Simple Summary
This review focuses on breast cancer, a common disease that remains difficult to treat due to its heterogeneity and resistance to therapy. Here, we aim to examine the different molecular subtypes of breast cancer and how their differences impact treatment success. Moreover, we highlight that combining therapies, rather than using a single drug, can be more effective in overcoming resistance and reducing the risk of cancer returning. By analyzing the biological characteristics of tumors using advanced techniques like single-cell sequencing, this study aims to elucidate the benefits of personalized treatment strategies. The findings contribute to the medical community by promoting precision medicine, offering insight into why some treatments fail and suggesting new ways to enhance cancer therapies.
Abstract
Breast cancer remains one of the most prevalent diseases worldwide, primarily affecting women. Its heterogeneous nature poses a significant challenge in the development of effective and targeted treatments. Molecular characterization has enabled breast cancer to be classified into four main subtypes: luminal A, luminal B, HER2-positive, and triple-negative breast cancer, based on hormone receptor expression and HER2 status. A deeper understanding of these molecular markers and their associated signaling pathways, such as MAPK and PI3K/AKT, is essential for improving prognosis and optimizing treatment strategies. Currently, several therapeutic agents are utilized in neoadjuvant and adjuvant therapies, often in combination with surgical interventions. However, emerging evidence highlights the growing challenge of drug resistance, which significantly limits the efficacy of existing treatments. