
From Bench to Bedside: Translational Approaches to Cardiotoxicity in Breast Cancer, Lung Cancer, and Lymphoma Therapies

 ,
, 
 ,
,  ,
,
Simple Summary
Abstract
1. Introduction
2. In Vitro Models
2.1. Primary Cardiomyocytes
2.2. Established Cell Lines
2.3. Human Pluripotent Stem Cell-Derived Cardiomyocytes
3. Preclinical Animal Model
3.1. Radiotherapy
Mechanisms of Radiation-Induced Cardiovascular Toxicity
3.2. Chemotherapy
Mechanisms of Chemotherapy-Induced Cardiac-Toxicity
3.3. Immunotherapy
4. Examples of Application of In Vitro and In Vivo Models
4.1. Examples of In Vitro and In Vivo Models to Study the Effects of Radiotherapy
Clinical Reports on Radiotherapy Effects
4.2. Examples of In Vitro and In Vivo Models to Study the Effects of Chemotherapy
Clinical Reports on Chemotherapy Effects
4.3. Examples of In Vitro and In Vivo Models to Study the Effects of Immunotherapy
Clinical Reports on Immunotherapy Effects
5. Practical Points
5.1. Collaboration and Selection of Animal Models
5.2. Comprehensive Evaluation of Cardiac Function
5.3. Mechanism Investigation and Validation
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Miura, K.; Sano, Y.; Niho, S.; Kawasumi, K.; Mochizuki, N.; Yoh, K.; Matsumoto, S.; Zenke, Y.; Ikeda, T.; Nosaki, K. Impact of concomitant medication on clinical outcomes in patients with advanced non-small cell lung cancer treated with immune checkpoint inhibitors: A retrospective study. Thorac. Cancer 2021, 12, 1983–1994. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.; Persano, M.; Dubois, M.; Spanu, D.; Donisi, C.; Pozzari, M.; Deias, G.; Saba, G.; Migliari, M.; Liscia, N.; et al. Drug-related toxicity in breast cancer patients: A new path towards tailored treatment?—A narrative review. Precis. Cancer Med. 2022, 5, 1–24. [Google Scholar] [CrossRef]
- Pluchart, H.; Chanoine, S.; Moro-Sibilot, D.; Chouaid, C.; Frey, G.; Villa, J.; Degano, B.; Giaj Levra, M.; Bedouch, P.; Toffart, A.-C. Lung cancer, comorbidities, and medication: The infernal trio. Front. Pharmacol. 2024, 14, 1016976. [Google Scholar] [CrossRef] [PubMed]
- Thandra, K.C.; Barsouk, A.; Saginala, K.; Aluru, J.S.; Barsouk, A. Epidemiology of lung cancer. Contemp. Oncol./Współczesna Onkol. 2021, 25, 45–52. [Google Scholar] [CrossRef]
- Cheng, T.-Y.D.; Cramb, S.M.; Baade, P.D.; Youlden, D.R.; Nwogu, C.; Reid, M.E. The international epidemiology of lung cancer: Latest trends, disparities, and tumor characteristics. J. Thorac. Oncol. 2016, 11, 1653–1671. [Google Scholar] [CrossRef]
- Barta, J.A.; Powell, C.A.; Wisnivesky, J.P. Global epidemiology of lung cancer. Ann. Glob. Health 2019, 85, 8. [Google Scholar] [CrossRef]
- Giaquinto, A.N.; Sung, H.; Miller, K.D.; Kramer, J.L.; Newman, L.A.; Minihan, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2022. CA A Cancer J. Clin. 2022, 72, 524–541. [Google Scholar] [CrossRef]
- Tao, Z.; Shi, A.; Lu, C.; Song, T.; Zhang, Z.; Zhao, J. Breast cancer: Epidemiology and etiology. Cell Biochem. Biophys. 2015, 72, 333–338. [Google Scholar] [CrossRef]
- Fahad Ullah, M. Breast cancer: Current perspectives on the disease status. Breast Cancer Metastasis Drug Resist. Chall. Prog. 2019, 1152, 51–64. [Google Scholar]
- Metzger, M.L.; Mauz-Körholz, C. Epidemiology, outcome, targeted agents and immunotherapy in adolescent and young adult non-Hodgkin and Hodgkin lymphoma. Br. J. Haematol. 2019, 185, 1142–1157. [Google Scholar] [CrossRef]
- Shanbhag, S.; Ambinder, R.F. Hodgkin lymphoma: A review and update on recent progress. CA Cancer J. Clin. 2018, 68, 116–132. [Google Scholar] [PubMed]
- Li, Y.; Sun, H.; Yan, Y.; Sun, T.; Wang, S.; Ma, H. Long-term survival rates of patients with stage III–IV Hodgkin lymphoma according to age, sex, race, and socioeconomic status, 1984–2013. Oncologist 2018, 23, 1328–1336. [Google Scholar] [PubMed]
- Shakir, D.K.; Rasul, K.I. Chemotherapy induced cardiomyopathy: Pathogenesis, monitoring and management. J. Clin. Med. Res. 2009, 1, 8. [Google Scholar]
- Seltzer, J.H.; Gintant, G.; Amiri-Kordestani, L.; Singer, J.; Koplowitz, L.P.; Moslehi, J.J.; Barac, A.; Anthony, F.Y. Assessing cardiac safety in oncology drug development. Am. Heart J. 2019, 214, 125. [Google Scholar]
- Wang, H.; Wei, J.; Zheng, Q.; Meng, L.; Xin, Y.; Yin, X.; Jiang, X. Radiation-induced heart disease: A review of classification, mechanism and prevention. Int. J. Biol. Sci. 2019, 15, 2128. [Google Scholar]
- Abu Rmilah, A.; Adham, A.; Ikram-Ul, H.; Alzu’bi, H.; Nandan, A.; Jouni, H.; Hirashi, S.; Owen, D.; Deswal, A.; Lin, S.H. Novel risk score for predicting acute cardiovascular and cerebrovascular events after chest radiotherapy in patients with breast or lung cancer. Eur. J. Prev. Cardiol. 2024, zwae323. [Google Scholar] [CrossRef]
- Takemura, G.; Fujiwara, H. Doxorubicin-induced cardiomyopathy: From the cardiotoxic mechanisms to management. Prog. Cardiovasc. Dis. 2007, 49, 330–352. [Google Scholar]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar]
- Touyz, R.M.; Herrmann, S.M.S.; Herrmann, J. Vascular toxicities with VEGF inhibitor therapies–focus on hypertension and arterial thrombotic events. J. Am. Soc. Hypertens. 2018, 12, 409–425. [Google Scholar]
- Haas, N.B.; Manola, J.; Ky, B.; Flaherty, K.T.; Uzzo, R.G.; Kane, C.J.; Jewett, M.; Wood, L.; Wood, C.G.; Atkins, M.B. Effects of adjuvant sorafenib and sunitinib on cardiac function in renal cell carcinoma patients without overt metastases: Results from ASSURE, ECOG 2805. Clin. Cancer Res. 2015, 21, 4048–4054. [Google Scholar]
- Heinzerling, L.; Ott, P.A.; Hodi, F.S.; Husain, A.N.; Tajmir-Riahi, A.; Tawbi, H.; Pauschinger, M.; Gajewski, T.F.; Lipson, E.J.; Luke, J.J. Cardiotoxicity associated with CTLA4 and PD1 blocking immunotherapy. J. Immunother. Cancer 2016, 4, 50. [Google Scholar]
- Michel, L.; Rassaf, T.; Totzeck, M. Cardiotoxicity from immune checkpoint inhibitors. IJC Heart Vasc. 2019, 25, 100420. [Google Scholar]
- Upadhrasta, S.; Elias, H.; Patel, K.; Zheng, L. Managing cardiotoxicity associated with immune checkpoint inhibitors. Chronic Dis. Transl. Med. 2019, 5, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Boerma, M.; Hauer-Jensen, M. Preclinical research into basic mechanisms of radiation-induced heart disease. Cardiol. Res. Pract. 2011, 2011, 858262. [Google Scholar] [CrossRef]
- Castle, K.D.; Chen, M.; Wisdom, A.J.; Kirsch, D.G. Genetically engineered mouse models for studying radiation biology. Transl. Cancer Res. 2017, 6 (Suppl. S5), S900. [Google Scholar] [CrossRef]
- Pogwizd, S.M.; Bers, D.M. Rabbit models of heart disease. Drug Discov. Today Dis. Models 2008, 5, 185–193. [Google Scholar]
- Milani-Nejad, N.; Janssen, P.M.L. Small and large animal models in cardiac contraction research: Advantages and disadvantages. Pharmacol. Ther. 2014, 141, 235–249. [Google Scholar]
- Hearse, D.J.; Sutherland, F.J. Experimental models for the study of cardiovascular function and disease. Pharmacol. Res. 2000, 41, 597–603. [Google Scholar]
- Schlaak, R.A.; SenthilKumar, G.; Boerma, M.; Bergom, C. Advances in preclinical research models of radiation-induced cardiac toxicity. Cancers 2020, 12, 415. [Google Scholar] [CrossRef]
- Mohamed, T.M.A.; Moslehi, J.; Satin, J. Recent Advances in Cardiotoxicity Testing. Front. Pharmacol. 2021, 12, 798189. [Google Scholar]
- Golden, H.B.; Gollapudi, D.; Gerilechaogetu, F.; Li, J.; Cristales, R.J.; Peng, X.; Dostal, D.E. Isolation of cardiac myocytes and fibroblasts from neonatal rat pups. Methods Mol. Biol. 2012, 843, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Hasinoff, B.B. The cardiotoxicity and myocyte damage caused by small molecule anticancer tyrosine kinase inhibitors is correlated with lack of target specificity. Toxicol. Appl. Pharmacol. 2010, 244, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Graham, E.L.; Balla, C.; Franchino, H.; Melman, Y.; del Monte, F.; Das, S. Isolation, culture, and functional characterization of adult mouse cardiomyoctyes. J. Vis. Exp. 2013, 79, 50289. [Google Scholar]
- Bistola, V.; Nikolopoulou, M.; Derventzi, A.; Kataki, A.; Sfyras, N.; Nikou, N.; Toutouza, M.; Toutouzas, P.; Stefanadis, C.; Konstadoulakis, M.M. Long-term primary cultures of human adult atrial cardiac myocytes: Cell viability, structural properties and BNP secretion in vitro. Int. J. Cardiol. 2008, 131, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Bénardeau, A.; Hatem, S.N.; Rücker-Martin, C.; Tessier, S.; Dinanian, S.; Samuel, J.-L.; Coraboeuf, E.; Mercadier, J.-J. Primary culture of human atrial myocytes is associated with the appearance of structural and functional characteristics of immature myocardium. J. Mol. Cell. Cardiol. 1997, 29, 1307–1320. [Google Scholar] [CrossRef]
- Li, R.-K.; Mickle, D.A.G.; Weisel, R.D.; Carson, S.; Omar, S.A.; Tumiati, L.C.; Wilson, G.J.; Williams, W.G. Human pediatric and adult ventricular cardiomyocytes in culture: Assessment of phenotypic changes with passaging. Cardiovasc. Res. 1996, 32, 362–373. [Google Scholar] [CrossRef]
- Janssen, P.M.L.; Lehnart, S.E.; Prestle, J.; Hasenfuss, G. Preservation of contractile characteristics of human myocardium in multi-day cell culture. J. Mol. Cell. Cardiol. 1999, 31, 1419–1427. [Google Scholar] [CrossRef]
- Timolati, F.; Ott, D.; Pentassuglia, L.; Giraud, M.-N.; Perriard, J.-C.; Suter, T.M.; Zuppinger, C. Neuregulin-1 beta attenuates doxorubicin-induced alterations of excitation–contraction coupling and reduces oxidative stress in adult rat cardiomyocytes. J. Mol. Cell. Cardiol. 2006, 41, 845–854. [Google Scholar] [CrossRef]
- Kim, S.-Y.; Kim, S.-J.; Kim, B.-J.; Rah, S.-Y.; Chung, S.M.; Im, M.-J.; Kim, U.-H. Doxorubicin-induced reactive oxygen species generation and intracellular Ca2+ increase are reciprocally modulated in rat cardiomyocytes. Exp. Mol. Med. 2006, 38, 535–545. [Google Scholar] [CrossRef]
- Dimitrakis, P.; Romay-Ogando, M.-I.; Timolati, F.; Suter, T.M.; Zuppinger, C. Effects of doxorubicin cancer therapy on autophagy and the ubiquitin-proteasome system in long-term cultured adult rat cardiomyocytes. Cell Tissue Res. 2012, 350, 361–372. [Google Scholar] [CrossRef]
- Zordoky, B.N.M.; El-Kadi, A.O.S. H9c2 cell line is a valuable in vitro model to study the drug metabolizing enzymes in the heart. J. Pharmacol. Toxicol. Methods 2007, 56, 317–322. [Google Scholar] [PubMed]
- Watkins, S.J.; Borthwick, G.M.; Arthur, H.M. The H9C2 cell line and primary neonatal cardiomyocyte cells show similar hypertrophic responses in vitro. Vitr. Cell. Dev. Biol. Anim. 2011, 47, 125–131. [Google Scholar]
- Hescheler, J.; Meyer, R.; Plant, S.; Krautwurst, D.; Rosenthal, W.; Schultz, G. Morphological, biochemical, and electrophysiological characterization of a clonal cell (H9c2) line from rat heart. Circ. Res. 1991, 69, 1476–1486. [Google Scholar] [PubMed]
- Kuznetsov, A.V.; Javadov, S.; Sickinger, S.; Frotschnig, S.; Grimm, M. H9c2 and HL-1 cells demonstrate distinct features of energy metabolism, mitochondrial function and sensitivity to hypoxia-reoxygenation. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2015, 1853, 276–284. [Google Scholar] [CrossRef]
- Kimes, B.W.; Brandt, B.L. Properties of a clonal muscle cell line from rat heart. Exp. Cell Res. 1976, 98, 367–381. [Google Scholar] [CrossRef]
- Elshenawy, O.H.; Anwar-Mohamed, A.; Abdelhamid, G.; El-Kadi, A.O.S. Murine atrial HL-1 cell line is a reliable model to study drug metabolizing enzymes in the heart. Vasc. Pharmacol. 2013, 58, 326–333. [Google Scholar]
- Asensio-López, M.C.; Soler, F.; Pascual-Figal, D.; Fernández-Belda, F.; Lax, A. Doxorubicin-induced oxidative stress: The protective effect of nicorandil on HL-1 cardiomyocytes. PLoS ONE 2017, 12, e0172803. [Google Scholar]
- Strigun, A.; Wahrheit, J.; Niklas, J.; Heinzle, E.; Noor, F. Doxorubicin increases oxidative metabolism in HL-1 cardiomyocytes as shown by 13C metabolic flux analysis. Toxicol. Sci. 2012, 125, 595–606. [Google Scholar]
- Davidson, M.M.; Nesti, C.; Palenzuela, L.; Walker, W.F.; Hernandez, E.; Protas, L.; Hirano, M.; Isaac, N.D. Novel cell lines derived from adult human ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 2005, 39, 133–147. [Google Scholar]
- Yoon, C.S.; Kim, H.K.; Mishchenko, N.P.; Vasileva, E.A.; Fedoreyev, S.A.; Stonik, V.A.; Han, J. Spinochrome D attenuates doxorubicin-induced cardiomyocyte death via improving glutathione metabolism and attenuating oxidative stress. Mar. Drugs 2018, 17, 2. [Google Scholar] [CrossRef]
- Burridge, P.W.; Li, Y.F.; Matsa, E.; Wu, H.; Ong, S.-G.; Sharma, A.; Holmström, A.; Chang, A.C.; Coronado, M.J.; Ebert, A.D. Human induced pluripotent stem cell–derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med. 2016, 22, 547–556. [Google Scholar] [PubMed]
- Sakamoto, K.; Sakatoku, K.; Sugimoto, S.; Iwasaki, N.; Sano, Y.; Yamaguchi, M.; Kurokawa, J. Continued exposure of anti-cancer drugs to human iPS cell-derived cardiomyocytes can unmask their cardiotoxic effects. J. Pharmacol. Sci. 2019, 140, 345–349. [Google Scholar] [PubMed]
- Doherty, K.R.; Wappel, R.L.; Talbert, D.R.; Trusk, P.B.; Moran, D.M.; Kramer, J.W.; Brown, A.M.; Shell, S.A.; Bacus, S. Multi-parameter in vitro toxicity testing of crizotinib, sunitinib, erlotinib, and nilotinib in human cardiomyocytes. Toxicol. Appl. Pharmacol. 2013, 272, 245–255. [Google Scholar] [PubMed]
- Magdy, T.; Schuldt, A.J.T.; Wu, J.C.; Bernstein, D.; Burridge, P.W. Human induced pluripotent stem cell (hiPSC)-derived cells to assess drug cardiotoxicity: Opportunities and problems. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 83–103. [Google Scholar]
- Konorev, E.A.; Vanamala, S.; Kalyanaraman, B. Differences in doxorubicin-induced apoptotic signaling in adult and immature cardiomyocytes. Free Radic. Biol. Med. 2008, 45, 1723–1728. [Google Scholar]
- Lim, C.C.; Zuppinger, C.; Guo, X.; Kuster, G.M.; Helmes, M.; Eppenberger, H.M.; Suter, T.M.; Liao, R.; Sawyer, D.B. Anthracyclines induce calpain-dependent titin proteolysis and necrosis in cardiomyocytes. J. Biol. Chem. 2004, 279, 8290–8299. [Google Scholar]
- Timolati, F.; Anliker, T.; Groppalli, V.; Perriard, J.-C.; Eppenberger, H.M.; Suter, T.M.; Zuppinger, C. The role of cell death and myofibrillar damage in contractile dysfunction of long-term cultured adult cardiomyocytes exposed to doxorubicin. Cytotechnology 2009, 61, 25–36. [Google Scholar]
- Claycomb, W.C.; Lanson, N.A., Jr.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J., Jr. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar]
- Anmann, T.; Guzun, R.; Beraud, N.; Pelloux, S.; Kuznetsov, A.V.; Kogerman, L.; Kaambre, T.; Sikk, P.; Paju, K.; Peet, N.; et al. Different kinetics of the regulation of respiration in permeabilized cardiomyocytes and in HL-1 cardiac cells: Importance of cell structure/organization for respiration regulation. Biochim. Biophys. Acta (BBA)-Bioenerg. 2006, 1757, 1597–1606. [Google Scholar] [CrossRef]
- Monge, C.; Beraud, N.; Tepp, K.; Pelloux, S.; Chahboun, S.; Kaambre, T.; Kadaja, L.; Roosimaa, M.; Piirsoo, A.; Tourneur, Y. Comparative analysis of the bioenergetics of adult cardiomyocytes and nonbeating HL-1 cells: Respiratory chain activities, glycolytic enzyme profiles, and metabolic fluxes. Can. J. Physiol. Pharmacol. 2009, 87, 318–326. [Google Scholar]
- Chen, I.Y.; Matsa, E.; Wu, J.C. Induced pluripotent stem cells: At the heart of cardiovascular precision medicine. Nat. Rev. Cardiol. 2016, 13, 333–349. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.S.; Kryshtal, D.O.; Feaster, T.K.; Sánchez-Freire, V.; Zhang, J.; Kamp, T.J.; Hong, C.C.; Wu, J.C.; Knollmann, B.C. Human induced pluripotent stem cell (hiPSC) derived cardiomyocytes to understand and test cardiac calcium handling: A glass half full. J. Mol. Cell. Cardiol. 2015, 89 Pt B, 379–380. [Google Scholar] [CrossRef]
- Poon, E.; Keung, W.; Liang, Y.; Ramalingam, R.; Yan, B.; Zhang, S.; Chopra, A.; Moore, J.; Herren, A.; Lieu, D.K. Proteomic analysis of human pluripotent stem Cell–Derived, fetal, and adult ventricular cardiomyocytes reveals pathways crucial for cardiac metabolism and maturation. Circ. Cardiovasc. Genet. 2015, 8, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Medhora, M.; Gao, F.; Gasperetti, T.; Narayanan, J.; Khan, A.H.; Jacobs, E.R.; Fish, B.L. Delayed effects of acute radiation exposure (DEARE) in juvenile and old rats: Mitigation by lisinopril. Health Phys. 2019, 116, 529–545. [Google Scholar] [CrossRef] [PubMed]
- Zakaria, Z.Z.; Benslimane, F.M.; Nasrallah, G.K.; Shurbaji, S.; Younes, N.N.; Mraiche, F.; Da’as, S.I.; Yalcin, H.C. Using zebrafish for investigating the molecular mechanisms of drug-induced cardiotoxicity. BioMed Res. Int. 2018, 2018, 1642684. [Google Scholar]
- Maciag, M.; Wnorowski, A.; Mierzejewska, M.; Plazinska, A. Pharmacological assessment of zebrafish-based cardiotoxicity models. Biomed. Pharmacother. 2022, 148, 112695. [Google Scholar]
- Han, Y.; Zhang, J.P.; Qian, J.Q.; Hu, C.Q. Cardiotoxicity evaluation of anthracyclines in zebrafish (Danio rerio). J. Appl. Toxicol. 2015, 35, 241–252. [Google Scholar]
- Lauk, S.; Trott, K.-R. Radiation induced heart disease in hypertensive rats. Int. J. Radiat. Oncol. Biol. Phys. 1988, 14, 109–114. [Google Scholar]
- Boerma, M.; Roberto, K.A.; Hauer-Jensen, M. Prevention and treatment of functional and structural radiation injury in the rat heart by pentoxifylline and alpha-tocopherol. Int. J. Radiat. Oncol. Biol. Phys. 2008, 72, 170–177. [Google Scholar] [CrossRef]
- Boerma, M. Experimental radiation-induced heart disease: Past, present, and future. Radiat. Res. 2012, 178, 1–6. [Google Scholar] [CrossRef]
- Zaragoza, C.; Gomez-Guerrero, C.; Martin-Ventura, J.L.; Blanco-Colio, L.; Lavin, B.; Mallavia, B.; Tarin, C.; Mas, S.; Ortiz, A.; Egido, J. Animal models of cardiovascular diseases. BioMed Res. Int. 2011, 2011, 497841. [Google Scholar]
- Gabriels, K.; Hoving, S.; Seemann, I.; Visser, N.L.; Gijbels, M.J.; Pol, J.F.; Daemen, M.J.; Stewart, F.A.; Heeneman, S. Local heart irradiation of ApoE−/− mice induces microvascular and endocardial damage and accelerates coronary atherosclerosis. Radiother. Oncol. 2012, 105, 358–364. [Google Scholar] [PubMed]
- Hoving, S.; Heeneman, S.; Gijbels, M.J.J.; Te Poele, J.A.M.; Visser, N.; Cleutjens, J.; Russell, N.S.; Daemen, M.J.A.P.; Stewart, F.A. Irradiation induces different inflammatory and thrombotic responses in carotid arteries of wildtype C57BL/6J and atherosclerosis-prone ApoE−/− mice. Radiother. Oncol. 2012, 105, 365–370. [Google Scholar] [PubMed]
- Gabriels, K.; Hoving, S.; Gijbels, M.J.; Pol, J.F.; te Poele, J.A.; Biessen, E.A.; Daemen, M.J.; Stewart, F.A.; Heeneman, S. Irradiation of existing atherosclerotic lesions increased inflammation by favoring pro-inflammatory macrophages. Radiother. Oncol. 2014, 110, 455–460. [Google Scholar] [CrossRef]
- Stewart, F.A.; Heeneman, S.; Te Poele, J.; Kruse, J.; Russell, N.S.; Gijbels, M.; Daemen, M. Ionizing radiation accelerates the development of atherosclerotic lesions in ApoE−/− mice and predisposes to an inflammatory plaque phenotype prone to hemorrhage. Am. J. Pathol. 2006, 168, 649–658. [Google Scholar]
- Yamamoto, Y.; Minami, M.; Yoshida, K.; Nagata, M.; Miyata, T.; Yang, T.; Takayama, N.; Suzuki, K.; Okawa, M.; Yamada, K. Irradiation Accelerates Plaque Formation and Cellular Senescence in Flow-Altered Carotid Arteries of Apolipoprotein E Knock-Out Mice. J. Am. Heart Assoc. 2021, 10, e020712. [Google Scholar]
- Lee, C.-L.; Moding, E.J.; Cuneo, K.C.; Li, Y.; Sullivan, J.M.; Mao, L.; Washington, I.; Jeffords, L.B.; Rodrigues, R.C.; Ma, Y. p53 functions in endothelial cells to prevent radiation-induced myocardial injury in mice. Sci. Signal. 2012, 5, ra52. [Google Scholar]
- Mitchel, R.E.J.; Hasu, M.; Bugden, M.; Wyatt, H.; Hildebrandt, G.; Chen, Y.X.; Priest, N.D.; Whitman, S.C. Low-dose radiation exposure and protection against atherosclerosis in ApoE–/–mice: The influence of P53 heterozygosity. Radiat. Res. 2013, 179, 190–199. [Google Scholar] [CrossRef]
- Lee, C.-L.; Min, H.; Befera, N.; Clark, D.; Qi, Y.; Das, S.; Johnson, G.A.; Badea, C.T.; Kirsch, D.G. Assessing cardiac injury in mice with dual energy-microCT, 4D-microCT, and microSPECT imaging after partial heart irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 686–693. [Google Scholar]
- Sievert, W.; Stangl, S.; Steiger, K.; Multhoff, G. Improved overall survival of mice by reducing lung side effects after high-precision heart irradiation using a small animal radiation research platform. Int. J. Radiat. Oncol. Biol. Phys. 2018, 101, 671–679. [Google Scholar]
- Dreyfuss, A.D.; Goia, D.; Shoniyozov, K.; Shewale, S.V.; Velalopoulou, A.; Mazzoni, S.; Avgousti, H.; Metzler, S.D.; Bravo, P.E.; Feigenberg, S.J. A novel mouse model of radiation-induced cardiac injury reveals biological and radiological biomarkers of cardiac dysfunction with potential clinical relevance. Clin. Cancer Res. 2021, 27, 2266–2276. [Google Scholar] [PubMed]
- Mayosi, B.M.; Kardos, A.; Davies, C.H.; Gumedze, F.; Hovnanian, A.; Burge, S.; Watkins, H. Heterozygous disruption of SERCA2a is not associated with impairment of cardiac performance in humans: Implications for SERCA2a as a therapeutic target in heart failure. Heart 2006, 92, 105–109. [Google Scholar] [PubMed]
- Bers, D.M. Altered cardiac myocyte Ca regulation in heart failure. Physiology 2006, 21, 380–387. [Google Scholar] [PubMed]
- Ponnaluri, A.V.S.; Perotti, L.E.; Liu, M.; Qu, Z.; Weiss, J.N.; Ennis, D.B.; Klug, W.S.; Garfinkel, A. Electrophysiology of heart failure using a rabbit model: From the failing myocyte to ventricular fibrillation. PLoS Comput. Biol. 2016, 12, e1004968. [Google Scholar]
- Quinn, T.A.; Kohl, P. Rabbit models of cardiac mechano-electric and mechano-mechanical coupling. Prog. Biophys. Mol. Biol. 2016, 121, 110–122. [Google Scholar]
- Robinson, C.G.; Samson, P.P.; Moore, K.M.S.; Hugo, G.D.; Knutson, N.; Mutic, S.; Goddu, S.M.; Lang, A.; Cooper, D.H.; Faddis, M. Phase I/II trial of electrophysiology-guided noninvasive cardiac radioablation for ventricular tachycardia. Circulation 2019, 139, 313–321. [Google Scholar]
- Stewart, J.R.; Fajardo, L.F.; Cohn, K.E.; Page, V. Experimental radiation-induced heart disease in rabbits. Radiology 1968, 91, 814–817. [Google Scholar]
- Peng, X. Transgenic rabbit models for studying human cardiovascular diseases. Comp. Med. 2012, 62, 472–479. [Google Scholar]
- Lu, R.; Yuan, T.; Wang, Y.; Zhang, T.; Yuan, Y.; Wu, D.; Zhou, M.; He, Z.; Lu, Y.; Chen, Y. Spontaneous severe hypercholesterolemia and atherosclerosis lesions in rabbits with deficiency of low-density lipoprotein receptor (LDLR) on exon 7. eBioMedicine 2018, 36, 29–38. [Google Scholar]
- Yan, R.; Song, J.; Wu, Z.; Guo, M.; Liu, J.; Li, J.; Hao, X.; Li, S. Detection of myocardial metabolic abnormalities by 18F-FDG PET/CT and corresponding pathological changes in beagles with local heart irradiation. Korean J. Radiol. 2015, 16, 919–928. [Google Scholar]
- Song, J.; Yan, R.; Wu, Z.; Li, J.; Yan, M.; Hao, X.; Liu, J.; Li, S. 13N-ammonia PET/CT detection of myocardial perfusion abnormalities in beagle dogs after local heart irradiation. J. Nucl. Med. 2017, 58, 605–610. [Google Scholar] [PubMed]
- El-Sherif, O.; Xhaferllari, I.; Sykes, J.; Butler, J.; deKemp, R.A.; Renaud, J.; Yin, H.; Wilk, B.; Sullivan, R.; Pickering, J.G. [18F] FDG cardiac PET imaging in a canine model of radiation-induced cardiovascular disease associated with breast cancer radiotherapy. Am. J. Physiol.-Heart Circ. Physiol. 2019, 316, H586–H595. [Google Scholar]
- Hasiwa, N.; Bailey, J.; Clausing, P.; Daneshian, M.; Eileraas, M.; Farkas, S.; Gyertyán, I.; Hubrecht, R.; Kobel, W.; Krummenacher, G.; et al. Critical evaluation of the use of dogs in biomedical research and testing in Europe. ALTEX 2011, 28, 326–340. [Google Scholar] [CrossRef] [PubMed]
- Spannbauer, A.; Traxler, D.; Zlabinger, K.; Gugerell, A.; Winkler, J.; Mester-Tonczar, J.; Lukovic, D.; Müller, C.; Riesenhuber, M.; Pavo, N. Large animal models of heart failure with reduced ejection fraction (HFrEF). Front. Cardiovasc. Med. 2019, 6, 117. [Google Scholar]
- Recchia, F.A.; Lionetti, V. Animal models of dilated cardiomyopathy for translational research. Vet. Res. Commun. 2007, 31, 35–41. [Google Scholar]
- Zalesak-Kravec, S.; Huang, W.; Wang, P.; Yu, J.; Liu, T.; Defnet, A.E.; Moise, A.R.; Farese, A.M.; MacVittie, T.J.; Kane, M.A. Multi-omic analysis of non-human primate heart after partial-body radiation with minimal bone marrow sparing. Health Phys. 2021, 121, 352–371. [Google Scholar]
- Patel, R.; Nagueh, S.F.; Tsybouleva, N.; Abdellatif, M.; Lutucuta, S.; Kopelen, H.A.; Quinones, M.A.; Zoghbi, W.A.; Entman, M.L.; Roberts, R. Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation 2001, 104, 317–324. [Google Scholar]
- Senthil, V.; Chen, S.N.; Tsybouleva, N.; Halder, T.; Nagueh, S.F.; Willerson, J.T.; Roberts, R.; Marian, A.J. Prevention of cardiac hypertrophy by atorvastatin in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circ. Res. 2005, 97, 285–292. [Google Scholar]
- Toyoda, Y.; Okada, M.; Kashem, M.A. A canine model of dilated cardiomyopathy induced by repetitive intracoronary doxorubicin administration. J. Thorac. Cardiovasc. Surg. 1998, 115, 1367–1373. [Google Scholar]
- Walker, L.; Baumgartner, L.; Keller, K.C.; Ast, J.; Trettner, S.; Zur Nieden, N.I. Non-human primate and rodent embryonic stem cells are differentially sensitive to embryotoxic compounds. Toxicol. Rep. 2015, 2, 165–174. [Google Scholar]
- Dixon, J.A.; Spinale, F.G. Large Animal Models of Heart Failure. Circ. Heart Fail. 2009, 2, 262–271. [Google Scholar] [PubMed]
- Taunk, N.K.; Haffty, B.G.; Kostis, J.B.; Goyal, S. Radiation-induced heart disease: Pathologic abnormalities and putative mechanisms. Front. Oncol. 2015, 5, 39. [Google Scholar] [CrossRef] [PubMed]
- Gujral, D.M.; Lloyd, G.; Bhattacharyya, S. Radiation-induced valvular heart disease. Heart 2016, 102, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Tapio, S. Pathology and biology of radiation-induced cardiac disease. J. Radiat. Res. 2016, 57, 439–448. [Google Scholar] [CrossRef]
- Wang, B.; Wang, H.; Zhang, M.; Ji, R.; Wei, J.; Xin, Y.; Jiang, X. Radiation-induced myocardial fibrosis: Mechanisms underlying its pathogenesis and therapeutic strategies. J. Cell. Mol. Med. 2020, 24, 7717–7729. [Google Scholar]
- Hinz, B.; Mastrangelo, D.; Iselin, C.E.; Chaponnier, C.; Gabbiani, G. Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. Am. J. Pathol. 2001, 159, 1009–1020. [Google Scholar] [CrossRef]
- Hinz, B.; Gabbiani, G. Mechanisms of force generation and transmission by myofibroblasts. Curr. Opin. Biotechnol. 2003, 14, 538–546. [Google Scholar]
- Slama, M.S.; Le Guludec, D.; Sebag, C.; Leenhardt, A.R.; Davy, J.M.; Pellerin, D.E.; Drieu, L.H.; Victor, J.; Brechenmacher, C.; Motté, G. Complete atrioventricular block following mediastinal irradiation: A report of six cases. Pacing Clin. Electrophysiol. 1991, 14, 1112–1118. [Google Scholar]
- Marks, L.B.; Yu, X.; Prosnitz, R.G.; Zhou, S.-M.; Hardenbergh, P.H.; Blazing, M.; Hollis, D.; Lind, P.; Tisch, A.; Wong, T.Z. The incidence and functional consequences of RT-associated cardiac perfusion defects. Int. J. Radiat. Oncol. Biol. Phys. 2005, 63, 214–223. [Google Scholar]
- Siaravas, K.C.; Katsouras, C.S.; Sioka, C. Radiation treatment mechanisms of cardiotoxicity: A systematic review. Int. J. Mol. Sci. 2023, 24, 6272. [Google Scholar] [CrossRef]
- Venkatesulu, B.P.; Mahadevan, L.S.; Aliru, M.L.; Yang, X.; Bodd, M.H.; Singh, P.K.; Yusuf, S.W.; Abe, J.-I.; Krishnan, S. Radiation-induced endothelial vascular injury: A review of possible mechanisms. JACC Basic Transl. Sci. 2018, 3, 563–572. [Google Scholar] [PubMed]
- Baselet, B.; Sonveaux, P.; Baatout, S.; Aerts, A. Pathological effects of ionizing radiation: Endothelial activation and dysfunction. Cell. Mol. Life Sci. 2019, 76, 699–728. [Google Scholar] [PubMed]
- Guipaud, O.; Jaillet, C.; Clément-Colmou, K.; François, A.; Supiot, S.; Milliat, F. The importance of the vascular endothelial barrier in the immune-inflammatory response induced by radiotherapy. Br. J. Radiol. 2018, 91, 20170762. [Google Scholar]
- Gao, M.; Shirato, H.; Miyasaka, K.; Kuwabara, M.; Koyama, T. Induction of growth factors in rat cardiac tissue by X irradiation. Radiat. Res. 2000, 153, 540–547. [Google Scholar] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 2019, 65, 70–99. [Google Scholar]
- Madan, R.; Benson, R.; Sharma, D.N.; Julka, P.K.; Rath, G.K. Radiation induced heart disease: Pathogenesis, management and review literature. J. Egypt. Natl. Cancer Inst. 2015, 27, 187–193. [Google Scholar]
- Czubryt, M.P. Cardiac fibroblast to myofibroblast phenotype conversion—An unexploited therapeutic target. J. Cardiovasc. Dev. Dis. 2019, 6, 28. [Google Scholar] [CrossRef]
- Ejaz, A.; Greenberger, J.S.; Rubin, P.J. Understanding the mechanism of radiation induced fibrosis and therapy options. Pharmacol. Ther. 2019, 204, 107399. [Google Scholar]
- Ma, C.-X.; Zhao, X.-K.; Li, Y.-D. New therapeutic insights into radiation-induced myocardial fibrosis. Ther. Adv. Chronic Dis. 2019, 10, 2040622319868383. [Google Scholar]
- Kai, H.; Mori, T.; Tokuda, K.; Takayama, N.; Tahara, N.; Takemiya, K.; Kudo, H.; Sugi, Y.; Fukui, D.; Yasukawa, H. Pressure overload–induced transient oxidative stress mediates perivascular inflammation and cardiac fibrosis through angiotensin II. Hypertens. Res. 2006, 29, 711–718. [Google Scholar]
- Firsanov, D.; Vasilishina, A.; Kropotov, A.; Mikhailov, V. Dynamics of γH2AX formation and elimination in mammalian cells after X-irradiation. Biochimie 2012, 94, 2416–2422. [Google Scholar] [PubMed]
- Sarosiek, K.A.; Chi, X.; Bachman, J.A.; Sims, J.J.; Montero, J.; Patel, L.; Flanagan, A.; Andrews, D.W.; Sorger, P.; Letai, A. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol. Cell 2013, 51, 751–765. [Google Scholar] [PubMed]
- Salata, C.; Ferreira-Machado, S.C.; De Andrade, C.B.V.; Mencalha, A.L.; Mandarim-De-Lacerda, C.A.; de Almeida, C.E. Apoptosis induction of cardiomyocytes and subsequent fibrosis after irradiation and neoadjuvant chemotherapy. Int. J. Radiat. Biol. 2014, 90, 284–290. [Google Scholar] [PubMed]
- Liu, L.K.; Ouyang, W.; Zhao, X.; Su, S.F.; Yang, Y.; Ding, W.J.; He, Z.X.; Lu, B. Pathogenesis and prevention of radiation-induced myocardial fibrosis. Asian Pac. J. Cancer Prev. APJCP 2017, 18, 583. [Google Scholar]
- Zheng, M.; Liu, Z.; He, Y. Radiation-induced fibrosis: Mechanisms and therapeutic strategies from an immune microenvironment perspective. Immunology 2024, 172, 533–546. [Google Scholar]
- Wu, B.; Zhao, S.; Zhang, J.; Liu, Y.; Bai, J.; Wang, G.; Wang, Y.; Jiang, H.; Hu, Y.; OuYang, W. PD-1 Inhibitor Aggravate Irradiation-Induced Myocardial Fibrosis by Regulating TGF-β1/Smads Signaling Pathway via GSDMD-Mediated Pyroptosis. Inflammation 2024, 48, 181–198. [Google Scholar]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.-L.; Gabbiani, G. The myofibroblast: One function, multiple origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Prunotto, M.; Desmoulière, A.; Varga, J.; De Wever, O.; Mareel, M.; Gabbiani, G. Recent developments in myofibroblast biology: Paradigms for connective tissue remodeling. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar]
- Desmoulière, A.; Chaponnier, C.; Gabbiani, G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen. 2005, 13, 7–12. [Google Scholar]
- Belzile-Dugas, E.; Eisenberg, M.J. Radiation-induced cardiovascular disease: Review of an underrecognized pathology. J. Am. Heart Assoc. 2021, 10, e021686. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, L.F. The unique physiology of endothelial cells and its implications in radiobiology. Radiat. Toler. Norm. Tissues 1989, 23, 96–112. [Google Scholar]
- Darby, S.C.; Ewertz, M.; McGale, P.; Bennet, A.M.; Blom-Goldman, U.; Brønnum, D.; Correa, C.; Cutter, D.; Gagliardi, G.; Gigante, B. Risk of ischemic heart disease in women after radiotherapy for breast cancer. N. Engl. J. Med. 2013, 368, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Yarnold, J.; Brotons, M.-C.V. Pathogenetic mechanisms in radiation fibrosis. Radiother. Oncol. 2010, 97, 149–161. [Google Scholar] [CrossRef]
- Stewart, F.A.; Seemann, I.; Hoving, S.; Russell, N.S. Understanding radiation-induced cardiovascular damage and strategies for intervention. Clin. Oncol. 2013, 25, 617–624. [Google Scholar] [CrossRef]
- Straub, J.M.; New, J.; Hamilton, C.D.; Lominska, C.; Shnayder, Y.; Thomas, S.M. Radiation-induced fibrosis: Mechanisms and implications for therapy. J. Cancer Res. Clin. Oncol. 2015, 141, 1985–1994. [Google Scholar] [CrossRef]
- Zhao, W.; Robbins, M.E.C. Inflammation and chronic oxidative stress in radiation-induced late normal tissue injury: Therapeutic implications. Curr. Med. Chem. 2009, 16, 130–143. [Google Scholar] [CrossRef]
- Tewey, K.M.; Rowe, T.C.; Yang, L.; Halligan, B.D.; Liu, L.F. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 1984, 226, 466–468. [Google Scholar] [CrossRef]
- Fragoulakis, V.; Roncato, R.; Dalle Fratte, C.; Ecca, F.; Bartsakoulia, M.; Innocenti, F.; Toffoli, G.; Cecchin, E.; Patrinos, G.P.; Mitropoulou, C. Estimating the effectiveness of DPYD genotyping in Italian individuals suffering from cancer based on the cost of chemotherapy-induced toxicity. Am. J. Hum. Genet. 2019, 104, 1158–1168. [Google Scholar] [CrossRef]
- Meulendijks, D.; Cats, A.; Beijnen, J.H.; Schellens, J.H.M. Improving safety of fluoropyrimidine chemotherapy by individualizing treatment based on dihydropyrimidine dehydrogenase activity–ready for clinical practice? Cancer Treat. Rev. 2016, 50, 23–34. [Google Scholar] [CrossRef]
- Gershwin, M.E.; Goetzl, E.J.; Steinberg, A.D. Cyclophosphamide: Use in practice. Ann. Intern. Med. 1974, 80, 531–540. [Google Scholar] [PubMed]
- Field, J.J.; Kanakkanthara, A.; Miller, J.H. Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorg. Med. Chem. 2014, 22, 5050–5059. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.H.; Booser, D.J.; Murray, J.L.; Ibrahim, N.K.; Rahman, Z.U.; Valero, V.; Theriault, R.L.; Rosales, M.F.; Rivera, E.; Frye, D. A detailed evaluation of cardiac toxicity: A phase II study of doxorubicin and one-or three-hour-infusion paclitaxel in patients with metastatic breast cancer. Clin. Cancer Res. 2002, 8, 3360–3368. [Google Scholar] [PubMed]
- Napoli, C.; Benincasa, G.; Donatelli, F.; Ambrosio, G. Precision medicine in distinct heart failure phenotypes: Focus on clinical epigenetics. Am. Heart J. 2020, 224, 113–128. [Google Scholar]
- Gianni, L.; Baselga, J.; Eiermann, W.; Guillem Porta, V.; Semiglazov, V.; Lluch, A.; Zambetti, M.; Sabadell, D.; Raab, G.; Llombart Cussac, A.; et al. Feasibility and tolerability of sequential doxorubicin/paclitaxel followed by cyclophosphamide, methotrexate, and fluorouracil and its effects on tumor response as preoperative therapy. Clin. Cancer Res. 2005, 11, 8715–8721. [Google Scholar] [CrossRef]
- Pritchard, K.I.; Shepherd, L.E.; O’Malley, F.P.; Andrulis, I.L.; Tu, D.; Bramwell, V.H.; Levine, M.N. HER2 and responsiveness of breast cancer to adjuvant chemotherapy. N. Engl. J. Med. 2006, 354, 2103–2111. [Google Scholar] [CrossRef]
- Piccart-Gebhart, M.J.; Procter, M.; Leyland-Jones, B.; Goldhirsch, A.; Untch, M.; Smith, I.; Gianni, L.; Baselga, J.; Bell, R.; Jackisch, C.; et al. Trastuzumab after Adjuvant Chemotherapy in HER2-Positive Breast Cancer. N. Engl. J. Med. 2005, 353, 1659–1672. [Google Scholar] [CrossRef]
- Bowles, E.J.A.; Wellman, R.; Feigelson, H.S.; Onitilo, A.A.; Freedman, A.N.; Delate, T.; Allen, L.A.; Nekhlyudov, L.; Goddard, K.A.B.; Davis, R.L. Risk of heart failure in breast cancer patients after anthracycline and trastuzumab treatment: A retrospective cohort study. J. Natl. Cancer Inst. 2012, 104, 1293–1305. [Google Scholar]
- Yang, B.; Papoian, T. Tyrosine kinase inhibitor (TKI)-induced cardiotoxicity: Approaches to narrow the gaps between preclinical safety evaluation and clinical outcome. J. Appl. Toxicol. 2012, 32, 945–951. [Google Scholar]
- Groarke, J.D.; Choueiri, T.K.; Slosky, D.; Cheng, S.; Moslehi, J. Recognizing and managing left ventricular dysfunction associated with therapeutic inhibition of the vascular endothelial growth factor signaling pathway. Curr. Treat. Options Cardiovasc. Med. 2014, 16, 335. [Google Scholar]
- Nazer, B.; Humphreys, B.D.; Moslehi, J. Effects of novel angiogenesis inhibitors for the treatment of cancer on the cardiovascular system: Focus on hypertension. Circulation 2011, 124, 1687–1691. [Google Scholar] [PubMed]
- Vigneau, C.; Lorcy, N.; Dolley-Hitze, T.; Jouan, F.; Arlot-Bonnemains, Y.; Laguerre, B.; Verhoest, G.; Goujon, J.M.; Belaud-Rotureau, M.-A.; Rioux-Leclercq, N. All anti-vascular endothelial growth factor drugs can induce ’pre-eclampsia-like syndrome’: A RARe study. Nephrol. Dial. Transplant. 2014, 29, 325–332. [Google Scholar] [PubMed]
- Keefe, D.L. Anthracycline-Induced cardiomyopathy. Semin. Oncol. 2001, 28, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Shan, K.; Lincoff, A.M.; Young, J.B. Anthracycline-induced cardiotoxicity. Ann. Intern. Med. 1997, 126, 827–828. [Google Scholar]
- Mordente, A.; Meucci, E.; Silvestrini, A.; Martorana, G.E.; Giardina, B. New developments in anthracycline-induced cardiotoxicity. Curr. Med. Chem. 2009, 16, 1656–1672. [Google Scholar]
- Jones, R.L.; Swanton, C.; Ewer, M.S. Anthracycline cardiotoxicity. Expert Opin. Drug Saf. 2006, 5, 791–809. [Google Scholar]
- Layoun, M.E.; Wickramasinghe, C.D.; Peralta, M.V.; Yang, E.H. Fluoropyrimidine-induced cardiotoxicity: Manifestations, mechanisms, and management. Curr. Oncol. Rep. 2016, 18, 35. [Google Scholar]
- Kanduri, J.; More, L.A.; Godishala, A.; Asnani, A. Fluoropyrimidine-associated cardiotoxicity. Cardiol. Clin. 2019, 37, 399–405. [Google Scholar]
- Depetris, I.; Marino, D.; Bonzano, A.; Cagnazzo, C.; Filippi, R.; Aglietta, M.; Leone, F. Fluoropyrimidine-induced cardiotoxicity. Crit. Rev. Oncol./Hematol. 2018, 124, 1–10. [Google Scholar]
- Saif, M.W.; Shah, M.M.; Shah, A.R. Fluoropyrimidine-associated cardiotoxicity: Revisited. Expert Opin. Drug Saf. 2009, 8, 191–202. [Google Scholar]
- Raber, I.; Warack, S.; Kanduri, J.; Pribish, A.; Godishala, A.; Abovich, A.; Orbite, A.; Dommaraju, S.; Frazer, M.; Peters, M.L. Fluoropyrimidine-associated cardiotoxicity: A retrospective case-control study. Oncologist 2020, 25, e606–e609. [Google Scholar] [PubMed]
- Jiang, T.; Gan, T.; Xu, X.; Cao, Y.; Li, H.; Yang, J. Effects of the Alkylating Agent Methyl Methanesulfonate on the Cardiovascular System in SD Rats. J. Physiol. Stud. 2019, 7, 17–24. [Google Scholar]
- Francisco, A.P.; Perry, M.D.J.; Moreira, R.; Mendes, E. Alkylating Agents. In Anticancer Therapeutics; John and Wiley and Sons: Hoboken, NJ, USA, 2008; pp. 133–158. [Google Scholar]
- Ranchoux, B.; Günther, S.; Quarck, R.; Chaumais, M.-C.; Dorfmüller, P.; Antigny, F.; Dumas, S.J.; Raymond, N.; Lau, E.; Savale, L. Chemotherapy-induced pulmonary hypertension: Role of alkylating agents. Am. J. Pathol. 2015, 185, 356–371. [Google Scholar] [PubMed]
- Pantazi, D.; Tselepis, A.D. Cardiovascular toxic effects of antitumor agents: Pathogenetic mechanisms. Thromb. Res. 2022, 213, S95–S102. [Google Scholar]
- Caporizzo, M.A.; Chen, C.Y.; Prosser, B.L. Cardiac microtubules in health and heart disease. Exp. Biol. Med. 2019, 244, 1255–1272. [Google Scholar]
- Chen, C.Y.; Caporizzo, M.A.; Bedi, K.; Vite, A.; Bogush, A.I.; Robison, P.; Heffler, J.G.; Salomon, A.K.; Kelly, N.A.; Babu, A. Suppression of detyrosinated microtubules improves cardiomyocyte function in human heart failure. Nat. Med. 2018, 24, 1225–1233. [Google Scholar]
- Saji, K.; Fukumoto, Y.; Suzuki, J.; Fukui, S.; Nawata, J.; Shimokawa, H. Colchicine, a microtubule depolymerizing agent, inhibits myocardial apoptosis in rats. Tohoku J. Exp. Med. 2007, 213, 139–148. [Google Scholar]
- Zile, M.R.; Koide, M.; Sato, H.; Ishiguro, Y.; Conrad, C.H.; Buckley, J.M.; Morgan, J.P.; Cooper, G. Role of microtubules in the contractile dysfunction of hypertrophied myocardium. J. Am. Coll. Cardiol. 1999, 33, 250–260. [Google Scholar]
- Cooper, G., IV. Cytoskeletal networks and the regulation of cardiac contractility: Microtubules, hypertrophy, and cardiac dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1003–H1014. [Google Scholar]
- Ewer, M.S.; O’Shaughnessy, J.A. Cardiac toxicity of trastuzumab-related regimens in HER2-overexpressing breast cancer. Clin. Breast Cancer 2007, 7, 22–29. [Google Scholar]
- Abouegylah, M.; Braunstein, L.Z.; Alm El-Din, M.A.; Niemierko, A.; Salama, L.; Elebrashi, M.; Edgington, S.K.; Remillard, K.; Napolitano, B.; Naoum, G.E. Evaluation of radiation-induced cardiac toxicity in breast cancer patients treated with Trastuzumab-based chemotherapy. Breast Cancer Res. Treat. 2019, 174, 179–185. [Google Scholar] [PubMed]
- Di Cosimo, S. Heart to heart with trastuzumab: A review on cardiac toxicity. Target. Oncol. 2011, 6, 189–195. [Google Scholar] [PubMed]
- Sparano, J.A. Cardiac toxicity of trastuzumab (Herceptin): Implications for the design of adjuvant trials. Semin. Oncol. 2001, 28, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Nowsheen, S.; Viscuse, P.V.; O’Sullivan, C.C.; Sandhu, N.P.; Haddad, T.C.; Blaes, A.; Klemp, J.; Nhola, L.; Herrmann, J.; Ruddy, K.J. Incidence, diagnosis, and treatment of cardiac toxicity from trastuzumab in patients with breast cancer. Curr. Breast Cancer Rep. 2017, 9, 173–182. [Google Scholar] [CrossRef]
- Keefe, D.L. Trastuzumab-associated cardiotoxicity. Cancer 2002, 95, 1592–1600. [Google Scholar]
- Pandey, A.K.; Singhi, E.K.; Arroyo, J.P.; Ikizler, T.A.; Gould, E.R.; Brown, J.; Beckman, J.A.; Harrison, D.G.; Moslehi, J. Mechanisms of VEGF (vascular endothelial growth factor) inhibitor–associated hypertension and vascular disease. Hypertension 2018, 71, e1–e8. [Google Scholar]
- Touyz, R.M.; Lang, N.N.; Herrmann, J.; Van Den Meiracker, A.H.; Danser, A.H.J. Recent advances in hypertension and cardiovascular toxicities with vascular endothelial growth factor inhibition. Hypertension 2017, 70, 220–226. [Google Scholar]
- Dabravolski, S.A.; Khotina, V.A.; Omelchenko, A.V.; Kalmykov, V.A.; Orekhov, A.N. The role of the VEGF family in atherosclerosis development and its potential as treatment targets. Int. J. Mol. Sci. 2022, 23, 931. [Google Scholar] [CrossRef]
- Neves, K.B.; Rios, F.J.; Van Der Mey, L.; Alves-Lopes, R.; Cameron, A.C.; Volpe, M.; Montezano, A.C.; Savoia, C.; Touyz, R.M. VEGFR (vascular endothelial growth factor receptor) inhibition induces cardiovascular damage via redox-sensitive processes. Hypertension 2018, 71, 638–647. [Google Scholar] [CrossRef]
- Winnik, S.; Lohmann, C.; Siciliani, G.; von Lukowicz, T.; Kuschnerus, K.; Kraenkel, N.; Brokopp, C.E.; Enseleit, F.; Michels, S.; Ruschitzka, F. Systemic VEGF inhibition accelerates experimental atherosclerosis and disrupts endothelial homeostasis–implications for cardiovascular safety. Int. J. Cardiol. 2013, 168, 2453–2461. [Google Scholar]
- Lesterhuis, W.J.; Haanen, J.B.A.G.; Punt, C.J.A. Cancer immunotherapy–revisited. Nat. Rev. Drug Discov. 2011, 10, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-W.; Zhu, Y.-J.; Wang, M.-N.; Xie, Y.; Chen, C.-Y.; Zhang, T.; Xia, F.; Ding, Z.-Y.; Liu, J.-Y. Immune checkpoint inhibitor-associated cardiotoxicity: Current understanding on its mechanism, diagnosis and management. Front. Pharmacol. 2019, 10, 1350. [Google Scholar]
- Baban, B.; Liu, J.Y.; Qin, X.; Weintraub, N.L.; Mozaffari, M.S. Upregulation of programmed death-1 and its ligand in cardiac injury models: Interaction with GADD153. PLoS ONE 2015, 10, e0124059. [Google Scholar]
- Kushnareva, E.; Kushnarev, V.; Artemyeva, A.; Mitrofanova, L.; Moiseeva, O. Myocardial PD-L1 expression in patients with ischemic and non-ischemic heart failure. Front. Cardiovasc. Med. 2022, 8, 759972. [Google Scholar]
- Xia, W.; Zou, C.; Chen, H.; Xie, C.; Hou, M. Immune checkpoint inhibitor induces cardiac injury through polarizing macrophages via modulating microRNA-34a/Kruppel-like factor 4 signaling. Cell Death Dis. 2020, 11, 575. [Google Scholar]
- Reuben, A.; Petaccia de Macedo, M.; McQuade, J.; Joon, A.; Ren, Z.; Calderone, T.; Conner, B.; Wani, K.; Cooper, Z.A.; Tawbi, H. Comparative immunologic characterization of autoimmune giant cell myocarditis with ipilimumab. Oncoimmunology 2017, 6, e1361097. [Google Scholar] [CrossRef]
- Ganatra, S.; Neilan, T.G. Immune checkpoint inhibitor-associated myocarditis. Oncology 2018, 23, 879–886. [Google Scholar]
- Ceschi, A.; Noseda, R.; Palin, K.; Verhamme, K. Immune checkpoint inhibitor-related cytokine release syndrome: Analysis of WHO global pharmacovigilance database. Front. Pharmacol. 2020, 11, 557. [Google Scholar]
- Baik, A.H.; Oluwole, O.O.; Johnson, D.B.; Shah, N.; Salem, J.-E.; Tsai, K.K.; Moslehi, J.J. Mechanisms of cardiovascular toxicities associated with immunotherapies. Circ. Res. 2021, 128, 1780–1801. [Google Scholar]
- Michel, L.; Helfrich, I.; Hendgen-Cotta, U.B.; Mincu, R.-I.; Korste, S.; Mrotzek, S.M.; Spomer, A.; Odersky, A.; Rischpler, C.; Herrmann, K. Targeting early stages of cardiotoxicity from anti-PD1 immune checkpoint inhibitor therapy. Eur. Heart J. 2022, 43, 316–329. [Google Scholar]
- Varricchi, G.; Galdiero, M.R.; Tocchetti, C.G. Novel actors on the stage of cardiac dysfunction induced by anti-PD1 oncological treatments. Eur. Heart J. 2022, 43, 330–332. [Google Scholar] [CrossRef]
- Irabor, O.C.; Nelson, N.; Shah, Y.; Niazi, M.K.; Poiset, S.; Storozynsky, E.; Singla, D.K.; Hooper, D.C.; Lu, B. Overcoming the cardiac toxicities of cancer therapy immune checkpoint inhibitors. Front. Oncol. 2022, 12, 940127. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Choi, S.W.; Park, Y.-G.; Kim, S.J.; Choi, C.H.; Cha, M.-J.; Chang, J.H. Impact of high-dose irradiation on human iPSC-derived cardiomyocytes using multi-electrode arrays: Implications for the antiarrhythmic effects of cardiac radioablation. Int. J. Mol. Sci. 2021, 23, 351. [Google Scholar]
- Mocan-Hognogi, D.L.; Trancǎ, S.; Farcaş, A.D.; Mocan-Hognogi, R.F.; Pârvu, A.V.; Bojan, A.S. Immune checkpoint inhibitors and the heart. Front. Cardiovasc. Med. 2021, 8, 726426. [Google Scholar]
- Jiménez-Alejandre, R.; Ruiz-Fernandez, I.; Martin, P. Pathophysiology of immune checkpoint inhibitor-induced myocarditis. Cancers 2022, 14, 4494. [Google Scholar] [CrossRef]
- Kadowaki, H.; Akazawa, H.; Ishida, J.; Komuro, I. Mechanisms and management of immune checkpoint inhibitor-related cardiac adverse events. JMA J. 2021, 4, 91–98. [Google Scholar]
- Suliman, B.A. Potential clinical implications of molecular mimicry-induced autoimmunity. Immun. Inflamm. Dis. 2024, 12, e1178. [Google Scholar] [CrossRef]
- Rose, N.R. Learning from myocarditis: Mimicry, chaos and black holes. F1000prime Rep. 2014, 6, 25. [Google Scholar] [CrossRef]
- Bracamonte-Baran, W.; Čiháková, D. Cardiac autoimmunity: Myocarditis. Immunol. Cardiovasc. Homeost. Pathol. 2017, 1003, 187–221. [Google Scholar]
- Rose, N.R.; Herskowitz, A.; Neumann, D.A.; Neu, N. Autoimmune myocarditis: A paradigm of post-infection autoimmune disease. Immunol. Today 1988, 9, 117–120. [Google Scholar]
- Root-Bernstein, R.; Fairweather, D. Unresolved issues in theories of autoimmune disease using myocarditis as a framework. J. Theor. Biol. 2015, 375, 101–123. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Barrett, D.; Teachey, D.T.; Grupp, S.A. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014, 20, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat. Rev. Immunol. 2022, 22, 85–96. [Google Scholar] [CrossRef]
- Acharya, U.H.; Dhawale, T.; Yun, S.; Jacobson, C.A.; Chavez, J.C.; Ramos, J.D.; Appelbaum, J.; Maloney, D.G. Management of cytokine release syndrome and neurotoxicity in chimeric antigen receptor (CAR) T cell therapy. Expert Rev. Hematol. 2019, 12, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Teachey, D.T.; Lacey, S.F.; Shaw, P.A.; Melenhorst, J.J.; Maude, S.L.; Frey, N.; Pequignot, E.; Gonzalez, V.E.; Chen, F.; Finklestein, J. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov. 2016, 6, 664–679. [Google Scholar] [CrossRef]
- Giavridis, T.; van der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell–induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef]
- Bronson, S.; Stirling, E.; Westwood, B.; Triozzi, P.; Soto-Pantoja, D. 806 PD-1 blockade affects inflammation and metabolic flexibility to potentially mediate cardiac immune-related adverse events. J. Immunother. Cancer 2021, 9, 806. [Google Scholar]
- Banerjee, A.; Narasimhulu, C.A.; Singla, D.K. Immune interactions in pembrolizumab (PD-1 inhibitor) cancer therapy and cardiovascular complications. Am. J. Physiol. Heart Circ. Physiol. 2023, 325, H751–H767. [Google Scholar] [CrossRef]
- Gergely, T.G.; Kucsera, D.; Tóth, V.E.; Kovács, T.; Sayour, N.V.; Drobni, Z.D.; Ruppert, M.; Petrovich, B.; Ágg, B.; Onódi, Z. Characterization of immune checkpoint inhibitor-induced cardiotoxicity reveals interleukin-17A as a driver of cardiac dysfunction after anti-PD-1 treatment. Br. J. Pharmacol. 2023, 180, 740–761. [Google Scholar] [CrossRef]
- Zhang, C.; Wei, F.; Ma, W.; Zhang, J. Immune-related cardiovascular toxicities of PD-1/PD-L1 inhibitors in solid tumors: An updated systematic review and meta-analysis. Front. Immunol. 2024, 15, 1255825. [Google Scholar] [CrossRef]
- Rosen, E.; Kryndushkin, D.; Aryal, B.; Gonzalez, Y.; Chehab, L.; Dickey, J.; Rao, V.A. Acute total body ionizing gamma radiation induces long-term adverse effects and immediate changes in cardiac protein oxidative carbonylation in the rat. PLoS ONE 2020, 15, e0233967. [Google Scholar]
- Yi, P.; Li, H.; Su, J.; Cai, J.; Xu, C.; Chen, J.; Cao, L.; Li, M. Trastuzumab aggravates radiation induced cardiotoxicity in mice. Am. J. Cancer Res. 2022, 12, 381. [Google Scholar]
- Nardone, V.; Bruni, A.; Franceschini, D.; Marini, B.; Vagge, S.; Ciammella, P.; Sepulcri, M.; Cappelli, A.; D’Angelo, E.; De Marco, G.; et al. Adjuvant modern radiotherapy in resected pN2 NSCLC patients: Results from a multicentre retrospective analysis on acute and late toxicity on behalf of AIRO thoracic oncology study group: The RAC-TAC study. La Radiol. Medica 2024, 129, 1700–1709. [Google Scholar]
- Nardone, V.; Reginelli, A.; De Marco, G.; Natale, G.; Patanè, V.; De Chiara, M.; Buono, M.; Russo, G.M.; Monti, R.; Balestrucci, G.; et al. Role of Cardiac Biomarkers in Non-Small Cell Lung Cancer Patients. Diagnostics 2023, 13, 400. [Google Scholar] [CrossRef]
- Nardone, V.; Belfiore, M.P.; De Chiara, M.; De Marco, G.; Patanè, V.; Balestrucci, G.; Buono, M.; Salvarezza, M.; Di Guida, G.; D’Angiolella, D.; et al. CARdioimaging in Lung Cancer PatiEnts Undergoing Radical RadioTherapy: CARE-RT Trial. Diagnostics 2023, 13, 1717. [Google Scholar] [CrossRef]
- Bucknell, N.W.; Belderbos, J.; Palma, D.A.; Iyengar, P.; Samson, P.; Chua, K.; Gomez, D.; McDonald, F.; Louie, A.V.; Faivre-Finn, C.; et al. Avoiding Toxicity with Lung Radiation Therapy: An IASLC Perspective. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2022, 17, 961–973. [Google Scholar]
- Zhou, H.; Rodriguez, M.; van den Haak, F.; Nelson, G.; Jogani, R.; Xu, J.; Zhu, X.; Xian, Y.; Tran, P.T.; Felsher, D.W.; et al. Development of a micro-computed tomography-based image-guided conformal radiotherapy system for small animals. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 297–305. [Google Scholar]
- Tini, P.; Nardone, V.; Pastina, P.; Pirtoli, L.; Correale, P.; Giordano, A. The effects of radiotherapy on the survival of patients with unresectable non-small cell lung cancer. Expert Rev. Anticancer Ther. 2018, 18, 593–602. [Google Scholar]
- Schlaak, R.A.; Frei, A.; Schottstaedt, A.M.; Tsaih, S.W.; Fish, B.L.; Harmann, L.; Liu, Q.; Gasperetti, T.; Medhora, M.; North, P.E.; et al. Mapping genetic modifiers of radiation-induced cardiotoxicity to rat chromosome 3. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1267–H1280. [Google Scholar]
- Dreyfuss, A.D.; Velalopoulou, A.; Avgousti, H.; Bell, B.I.; Verginadis, I.I. Preclinical models of radiation-induced cardiac toxicity: Potential mechanisms and biomarkers. Front. Oncol. 2022, 12, 920867. [Google Scholar]
- Zei, P.C.; Wong, D.; Gardner, E.; Fogarty, T.; Maguire, P. Safety and efficacy of stereotactic radioablation targeting pulmonary vein tissues in an experimental model. Heart Rhythm. 2018, 15, 1420–1427. [Google Scholar] [PubMed]
- Lehmann, H.I.; Deisher, A.J.; Takami, M.; Kruse, J.J.; Song, L.; Anderson, S.E.; Cusma, J.T.; Parker, K.D.; Johnson, S.B.; Asirvatham, S.J.; et al. External Arrhythmia Ablation Using Photon Beams: Ablation of the Atrioventricular Junction in an Intact Animal Model. Circ. Arrhythmia Electrophysiol. 2017, 10, e004304. [Google Scholar]
- Tomlinson, L.; Lu, Z.Q.; Bentley, R.A.; Colley, H.E.; Murdoch, C.; Webb, S.D.; Cross, M.J.; Copple, I.M.; Sharma, P. Attenuation of doxorubicin-induced cardiotoxicity in a human in vitro cardiac model by the induction of the NRF-2 pathway. Biomed. Pharmacother. 2019, 112, 108637. [Google Scholar]
- Herman, E.H.; Ferrans, V.J. Preclinical animal models of cardiac protection from anthracycline-induced cardiotoxicity. Semin. Oncol. 1998, 25, 15–21. [Google Scholar] [PubMed]
- Herman, E.H.; El-Hage, A.N.; Ferrans, V.J.; Ardalan, B. Comparison of the severity of the chronic cardiotoxicity produced by doxorubicin in normotensive and hypertensive rats. Toxicol. Appl. Pharmacol. 1985, 78, 202–214. [Google Scholar]
- Zhu, W.; Shou, W.; Payne, R.M.; Caldwell, R.; Field, L.J. A mouse model for juvenile doxorubicin-induced cardiac dysfunction. Pediatr. Res. 2008, 64, 488–494. [Google Scholar]
- Moulin, M.; Piquereau, J.; Mateo, P.; Fortin, D.; Rucker-Martin, C.; Gressette, M.; Lefebvre, F.; Gresikova, M.; Solgadi, A.; Veksler, V. Sexual dimorphism of doxorubicin-mediated cardiotoxicity: Potential role of energy metabolism remodeling. Circ. Heart Fail. 2015, 8, 98–108. [Google Scholar]
- Oh, S.; Jung, J. Sex-dependent liver cancer xenograft models for predicting clinical data in the evaluation of anticancer drugs. Lab. Anim. Res. 2021, 37, 10. [Google Scholar] [CrossRef]
- Baker, K.; Warren, K.S.; Yellen, G.; Fishman, M.C. Defective “pacemaker” current (I h) in a zebrafish mutant with a slow heart rate. Proc. Natl. Acad. Sci. USA 1997, 94, 4554–4559. [Google Scholar] [CrossRef]
- Lane, S.; More, L.A.; Asnani, A. Zebrafish models of cancer therapy-induced cardiovascular toxicity. J. Cardiovasc. Dev. Dis. 2021, 8, 8. [Google Scholar] [CrossRef]
- Lam, P.Y.; Kutchukian, P.; Anand, R.; Imbriglio, J.; Andrews, C.; Padilla, H.; Vohra, A.; Lane, S.; Parker, D.L., Jr.; Cornella Taracido, I. Cyp1 inhibition prevents doxorubicin-induced cardiomyopathy in a zebrafish heart-failure model. Chembiochem 2020, 21, 1905–1910. [Google Scholar] [PubMed]
- Legi, A.; Rodriguez, E.; Eckols, T.K.; Mistry, C.; Robinson, P. Substance P antagonism prevents chemotherapy-induced cardiotoxicity. Cancers 2021, 13, 1732. [Google Scholar] [CrossRef] [PubMed]
- Galan-Arriola, C.; Vílchez-Tschischke, J.P.; Lobo, M.; Lopez, G.J.; de Molina-Iracheta, A.; Pérez-Martínez, C.; Villena-Gutierrez, R.; Macías, Á.; Díaz-Rengifo, I.A.; Oliver, E. Coronary microcirculation damage in anthracycline cardiotoxicity. Cardiovasc. Res. 2022, 118, 531–541. [Google Scholar] [CrossRef]
- Salloum, F.N.; Tocchetti, C.G.; Ameri, P.; Ardehali, H.; Asnani, A.; de Boer, R.A.; Burridge, P.; Cabrera, J.; de Castro, J.; Córdoba, R.; et al. Priorities in Cardio-Oncology Basic and Translational Science: GCOS 2023 Symposium Proceedings: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol. 2023, 5, 715–731. [Google Scholar] [CrossRef]
- Berthiaume, J.M.; Wallace, K.B. Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell Biol. Toxicol. 2007, 23, 15–25. [Google Scholar] [PubMed]
- Galán-Arriola, C.; Villena-Gutiérrez, R.; Higuero-Verdejo, M.I.; Díaz-Rengifo, I.A.; Pizarro, G.; López, G.J.; Molina-Iracheta, A.; Pérez-Martínez, C.; García, R.D.; González-Calle, D.; et al. Remote ischaemic preconditioning ameliorates anthracycline-induced cardiotoxicity and preserves mitochondrial integrity. Cardiovasc. Res. 2021, 117, 1132–1143. [Google Scholar] [CrossRef]
- Gulluni, F.; Prever, L.; Li, H.; Krafcikova, P.; Corrado, I.; Lo, W.T.; Margaria, J.P.; Chen, A.; De Santis, M.C.; Cnudde, S.J.; et al. PI(3,4)P2-mediated cytokinetic abscission prevents early senescence and cataract formation. Science 2021, 374, eabk0410. [Google Scholar]
- Ibáñez, B.; Gomes-Silva, M. Remote Ischemic Conditioning for Anthracycline Cardiotoxicity: The Need to Protect the Most Vulnerable. JACC CardioOncol. 2023, 5, 356–359. [Google Scholar]
- Galán-Arriola, C.; Lobo, M.; Vílchez-Tschischke, J.P.; López, G.J.; de Molina-Iracheta, A.; Pérez-Martínez, C.; Agüero, J.; Fernández-Jiménez, R.; Martín-García, A.; Oliver, E.; et al. Serial Magnetic Resonance Imaging to Identify Early Stages of Anthracycline-Induced Cardiotoxicity. J. Am. Coll. Cardiol. 2019, 73, 779–791. [Google Scholar]
