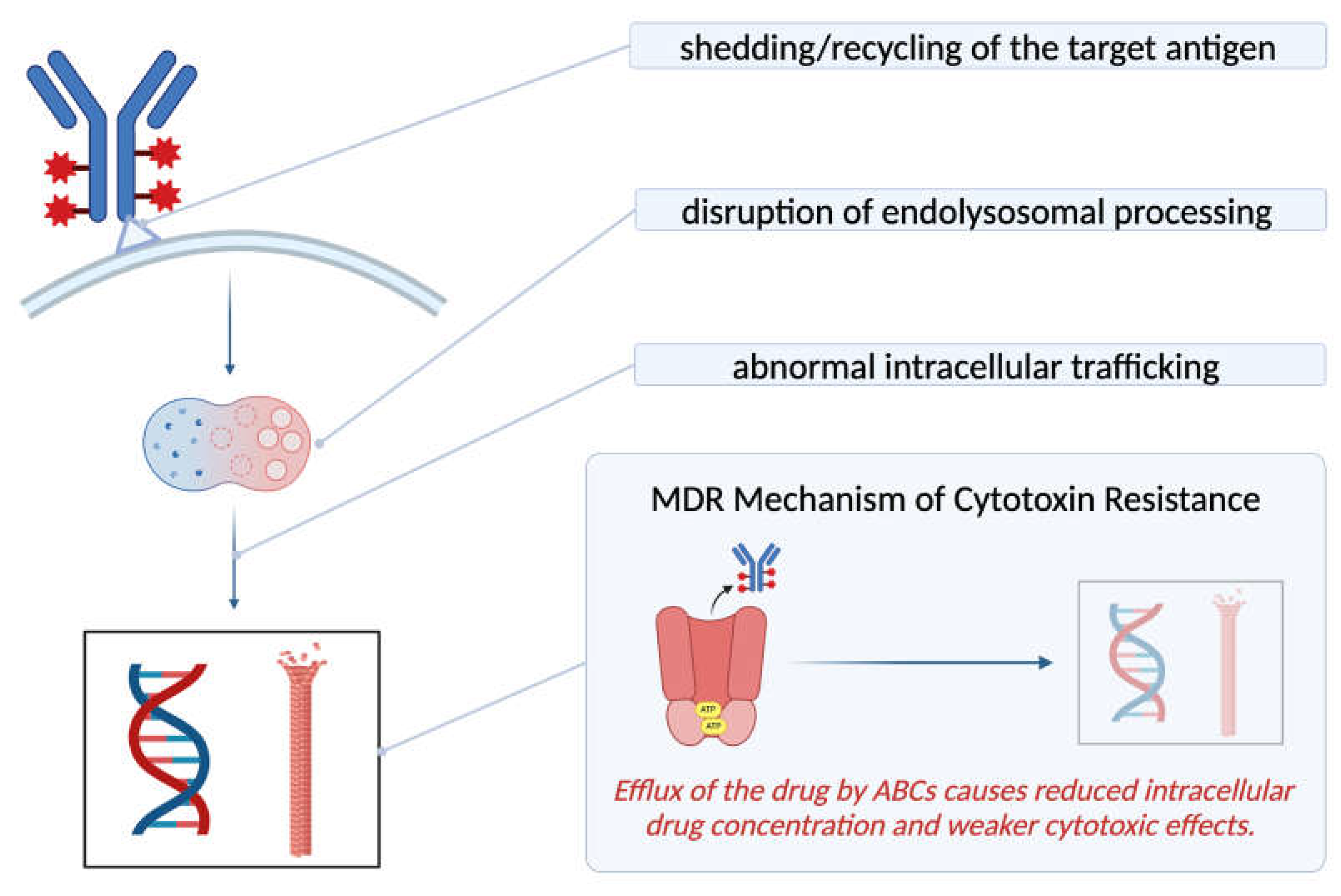
4.2. Mechanism of Resistance
5. Discussion and Future Perspective
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| TLA | Three-letter acronym |
| LD | Linear dichroism |
References
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27, 87–97. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, E.F. The Toxins of William B. Coley and the Treatment of Bone and Soft-Tissue Sarcomas. Iowa Orthop. J. 2006, 26, 154–158. [Google Scholar] [PubMed]
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 Years of Progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef] [PubMed]
- The Nobel Prize in Physiology or Medicine 1908 NobelPrize.org. Available online: https://www.nobelprize.org/prizes/medicine/1908/ehrlich/biographical/ (accessed on 10 February 2024).
- Shefet-Carasso, L.; Benhar, I. Antibody-targeted drugs and drug resistance—Challenges and solutions. Drug Resist. Updates 2015, 18, 36–46. [Google Scholar] [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef]
- Baah, S.; Laws, M.; Rahman, K.M. Antibody–drug conjugates—A tutorial review. Molecules 2021, 26, 2943. [Google Scholar] [CrossRef]
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef]
- Maecker, H.; Jonnalagadda, V.; Bhakta, S.; Jammalamadaka, V.; Junutula, J.R. Exploration of the antibody–drug conjugate clinical landscape. mAbs 2023, 15, 2229101. [Google Scholar] [CrossRef]
- Karpel, H.C.; Stonefeld Powell, S.; Pothuri, B. Antibody-drug conjugates in gynecologic cancer. Am. Soc. Clin. Oncol. Educ. Book 2023, 43, e390772. [Google Scholar] [CrossRef]
- Yoshino, T.; Di Bartolomeo, M.; Raghav, K.; Masuishi, T.; Loupakis, F.; Kawakami, H.; Yamaguchi, K.; Nishina, T.; Wainberg, Z.; Elez, E.; et al. Final results of destiny-CRC01 investigating trastuzumab deruxtecan in patients with HER2-expressing metastatic colorectal cancer. Nat. Commun. 2023, 14, 3332. [Google Scholar] [CrossRef]
- Birrer, M.J.; Moore, K.N.; Betella, I.; Bates, R.C. Antibody-Drug Conjugate-Based Therapeutics: State of the Science. JNCI J. Natl. Cancer Inst. 2019, 111, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Brezski, R.J.; Georgiou, G. Immunoglobulin isotype knowledge and application to Fc engineering. Curr. Opin. Immunol. 2016, 40, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.H.; Jung, S.T. Reprogramming the Constant Region of Immunoglobulin G Subclasses for Enhanced Therapeutic Potency against Cancer. Biomolecules 2020, 10, 382. [Google Scholar] [CrossRef] [PubMed]
- Natsume, A. Improving effector functions of antibodies for cancer treatment: Enhancing ADCC and CDC. Drug Des. Devel. Ther. 2008, 3, 7–16. [Google Scholar] [CrossRef]
- Singh, D.; Dheer, D.; Samykutty, A.; Shankar, R. Antibody drug conjugates in gastrointestinal cancer: From lab to clinical development. J. Control Release 2021, 340, 1–34. [Google Scholar] [CrossRef]
- Riccardi, F.; Dal Bo, M.; Macor, P.; Toffoli, G. A comprehensive overview on antibody-drug conjugates: From the conceptualization to cancer therapy. Front. Pharmacol. 2023, 14, 1274088. [Google Scholar] [CrossRef]
- Zhang, J.; Woods, C.; He, F.; Han, M.; Treuheit, M.J.; Volkin, D.B. Structural Changes and Aggregation Mechanisms of Two Different Dimers of an IGG2 Monoclonal Antibody. Biochemistry 2018, 57, 5466–5479. [Google Scholar] [CrossRef]
- Stapleton, N.M.; Andersen, J.T.; Stemerding, A.M.; Bjarnarson, S.P.; Verheul, R.C.; Gerritsen, J.; Zhao, Y.; Kleijer, M.; Sandlie, I.; de Haas, M.; et al. Competition for FcRn-mediated transport gives rise to short half-life of human IGG3 and offers therapeutic potential. Nat. Commun. 2011, 2, 599. [Google Scholar] [CrossRef]
- Teicher, B.A.; Morris, J. Antibody-drug Conjugate Targets, Drugs, and Linkers. Curr. Cancer Drug Targets 2022, 22, 463–529. [Google Scholar] [CrossRef]
- Samantasinghar, A.; Sunildutt, N.P.; Ahmed, F.; Soomro, A.M.; Salih, A.R.; Parihar, P.; Memon, F.H.; Kim, K.H.; Kang, I.S.; Choi, K.H. A comprehensive review of key factors affecting the efficacy of antibody drug conjugate. Biomed. Pharmacother. 2023, 161, 114408. [Google Scholar] [CrossRef]
