

Presence of On-Target Resistant Mutation in Pre-Treatment Samples of ALK Fusion Gene Positive Lung Cancer Patients
 , , , , ,
, , , , ,  and
and 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Selection, Tumor and Control Samples
2.2. Generation of Positive Controls for ddPCR
2.3. Droplet Digital (dd)PCR
2.4. Statistical Analysis
3. Results
3.1. Patient Selection and Clinical Characteristics
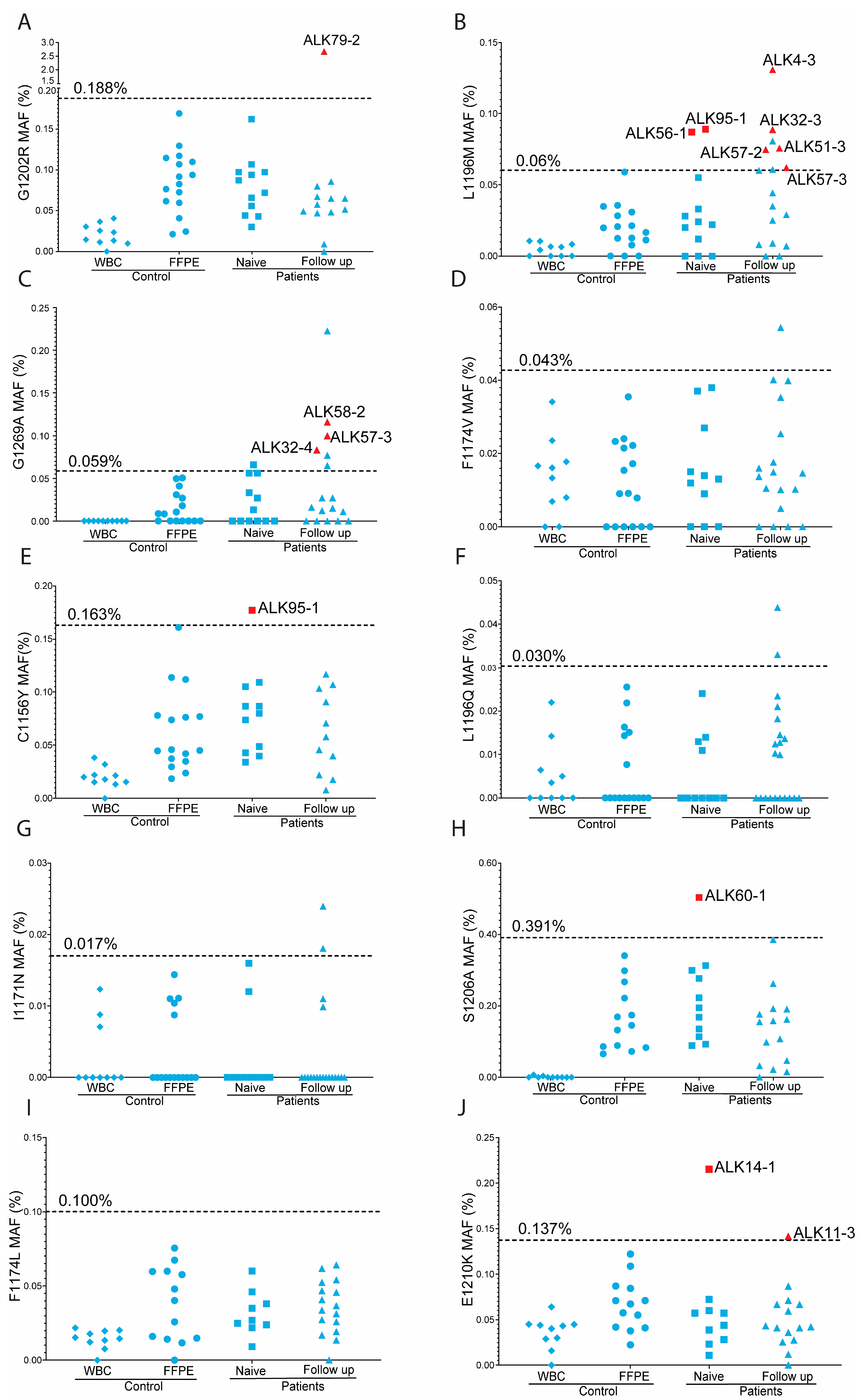
3.2. Positive Controls and Cut-Off Values per Assay
3.3. Overview of Overall ddPCR Results
3.4. On-Target ALKi Resistant Mutations in Treatment-Naïve Samples
3.5. On-Target ALKi Resistant Mutations in Relapsed Samples
3.6. Predictive Value of Minor Clones with On-Target ALKi Resistant Mutations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar]
- Allouche, M. ALK is a novel dependence receptor: Potential implications in development and cancer. Cell Cycle 2007, 6, 1533–1538. [Google Scholar]
- McDermott, U.; Iafrate, A.J.; Gray, N.S.; Shioda, T.; Classon, M.; Maheswaran, S.; Zhou, W.; Choi, H.G.; Smith, S.L.; Dowell, L.; et al. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res. 2008, 68, 3389–3395. [Google Scholar] [CrossRef]
- Kelleher, F.C.; McDermott, R. The emerging pathogenic and therapeutic importance of the anaplastic lymphoma kinase gene. Eur. J. Cancer 2010, 46, 2357–2368. [Google Scholar]
- Mano, H. Non-solid oncogenes in solid tumors: EML4-ALK fusion genes in lung cancer. Cancer Sci. 2008, 99, 2349–2355. [Google Scholar]
- FDA Approves Xalkori with Companion Diagnostic for a Type of Late-Stage Lung Cancer. Available online: https://www.prnewswire.com/news-releases/fda-approves-xalkori-with-companion-diagnostic-for-a-type-of-late-stage-lung-cancer-128484413.html (accessed on 21 January 2024).
- Kwak, E.L.; Bang, Y.J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.H.; Dezube, B.J.; Janne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar]
- Solomon, B.J.; Mok, T.; Kim, D.W.; Wu, Y.L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar]
- Recondo, G.; Facchinetti, F.; Olaussen, K.A.; Besse, B.; Friboulet, L. Making the first move in EGFR-driven or ALK-driven NSCLC: First-generation or next-generation TKI? Nat. Rev. Clin. Oncol. 2018, 15, 694–708. [Google Scholar]
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crino, L.; Ahn, M.J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar]
- Lin, J.J.; Riely, G.J.; Shaw, A.T. Targeting ALK: Precision Medicine Takes on Drug Resistance. Cancer Discov. 2017, 7, 137–155. [Google Scholar] [CrossRef]