- Gómez-Talavera, S.; Fernandez-Jimenez, R.; Fuster, V.; Nothnagel, N.D.; Kouwenhoven, M.; Clemence, M.; García-Lunar, I.; Gómez-Rubín, M.C.; Navarro, F.; Pérez-Asenjo, B.; et al. Clinical Validation of a 3-Dimensional Ultrafast Cardiac Magnetic Resonance Protocol Including Single Breath-Hold 3-Dimensional Sequences. JACC Cardiovasc. Imaging 2021, 14, 1742–1754. [Google Scholar]
- Del Buono, M.G.; Montone, R.A.; Camilli, M.; Carbone, S.; Narula, J.; Lavie, C.J.; Niccoli, G.; Crea, F. Coronary Microvascular Dysfunction Across the Spectrum of Cardiovascular Diseases: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 78, 1352–1371. [Google Scholar] [PubMed]
- Luu, A.Z.; Chowdhury, B.; Al-Omran, M.; Teoh, H.; Hess, D.A.; Verma, S. Role of Endothelium in Doxorubicin-Induced Cardiomyopathy. JACC Basic Transl. Sci. 2018, 3, 861–870. [Google Scholar] [PubMed]
- Quagliariello, V.; Passariello, M.; Rea, D.; Barbieri, A.; Iovine, M.; Bonelli, A.; Caronna, A.; Botti, G.; De Lorenzo, C.; Maurea, N. Evidences of CTLA-4 and PD-1 blocking agents-induced cardiotoxicity in cellular and preclinical models. J. Pers. Med. 2020, 10, 179. [Google Scholar] [CrossRef] [PubMed]
- Quagliariello, V.; Passariello, M.; Coppola, C.; Rea, D.; Barbieri, A.; Scherillo, M.; Monti, M.G.; Iaffaioli, R.V.; De Laurentiis, M.; Ascierto, P.A. Cardiotoxicity and pro-inflammatory effects of the immune checkpoint inhibitor Pembrolizumab associated to Trastuzumab. Int. J. Cardiol. 2019, 292, 171–179. [Google Scholar]
- Wei, S.C.; Meijers, W.C.; Axelrod, M.L.; Anang, N.A.S.; Screever, E.M.; Wescott, E.C.; Johnson, D.B.; Whitley, E.; Lehmann, L.; Courand, P.Y.; et al. A Genetic Mouse Model Recapitulates Immune Checkpoint Inhibitor-Associated Myocarditis and Supports a Mechanism-Based Therapeutic Intervention. Cancer Discov. 2021, 11, 614–625. [Google Scholar]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar]
- Moslehi, J.J.; Salem, J.E.; Sosman, J.A.; Lebrun-Vignes, B.; Johnson, D.B. Increased reporting of fatal immune checkpoint inhibitor-associated myocarditis. Lancet 2018, 391, 933. [Google Scholar]
- Axelrod, M.L.; Meijers, W.C.; Screever, E.M.; Qin, J.; Carroll, M.G.; Sun, X.; Tannous, E.; Zhang, Y.; Sugiura, A.; Taylor, B.C.; et al. T cells specific for α-myosin drive immunotherapy-related myocarditis. Nature 2022, 611, 818–826. [Google Scholar]
- Thomas, D.; Choi, S.; Alamana, C.; Parker, K.K.; Wu, J.C. Cellular and Engineered Organoids for Cardiovascular Models. Circ. Res. 2022, 130, 1780–1802. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Results of the Study | References |
|---|---|---|
| Primary Cardiomyocytes | Doxorubicin impairs contractility, increases oxidative stress, and induces protein accumulation in cardiomyocytes, causing cardiac dysfunction | [38,39,40,55,56,57] |
| Established Cell Lines | H9C2 cells, resembling cardiomyocytes with skeletal traits, are a better model than HL-1 cells for studying antineoplastic cardiotoxicity | [41,43,44,45,58,59,60] |
| Human Pluripotent Stem Cells | Doxorubicin is more cardiotoxic than erlotinib; TKIs have varying effects, and severe cardiac dysfunction increases trastuzumab risk | [51,53,61,62,63] |
| Animal | Description | References |
|---|---|---|
| Mice/Rats | Explored RIHD pathways, radiation-induced heart disease, and inflammation-related injury | [68,69,70,72,73,75,77,78,79,80,81] |
| Rabbits | Model for cardiovascular and radiation studies with distinct physiology from mice | [84,85,87,88,89,97,98] |
| Canines | Useful for cardiac radiation studies but limited by cost, regulations, and ethical concerns | [90,91,92,93,99] |
| Pigs/Nonhuman Primates | Common RIHD effects, but limited by higher costs | [94,95,96,100,101] |
| Mechanism | Description | References |
|---|---|---|
| Radiation-Induced Myocardial Fibrosis (RIMF) | Chronic condition with excess collagen in heart tissue causes stiffening, reduced function, and symptoms such as shortness of breath, fatigue, and chest pain | [103,104,105,124,125,126] |
| Deposition of Collagen and Actin Stress Fibers | Increased myocardial thickness results from collagen deposition and actin stress fibers in collagen-producing myofibroblasts | [106,107,127,128,129,130] |
| Inflammation and Tissue Damage | Radiation exposure causes oxidative stress, inflammation, and immune cell activation, leading to tissue fibrosis and impaired heart function | [110,111,112,131,132,133,134,135] |
| Fibrosis and Impaired Heart Function | Myofibroblasts induce fibrosis, stiffening the heart and reducing efficiency, potentially causing cardiac failure | [117,118,119,136,137] |
| Agent | Description | References |
|---|---|---|
| Anthracyclines | Anthracyclines can cause cardiopathy through oxidative stress, DNA damage, and inhibition of DNA repair | [138,153,154,155,156] |
| Fluoropyrimidines | Fluoropyrimidines can cause cardiotoxicity in about 30% of patients by inhibiting pyrimidine nucleotide biosynthesis | [140,157,158,159,160,161] |
| Alkylating agents | Alkylating agents can cause cardiac damage, particularly at higher doses, by affecting DNA transcription and protein synthesis | [162,163,164,165] |
| Microtubular polymerization inhibitors | Microtubular polymerization inhibitors can increase the risk of heart failure in patients undergoing anthracycline therapy | [142,166,167,168,169,170] |
| Anti-HER2 therapy | Trastuzumab-based anti-HER2 therapy can cause cardiac toxicity, particularly when combined with anthracyclines | [146,147,148,171,172,173,174,175,176] |
| VEGF inhibitors | VEGF inhibitors can cause hypertension and atherosclerosis by affecting angiogenesis | [149,151,152,177,178,179,180,181] |
| Mechanism | Description | References |
|---|---|---|
| Direct cellular destruction of cardiac tissue | ICI therapy disrupts cardiac immune balance, causing autoimmune toxicity with increased PD-L1 expression and T lymphocyte infiltration in myocarditis | [184,185,186,188,189,191,192,193,195,196,197] |
| Cardiac antigen immune reactivity | Myocarditis can result from disrupted molecular mimicry and autoantibodies induced by ICI therapy, leading to myocardial dysfunction | [188,189,198,199,200,201,202] |
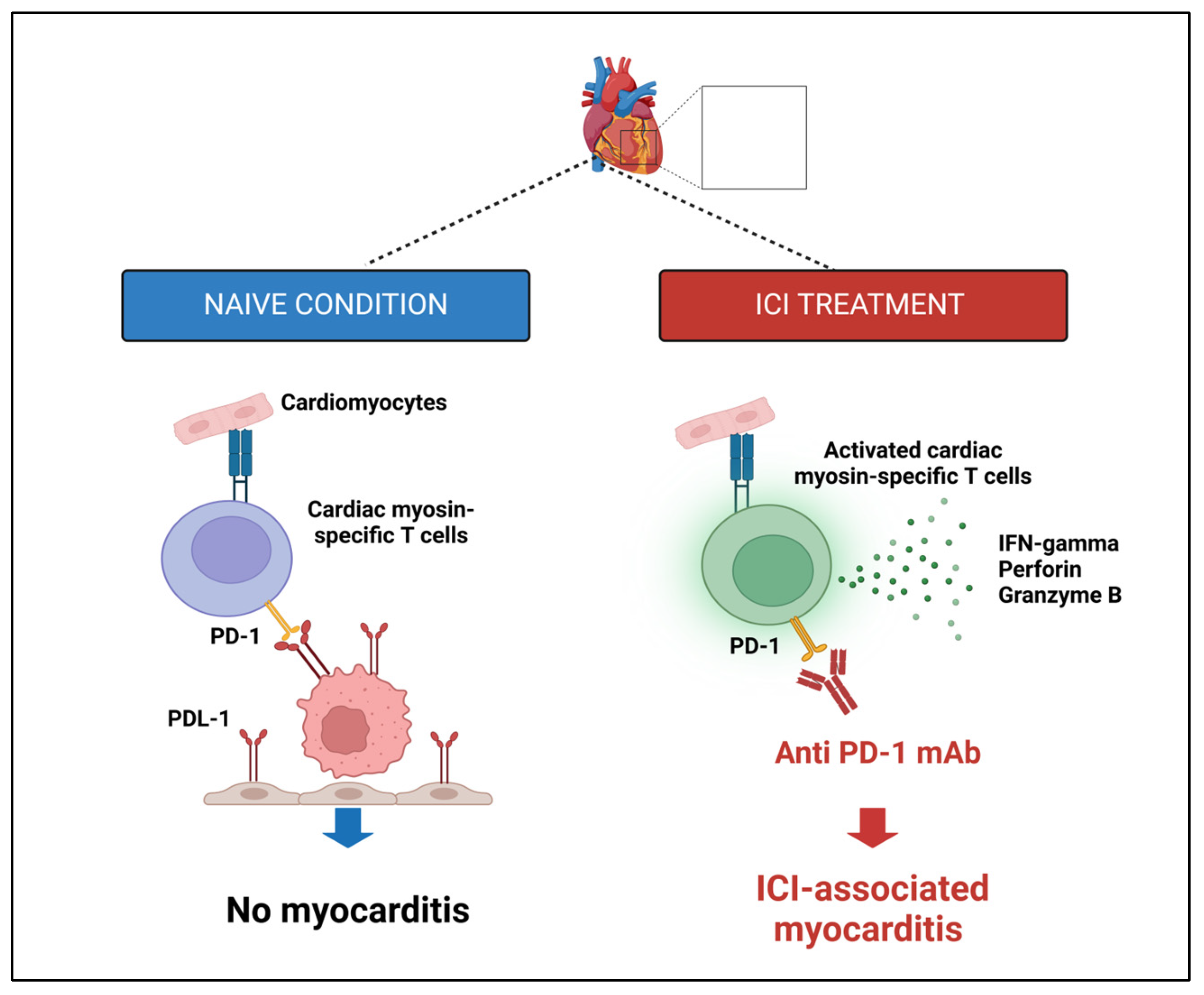
| ICI-induced cytokines release | Therapies activating T-cell subsets can cause cytokine release syndrome (CRS), with pro-inflammatory cytokines and radicals damaging cardiac myocytes and causing cardiac anomalies | [190,203,204,205,206,207] |
| Dysregulation of myocardial metabolism | Anti-PD1 therapy can disrupt cardiomyocyte metabolism, affecting lipid/glucose metabolism mitochondrial function, and contributing to myocardial dysfunction and cell death | [192,193,194,208,209,210,211] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nardone, V.; Ruggiero, D.; Chini, M.G.; Bruno, I.; Lauro, G.; Terracciano, S.; Nebbioso, A.; Bifulco, G.; Cappabianca, S.; Reginelli, A. From Bench to Bedside: Translational Approaches to Cardiotoxicity in Breast Cancer, Lung Cancer, and Lymphoma Therapies. Cancers 2025, 17, 1059. https://doi.org/10.3390/cancers17071059
Nardone V, Ruggiero D, Chini MG, Bruno I, Lauro G, Terracciano S, Nebbioso A, Bifulco G, Cappabianca S, Reginelli A. From Bench to Bedside: Translational Approaches to Cardiotoxicity in Breast Cancer, Lung Cancer, and Lymphoma Therapies. Cancers. 2025; 17(7):1059. https://doi.org/10.3390/cancers17071059
Chicago/Turabian StyleNardone, Valerio, Dafne Ruggiero, Maria Giovanna Chini, Ines Bruno, Gianluigi Lauro, Stefania Terracciano, Angela Nebbioso, Giuseppe Bifulco, Salvatore Cappabianca, and Alfonso Reginelli. 2025. "From Bench to Bedside: Translational Approaches to Cardiotoxicity in Breast Cancer, Lung Cancer, and Lymphoma Therapies" Cancers 17, no. 7: 1059. https://doi.org/10.3390/cancers17071059
APA StyleNardone, V., Ruggiero, D., Chini, M. G., Bruno, I., Lauro, G., Terracciano, S., Nebbioso, A., Bifulco, G., Cappabianca, S., & Reginelli, A. (2025). From Bench to Bedside: Translational Approaches to Cardiotoxicity in Breast Cancer, Lung Cancer, and Lymphoma Therapies. Cancers, 17(7), 1059. https://doi.org/10.3390/cancers17071059