- Xiao, Y.; Yu, D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol. Ther. 2021, 221, 107753. [Google Scholar] [CrossRef] [PubMed]
- Visintin, A.; Knowlton, K.; Tyminski, E.; Lin, C.I.; Zheng, X.; Marquette, K.; Jain, S.; Tchistiakova, L.; Li, D.; O’Donnell, C.J.; et al. Novel Anti-TM4SF1 Antibody–Drug Conjugates with Activity against Tumor Cells and Tumor Vasculature. Mol. Cancer Ther. 2015, 14, 1868–1876. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Reichert, J.M.; Senter, P.D.; Lambert, J.M.; Beck, A. Antibody–drug conjugates come of age in oncology. Nat. Rev. Drug Discov. 2023, 22, 641–661. [Google Scholar] [CrossRef] [PubMed]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2016, 9, 33–46. [Google Scholar] [CrossRef]
- Pettinato, M.C. Introduction to Antibody-Drug Conjugates. Antibodies 2021, 10, 42. [Google Scholar] [CrossRef]
- Sheyi, R.; de la Torre, B.G.; Albericio, F. Linkers: An Assurance for Controlled Delivery of Antibody-Drug Conjugate. Pharmaceutics 2022, 14, 396. [Google Scholar] [CrossRef]
- Kostova, V.; Désos, P.; Starck, J.B.; Kotschy, A. The Chemistry Behind ADCs. Pharmaceuticals 2021, 14, 442. [Google Scholar] [CrossRef]
- Gamcsik, M.P.; Kasibhatla, M.S.; Teeter, S.D.; Colvin, O.M. Glutathione levels in human tumors. Biomarkers 2012, 17, 671–691. [Google Scholar] [CrossRef]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.J.; Lutz, R.J.; et al. Targeting HER2-Positive Breast Cancer with Trastuzumab-DM1, an Antibody–Cytotoxic Drug Conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef]
- Dorywalska, M.; Strop, P.; Melton-Witt, J.A.; Hasa-Moreno, A.; Farias, S.E.; Galindo Casas, M.; Delaria, K.; Lui, V.; Poulsen, K.; Sutton, J.; et al. Site-Dependent Degradation of a Non-Cleavable Auristatin-Based Linker-Payload in Rodent Plasma and Its Effect on ADC Efficacy. PLoS ONE 2015, 10, e0132282. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Chari, R.V.J. Antibody Conjugate Therapeutics: Challenges and Potential. Clin. Cancer Res. 2011, 17, 6389–6397. [Google Scholar] [CrossRef] [PubMed]
- Wiedemeyer, W.R.; Gavrilyuk, J.; Schammel, A.; Zhao, X.; Sarvaiya, H.; Pysz, M.; Gu, C.; You, M.; Isse, K.; Sullivan, T.; et al. ABBV-011, A Novel, Calicheamicin-Based Antibody–Drug Conjugate, Targets SEZ6 to Eradicate Small Cell Lung Cancer Tumors. Mol. Cancer Ther. 2022, 21, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Könning, D.; Rasche, N.; Hart, F.; Sensbach, J.; Krug, C.; Raab-Westphal, S.; Richter, K.; Unverzagt, C.; Hecht, S.; et al. Generation and Characterization of Iduronidase-Cleavable ADCs. Bioconjugate Chem. 2023, 34, 2221–2233. [Google Scholar] [CrossRef]
- Sakata, J.; Tatsumi, T.; Sugiyama, A.; Shimizu, A.; Inagaki, Y.; Katoh, H.; Yamashita, T.; Takahashi, K.; Aki, S.; Kaneko, Y.; et al. Antibody-mimetic drug conjugate with efficient internalization activity using anti-HER2 VHH and duocarmycin. Protein Expr. Purif. 2024, 214, 106375. [Google Scholar] [CrossRef]
- Brignole, C.; Calarco, E.; Bensa, V.; Giusto, E.; Perri, P.; Ciampi, E.; Corrias, M.V.; Astigiano, S.; Cilli, M.; Loo, D.; et al. Antitumor activity of the investigational B7-H3 antibody-drug conjugate, vobramitamab duocarmazine, in preclinical models of neuroblastoma. J. Immunother. Cancer 2023, 11, e007174. [Google Scholar] [CrossRef]
- Zammarchi, F.; Havenith, K.E.G.; Chivers, S.; Hogg, P.; Bertelli, F.; Tyrer, P.; Janghra, N.; Reinert, H.W.; Hartley, J.A.; van Berkel, P.H. Preclinical Development of ADCT-601, a Novel Pyrrolobenzodiazepine Dimer-Based Antibody–Drug Conjugate Targeting AXL-Expressing Cancers. Mol. Cancer Ther. 2022, 21, 582–593. [Google Scholar] [CrossRef]
- Lewis, T.; Corcoran, D.B.; Thurston, D.E.; Giles, P.J.; Ashelford, K.; Walsby, E.J.; Fegan, C.D.; Pepper, A.G.S.; Rahman, K.M.; Pepper, C. Novel pyrrolobenzodiazepine benzofused hybrid molecules inhibit NF-ΚB activity and synergise with bortezomib and ibrutinib in hematological cancers. Haematologica 2020, 106, 958–967. [Google Scholar] [CrossRef]
- Wang, Z.; Li, H.; Gou, L.; Li, W.; Wang, Y. Antibody–drug conjugates: Recent advances in payloads. Acta Pharm. Sin. B 2023, 13, 4025–4059. [Google Scholar] [CrossRef]
- Chen, H.; Lin, Z.; Arnst, K.E.; Miller, D.D.; Li, W. Tubulin Inhibitor-Based Antibody-Drug Conjugates for Cancer Therapy. Molecules 2017, 22, 1281. [Google Scholar] [CrossRef]
- Staben, L.R.; Yu, S.F.; Chen, J.; Yan, G.; Xu, Z.; Del Rosario, G.; Lau, J.T.; Liu, L.; Guo, J.; Zheng, B.; et al. Stabilizing a Tubulysin Antibody–Drug conjugate to Enable Activity Against Multidrug-Resistant Tumors. ACS Med. Chem. Lett. 2017, 8, 1037–1041. [Google Scholar] [CrossRef] [PubMed]
- Tumey, L.N.; Leverett, C.A.; Vetelino, B.; Li, F.; Rago, B.; Han, X.; Loganzo, F.; Musto, S.; Bai, G.; Sukuru, S.C.; et al. Optimization of Tubulysin Antibody–Drug Conjugates: A Case Study in Addressing ADC Metabolism. ACS Med. Chem. Lett. 2016, 7, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Seki, H. The Development of Vinylheteroarene Linkers for Proteinogenic Cysteine Modification and Studies Towards Applying (+)-Discodermolide as a Novel Payload in Antibody-Drug Conjugates. Ph.D. Thesis, University of Cambridge, Cambridge, UK, 2021. [Google Scholar]
- Glatt, D.M.; Beckford Vera, D.R.; Prabhu, S.S.; Mumper, R.J.; Luft, J.C.; Benhabbour, S.R.; Parrott, M.C. Synthesis and Characterization of Cetuximab–Docetaxel and Panitumumab–Docetaxel Antibody–Drug Conjugates for EGFR-Overexpressing Cancer Therapy. Mol. Pharm. 2018, 15, 5089–5102. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.M.; Eskander, R.N.; Binder, P.S. Recent Therapeutic Advances in Gynecologic Oncology: A Review. Cancers 2024, 16, 770. [Google Scholar] [CrossRef]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Hegg, R.; Chung, W.P.; Im, S.A.; Jacot, W.; Ganju, V.; Chiu, J.W.; Xu, B.; Hamilton, E.; Madhusudan, S.; et al. Trastuzumab deruxtecan versus trastuzumab emtansine in patients with HER2-positive metastatic breast cancer: Updated results from Destiny-Breast03, a randomised, open-label, phase 3 trial. Lancet 2023, 401, 105–117. [Google Scholar] [CrossRef]
- Yamane, H.; Sugiyama, Y.; Komo, T.; Shibata, K.; Tazaki, T.; Koyama, M.; Sasaki, M. Long-Term Complete Response to Trastuzumab Deruxtecan in a Case of Unresectable Gastric Cancer. Case Rep. Oncol. 2024, 17, 463–470. [Google Scholar] [CrossRef]
- Bardia, A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Rugo, H.S.; Brufsky, A.; Kalinsky, K.; Cortés, J.; O’Shaughnessy, J.; et al. LBA17 ASCENT: A randomized phase III study of sacituzumab govitecan (SG) vs treatment of Physician’s Choice (TPC) in patients (PTS) with previously treated metastatic triple-negative breast cancer (mTNBC). Ann. Oncol. 2020, 31, S1149–S1150. [Google Scholar] [CrossRef]
- Gallo, F.; Korsak, B.; Müller, C.; Hechler, T.; Yanakieva, D.; Avrutina, O.; Kolmar, H.; Pahl, A. Enhancing the pharmacokinetics and antitumor activity of an α-amanitin-based small-molecule drug conjugate via conjugation with an FC domain. J. Med. Chem. 2021, 64, 4117–4129. [Google Scholar] [CrossRef]
- Ritchie, M.; Tchistiakova, L.; Scott, N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. mAbs 2013, 5, 13–21. [Google Scholar] [CrossRef]
- Hammood, M.; Craig, A.W.; Leyton, J.V. Impact of Endocytosis Mechanisms for the Receptors Targeted by the Currently Approved Antibody-Drug Conjugates (ADCs)—A Necessity for Future ADC Research and Development. Pharmaceuticals 2021, 14, 674. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zhou, D.; Li, W.; Duan, Y.; Xu, M.; Liu, J.; Cheng, J.; Xiao, Y.; Xiao, H.; Gan, T.; et al. Therapeutic efficacy of a MMAE-based anti-DR5 drug conjugate OBA01 in preclinical models of pancreatic cancer. Cell Death Dis. 2023, 14, 295. [Google Scholar] [CrossRef] [PubMed]
- Bergman, I.; Basse, P.H.; Barmada, M.A.; Griffin, J.A.; Cheung, N.K.V. Comparison of in vitro antibody-targeted cytotoxicity using mouse, rat and human effectors. Cancer Immunol. Immunother. 2000, 49, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Sun, L.; Huang, L.; Chen, Y. Nanodrug Delivery Systems Modulate Tumor Vessels to Increase the Enhanced Permeability and Retention Effect. J. Pers. Med. 2021, 11, 124. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ting, K.K.; Coleman, P.; Qi, Y.; Chen, J.; Vadas, M.; Gamble, J. The Tumour Vasculature as a Target to Modulate Leucocyte Trafficking. Cancers 2021, 13, 1724. [Google Scholar] [CrossRef]
- Jin, Y.; Schladetsch, M.A.; Huang, X.; Balunas, M.J.; Wiemer, A.J. Stepping forward in antibody-drug conjugate development. Pharmacol. Ther. 2022, 229, 107917. [Google Scholar] [CrossRef]
- Bordeau, B.M.; Yang, Y.; Balthasar, J.P. Transient Competitive Inhibition Bypasses the Binding Site Barrier to Improve Tumor Penetration of Trastuzumab and Enhance T-DM1 Efficacy. Cancer Res. 2021, 81, 4145–4154. [Google Scholar] [CrossRef]
- Singh, A.P.; Guo, L.; Verma, A.; Wong, G.G.L.; Thurber, G.M.; Shah, D.K. Antibody Coadministration as a Strategy to Overcome Binding-Site Barrier for ADCs: A Quantitative Investigation. AAPS J. 2020, 22, 28. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Bordeau, B.M.; Balthasar, J.P. Mechanisms of ADC Toxicity and Strategies to Increase ADC Tolerability. Cancers 2023, 15, 713. [Google Scholar] [CrossRef]
- Raghav, K.P.; Siena, S.; Takashima, A.; Kato, T.; Van Den Eynde, M.; Di Bartolomeo, M.; Komatsu, Y.; Kawakami, H.; Peeters, M.; Andre, T.; et al. Trastuzumab deruxtecan (T-DXD) in patients (PTS) with HER2-overexpressing/amplified (her2+) metastatic colorectal cancer (mcrc): Primary results from the Multicenter, randomized, phase 2 Destiny-CRC02 study. J. Clin. Oncol. 2023, 41, 3501. [Google Scholar] [CrossRef]
- Chen, Y.F.; Xu, Y.Y.; Shao, Z.M.; Yu, K.D. Resistance to antibody-drug conjugates in breast cancer: Mechanisms and solutions. Cancer Commun. 2022, 43, 297–337. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, F.; Zhang, T.; Yu, M.; Sun, Y. Recent advances in anti-multidrug resistance for nano-drug delivery system. Drug Deliv. 2022, 29, 1684–1697. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, C.M.; Yamaguchi, A.; Anami, Y.; Xiong, W.; Otani, Y.; Lee, J.; Ueno, N.T.; Zhang, N.; An, Z.; Tsuchikama, K. Antibody-drug conjugates with dual payloads for combating breast tumor heterogeneity and drug resistance. Nat. Commun. 2021, 12, 3528. [Google Scholar] [CrossRef] [PubMed]
- Healey, G.D.; Pan-Castillo, B.; Garcia-Parra, J.; Davies, J.; Roberts, S.; Jones, E.; Dhar, K.; Nandanan, S.; Tofazzal, N.; Piggott, L.; et al. Antibody drug conjugates against the receptor for advanced glycation end products (RAGE), a novel therapeutic target in endometrial cancer. J. Immunother. Cancer 2019, 7, 280. [Google Scholar] [CrossRef]
- Guo, P.; Huang, J.; Zhu, B.; Huang, A.C.; Jiang, L.; Fang, J.; Moses, M.A. A rationally designed ICAM1 antibody drug conjugate eradicates late-stage and refractory triple-negative breast tumors in vivo. Sci. Adv. 2023, 9, eabq7866. [Google Scholar] [CrossRef]
- Verma, S.; Breadner, D.; Raphael, J. ‘Targeting’ Improved Outcomes with Antibody-Drug Conjugates in Non-Small Cell Lung Cancer—An Updated Review. Curr. Oncol. 2023, 30, 4329–4350. [Google Scholar] [CrossRef]
- Sasso, J.M.; Tenchov, R.; Bird, R.; Iyer, K.A.; Ralhan, K.; Rodriguez, Y.; Zhou, Q.A. The Evolving Landscape of Antibody–Drug Conjugates: In Depth Analysis of Recent Research Progress. Bioconjug. Chem. 2023, 34, 1951–2000. [Google Scholar] [CrossRef]
- Schoenfeld, K.; Harwardt, J.; Habermann, J.; Elter, A.; Kolmar, H. Conditional activation of an anti-IgM antibody-drug conjugate for precise B cell lymphoma targeting. Front. Immunol. 2023, 14, 1258700. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Bordeau, B.M.; Balthasar, J.P. Use of Payload Binding Selectivity Enhancers to Improve Therapeutic Index of Maytansinoid–Antibody–Drug Conjugates. Mol. Cancer Ther. 2023, 22, 1332–1342. [Google Scholar] [CrossRef]
- Bordeau, B.M.; Nguyen, T.D.; Polli, J.R.; Chen, P.; Balthasar, J.P. Payload-Binding Fab Fragments Increase the Therapeutic Index of MMAE Antibody–Drug Conjugates. Mol. Cancer Ther. 2023, 22, 459–470. [Google Scholar] [CrossRef]
- Cilliers, C.; Menezes, B.; Nessler, I.; Linderman, J.; Thurber, G.M. Improved Tumor Penetration and Single-Cell Targeting of Antibody–Drug Conjugates Increases Anticancer Efficacy and Host Survival. Cancer Res. 2018, 78, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Menezes, B.; Khera, E.; Calopiz, M.; Smith, M.D.; Ganno, M.L.; Cilliers, C.; Abu-Yousif, A.O.; Linderman, J.J.; Thurber, G.M. Pharmacokinetics and Pharmacodynamics of TAK-164 Antibody Drug Conjugate Coadministered with Unconjugated Antibody. AAPS J. 2022, 24, 107. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Li, P.; Yang, T.; Zhu, J.; Sun, L.; Zhang, Z.; Wang, L.; Tian, X.; Chen, J.; Hu, C.; et al. The promise and challenges of combination therapies with antibody-drug conjugates in solid tumors. J. Hematol. Oncol. 2024, 17, 1. [Google Scholar] [CrossRef] [PubMed]
- Lankoff, A.; Czerwińska, M.; Kruszewski, M. Nanoparticle-Based Radioconjugates for Targeted Imaging and Therapy of Prostate Cancer. Molecules 2023, 28, 4122. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, R.; Pinheiro, D.P.; de Cássia Evangelista de Oliveira, F.; Galvão, G.F.; Marques, L.G.; Lopez, R.F.; Pessoa, C.; Eloy, J.O. Immunoconjugates for Cancer Targeting: A Review of Antibody-Drug Conjugates and Antibody-Functionalized Nanoparticles. Curr. Med. Chem. 2021, 28, 2485–2520. [Google Scholar] [CrossRef]
- Bruins, W.S.; Zweegman, S.; Mutis, T.; van de Donk, N.W. Targeted Therapy With Immunoconjugates for Multiple Myeloma. Front. Immunol. 2020, 11, 1155. [Google Scholar] [CrossRef]
- Dragovich, P.S. Degrader-antibody conjugates. Chem. Soc. Rev. 2022, 51, 3886–3897. [Google Scholar] [CrossRef]
- Jin, M.Z.; Jin, W.L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef]
- Mo, R.; Gu, Z. Tumor microenvironment and intracellular signal-activated nanomaterials for anticancer drug delivery. Mater. Today 2016, 19, 274–283. [Google Scholar] [CrossRef]
- Sahin, I.H.; Goyal, S.; Pumpalova, Y.; Sonbol, M.B.; Das, S.; Haraldsdottir, S.; Ahn, D.; Ciombor, K.K.; Chen, Z.; Draper, A.; et al. Mismatch Repair (MMR) gene alteration and Braf V600E mutation are potential predictive biomarkers of immune checkpoint inhibitors in MMR-deficient colorectal cancer. Oncologist 2021, 26, 668–675. [Google Scholar] [CrossRef]
- Ciner, A.T.; Jones, K.; Muschel, R.J.; Brodt, P. The unique immune microenvironment of liver metastases: Challenges and opportunities. Semin. Cancer Biol. 2021, 71, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Stern, O.; Vallis, Y.; Dhillon, J.; Buchanan, A.; McMahon, H. Cell surface protein aggregation triggers endocytosis to maintain plasma membrane proteostasis. Nat. Commun. 2023, 14, 947. [Google Scholar] [CrossRef] [PubMed]
- Pozniak, M.; Sokolowska-Wedzina, A.; Jastrzebski, K.; Szymczyk, J.; Porebska, N.; Krzyscik, M.A.; Zakrzewska, M.; Miaczynska, M.; Otlewski, J.; Opalinski, L. FGFR1 clustering with engineered tetravalent antibody improves the efficiency and modifies the mechanism of receptor internalization. Mol. Oncol. 2020, 14, 1998–2021. [Google Scholar] [CrossRef] [PubMed]
- Porębska, N.; Ciura, K.; Chorążewska, A.; Zakrzewska, M.; Otlewski, J.; Opaliński, Ł. Multivalent protein-drug conjugates—An emerging strategy for the upgraded precision and efficiency of drug delivery to cancer cells. Biotechnol. Adv. 2023, 67, 108213. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ADC | Commercial Name | Warhead + [Linker Type] | Target | Status | Indication |
|---|---|---|---|---|---|
| Gemtuzumab ozogamicin | Mylotarg® | Calicheamicin [cleavable] | CD33 | Reapproved in 2017; initially in 2000 | CD33+ AML |
| Brentuximab vedotin | Adcetris® | MMAE [cleavable] | CD30 | Approved in 2011 | R/R CD30+ HL and systemic ALCL |
| Inotuzumab ozogamicin | Besponsa® | Calicheamicin [cleavable] | CD22 | Approved in 2011 | R/R B-cell precursor ALL |
| Moxetumomab pasudotox | Lumoxiti® | PE38 (immunotoxin) [cleavable] | CD22 | Approved in 2018 | R/R HCL |
| Polatuzumab vedotin | Polivy® | MMAE [cleavable] | CD79B | Approved in 2019 | R/R DLBCL |
| Belantamab mafodotin | Blenrep® | MMAF [non-cleavable] | BCMA | Approved in 2020 | R/R MM |
| Loncastuximab tesirine | Zynlonta® | PBD [cleavable] | CD19 | Approved in 2021 | R/R Large B-Cell Lymphoma, DLBCL |
| Trastuzumab emtansine | Kadcyla® | DM1 (maytansinoid) [non-cleavable] | HER2 | Approved in 2013 | HER2+ Early or Metastatic Breast Cancer |
| Enfortumab vedotine | Padcev® | MMAE [cleavable] | Nectin-4 | Approved in 2019 | Metastatic Urothelial Cancer |
| Trastuzumab deruxtecan | Enhertu® | Deruxtecan (topoisomerase-1 inhibitor) [cleavable] | HER2 | Approved in 2019 | HER2+, HER2-low Breast Cancer, NSCLC, GC/GEJ Adenocarcinoma |