- Dhillon, S.; Clark, M. Ceritinib: First global approval. Drugs 2014, 74, 1285–1291. [Google Scholar]
- Gettinger, S.N.; Bazhenova, L.A.; Langer, C.J.; Salgia, R.; Gold, K.A.; Rosell, R.; Shaw, A.T.; Weiss, G.J.; Tugnait, M.; Narasimhan, N.I.; et al. Activity and safety of brigatinib in ALK-rearranged non-small-cell lung cancer and other malignancies: A single-arm, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 1683–1696. [Google Scholar]
- Syed, Y.Y. Lorlatinib: First Global Approval. Drugs 2019, 79, 93–98. [Google Scholar]
- Solomon, B.J.; Liu, G.; Felip, E.; Mok, T.S.K.; Soo, R.A.; Mazieres, J.; Shaw, A.T.; de Marinis, F.; Goto, Y.; Wu, Y.L.; et al. Lorlatinib Versus Crizotinib in Patients With Advanced ALK-Positive Non-Small Cell Lung Cancer: 5-Year Outcomes From the Phase III CROWN Study. J. Clin. Oncol. 2024, 42, 3400–3409. [Google Scholar]
- Tabbo, F.; Reale, M.L.; Bironzo, P.; Scagliotti, G.V. Resistance to anaplastic lymphoma kinase inhibitors: Knowing the enemy is half the battle won. Transl. Lung Cancer Res. 2020, 9, 2545–2556. [Google Scholar]
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci. Transl. Med. 2012, 4, 120ra117. [Google Scholar]
- Doebele, R.C.; Pilling, A.B.; Aisner, D.L.; Kutateladze, T.G.; Le, A.T.; Weickhardt, A.J.; Kondo, K.L.; Linderman, D.J.; Heasley, L.E.; Franklin, W.A.; et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 1472–1482. [Google Scholar] [CrossRef]
- Li, W.; Kok, K.; Tan, G.W.; Meng, P.; Mastik, M.; Rifaela, N.; Scherpen, F.; Hiltermann, T.J.N.; Groen, H.J.M.; Wekken, A.J.V.; et al. The Presence of EGFR T790M in TKI-Naive Lung Cancer Samples of Patients Who Developed a T790M-Positive Relapse on First or Second Generation TKI Is Rare. Cancers 2022, 14, 3511. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Sidorenkov, G.; Nagel, J.; Meijer, C.; Duker, J.J.; Groen, H.J.M.; Halmos, G.B.; Oonk, M.H.M.; Oostergo, R.J.; van der Vegt, B.; Witjes, M.J.H.; et al. The OncoLifeS data-biobank for oncology: A comprehensive repository of clinical data, biological samples, and the patient’s perspective. J. Transl. Med. 2019, 17, 374. [Google Scholar]
- Koopman, B.; Groen, H.J.M.; Schuuring, E.; Hiltermann, T.J.N.; Timens, W.; den Dunnen, W.F.A.; van den Berg, A.; Ter Elst, A.; van Kruchten, M.; Kluiver, J.L.; et al. Actionability of on-target alK Resistance Mutations in Patients With Non-Small Cell Lung Cancer: Local Experience and Review of the Literature. Clin. Lung Cancer 2022, 23, e104–e115. [Google Scholar]
- Jamme, P.; Descarpentries, C.; Gervais, R.; Dansin, E.; Wislez, M.; Gregoire, V.; Richard, N.; Baldacci, S.; Rabbe, N.; Kyheng, M.; et al. Relevance of Detection of Mechanisms of Resistance to ALK Inhibitors in ALK-Rearranged NSCLC in Routine Practice. Clin. Lung Cancer 2019, 20, 297–304.e291. [Google Scholar]
- O’Leary, B.; Hrebien, S.; Beaney, M.; Fribbens, C.; Garcia-Murillas, I.; Jiang, J.; Li, Y.; Huang Bartlett, C.; Andre, F.; Loibl, S.; et al. Comparison of BEAMing and Droplet Digital PCR for Circulating Tumor DNA Analysis. Clin. Chem. 2019, 65, 1405–1413. [Google Scholar]
- Milosevic, D.; Mills, J.R.; Campion, M.B.; Vidal-Folch, N.; Voss, J.S.; Halling, K.C.; Highsmith, W.E.; Liu, M.C.; Kipp, B.R.; Grebe, S.K.G. Applying Standard Clinical Chemistry Assay Validation to Droplet Digital PCR Quantitative Liquid Biopsy Testing. Clin. Chem. 2018, 64, 1732–1742. [Google Scholar]
- Arnolda, R.; Howlett, K.; Chan, T.; Raleigh, J.; Hatzimihalis, A.; Bell, A.; Fellowes, A.; Sandhu, S.; McArthur, G.A.; Fox, S.B.; et al. Clinical validation and implementation of droplet digital PCR for the detection of BRAF mutations from cell-free DNA. Pathology 2022, 54, 772–778. [Google Scholar] [CrossRef]
- Lettig, L.; Sahnane, N.; Pepe, F.; Cerutti, R.; Albeni, C.; Franzi, F.; Veronesi, G.; Ogliari, F.; Pastore, A.; Tuzi, A.; et al. EGFR T790M detection rate in lung adenocarcinomas at baseline using droplet digital PCR and validation by ultra-deep next generation sequencing. Transl. Lung Cancer Res. 2019, 8, 584–592. [Google Scholar] [CrossRef]
- Pan, Y.; Deng, C.; Qiu, Z.; Cao, C.; Wu, F. The Resistance Mechanisms and Treatment Strategies for ALK-Rearranged Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 713530. [Google Scholar]
- Dagogo-Jack, I.; Shaw, A.T. Crizotinib resistance: Implications for therapeutic strategies. Ann. Oncol. 2016, 27 (Suppl. S3), iii42–iii50. [Google Scholar]
- Diaz-Cano, S.J. Tumor heterogeneity: Mechanisms and bases for a reliable application of molecular marker design. Int. J. Mol. Sci. 2012, 13, 1951–2011. [Google Scholar] [CrossRef]
- Nakahara, Y.; Shimokawa, T.; Misumi, Y.; Nogami, N.; Shinkai, T.; Seki, N.; Hosomi, Y.; Hida, N.; Okamoto, H. Phase I/II study of erlotinib plus S-1 for patients with previously treated non-small cell lung cancer: Thoracic Oncology Research Group (TORG) 0808/0913. Investig. New Drugs 2021, 39, 202–209. [Google Scholar]
- Lodrini, M.; Wunschel, J.; Thole-Kliesch, T.M.; Grimaldi, M.; Sprussel, A.; Linke, R.B.; Hollander, J.F.; Tiburtius, D.; Kunkele, A.; Schulte, J.H.; et al. Circulating Cell-Free DNA Assessment in Biofluids from Children with Neuroblastoma Demonstrates Feasibility and Potential for Minimally Invasive Molecular Diagnostics. Cancers 2022, 14, 2080. [Google Scholar] [CrossRef] [PubMed]