| Sacituzumab govitecan | Trodelvy® | SN-38 (topoisomerase-1 inhibitor) [cleavable] | Trop-2 | Approved in 2020 | TNBC, Metastatic Urothelial Cancer |
| Disitamab vedotin | Aidixi® | MMAE [cleavable] | HER2 | Approved in 2021 | Gastric Cancer |
| Tisotumab vedotin | Tivdak® | MMAE [cleavable] | TF | Approved in 2021 | Cervical Cancer |
| Conjugate | Warhead | Target | Status | Patient Population |
|---|---|---|---|---|
| XMT-1536 | Auristatin F-hydroxypropylamide | NaPi2b | Phase III | Ovarian cancer |
| SHR-A1811 | Rezetecan | HER2 | Phase III | HER2+ Breast cancer |
| ARX788 | Amberstatin 269 | HER2 | Phase III | HER2+ Breast cancer |
| ABBV-399 | Monomethyl auristatin E | MET | Phase III | Non-small-cell lung cancer |
| U3-1402 | DXD | HER-3 | Phase III | Non-small-cell lung cancer |
| SAR408701 | DM4 | CEACAM5 | Phase III | Non-small-cell lung cancer |
| DS-1062 | DXD | TROP2 | Phase III | Breast cancer |
| SKB264 | Belotecan | TROP2 | Phase III | Triple-negative breast cancer |
| MK-2140 | Monomethyl auristatin E | ROR1 | Phase II/III | Diffuse large cell B-lymphoma |
| MGC018 | Duocarmycin | B7-H3 | Phase II/III | Prostate cancer |
| ADCT-301 | PBD SG3199 | CD25 | Phase II | Hodgkin’s lymphoma, acute myeloid leukemia |
| IMGN632 | DGN549 IGN | CD123 | Phase II | Blastic plasmacytoid dendritic cell neoplasm |
| CX-2009 | DM4 | ALCAM | Phase II | Breast cancer |
| DS-7300a | DXD | B7-H3 | Phase II | Small cell lung cancer |
| SGN-LIV1A | Monomethyl auristatin E | LIV-1 | Phase II | Lung cancer |
| BA3011 | Monomethyl auristatin E | AXL receptor tyrosine kinase | Phase II | Ovarian cancer, Non-small-cell lung cancer |
| BA3021 | Monomethyl auristatin E | ROR2 | Phase II | Head and neck squamous cell carcinoma, Non-small-cell lung cancer, ovarian cancer |
| MRG003 | Monomethyl auristatin E | EGFR | Phase II | Nasopharyngeal carcinoma, Biliary tract cancer, Non-small-cell lung cancer, Head and neck squamous cell carcinoma |
| MRG002 | Monomethyl auristatin E | HER2 | Phase II | Breast cancer, Non-small-cell lung cancer, Urothelium cancer, Biliary tract cancer |
| DX126-262 | Tub114 (Tubulysin B analogue) | HER2 | Phase II | HER2+ breast cancer |
| MORAb-202 | Eribulin | FR-α | Phase II | Non-small-cell lung cancer, Ovarian cancer |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buyukgolcigezli, I.; Tenekeci, A.K.; Sahin, I.H. Opportunities and Challenges in Antibody–Drug Conjugates for Cancer Therapy: A New Era for Cancer Treatment. Cancers 2025, 17, 958. https://doi.org/10.3390/cancers17060958
Buyukgolcigezli I, Tenekeci AK, Sahin IH. Opportunities and Challenges in Antibody–Drug Conjugates for Cancer Therapy: A New Era for Cancer Treatment. Cancers. 2025; 17(6):958. https://doi.org/10.3390/cancers17060958
Chicago/Turabian StyleBuyukgolcigezli, Idil, Ates Kutay Tenekeci, and Ibrahim Halil Sahin. 2025. "Opportunities and Challenges in Antibody–Drug Conjugates for Cancer Therapy: A New Era for Cancer Treatment" Cancers 17, no. 6: 958. https://doi.org/10.3390/cancers17060958
APA StyleBuyukgolcigezli, I., Tenekeci, A. K., & Sahin, I. H. (2025). Opportunities and Challenges in Antibody–Drug Conjugates for Cancer Therapy: A New Era for Cancer Treatment. Cancers, 17(6), 958. https://doi.org/10.3390/cancers17060958