- Leest, P.V.; Janning, M.; Rifaela, N.; Azpurua, M.L.A.; Kropidlowski, J.; Loges, S.; Lozano, N.; Sartori, A.; Irwin, D.; Lamy, P.J.; et al. Detection and Monitoring of Tumor-Derived Mutations in Circulating Tumor DNA Using the UltraSEEK Lung Panel on the MassARRAY System in Metastatic Non-Small Cell Lung Cancer Patients. Int. J. Mol. Sci. 2023, 24, 13390. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Spiegl, B.; Perakis, S.O.; Ulz, C.M.; Abuja, P.M.; Kashofer, K.; Leest, P.V.; Azpurua, M.A.; Tamminga, M.; Brudzewsky, D.; et al. Technical Evaluation of Commercial Mutation Analysis Platforms and Reference Materials for Liquid Biopsy Profiling. Cancers 2020, 12, 1588. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Patient Characteristics | Number of Patients |
|---|---|
| Male/Female | 4/13 |
| Median Age (range) | 64 (34–74) |
| Smoking history | |
| Never | 13 |
| Former | 3 |
| Unknown | 1 |
| ALK fusion | |
| EML4:ALK20 | 9 |
| IHC or FISH positive | 8 |
| Initial stage at diagnosis | |
| IIIA | 2 |
| IIIB | 1 |
| IVA | 4 |
| IVB | 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Zwierenga, F.; Andini, K.D.; Bucher, J.M.; Scherpen, F.; Hiltermann, T.J.N.; Groen, H.J.M.; van der Wekken, A.J.; Kok, K.; Berg, A.v.d. Presence of On-Target Resistant Mutation in Pre-Treatment Samples of ALK Fusion Gene Positive Lung Cancer Patients. Cancers 2025, 17, 1090. https://doi.org/10.3390/cancers17071090
Li W, Zwierenga F, Andini KD, Bucher JM, Scherpen F, Hiltermann TJN, Groen HJM, van der Wekken AJ, Kok K, Berg Avd. Presence of On-Target Resistant Mutation in Pre-Treatment Samples of ALK Fusion Gene Positive Lung Cancer Patients. Cancers. 2025; 17(7):1090. https://doi.org/10.3390/cancers17071090
Chicago/Turabian StyleLi, Weiting, Fenneke Zwierenga, Katarina D. Andini, Justyna M. Bucher, Frank Scherpen, T. Jeroen N. Hiltermann, Harry J. M. Groen, Anthonie J. van der Wekken, Klaas Kok, and Anke van den Berg. 2025. "Presence of On-Target Resistant Mutation in Pre-Treatment Samples of ALK Fusion Gene Positive Lung Cancer Patients" Cancers 17, no. 7: 1090. https://doi.org/10.3390/cancers17071090
APA StyleLi, W., Zwierenga, F., Andini, K. D., Bucher, J. M., Scherpen, F., Hiltermann, T. J. N., Groen, H. J. M., van der Wekken, A. J., Kok, K., & Berg, A. v. d. (2025). Presence of On-Target Resistant Mutation in Pre-Treatment Samples of ALK Fusion Gene Positive Lung Cancer Patients. Cancers, 17(7), 1090. https://doi.org/10.3390/cancers17071090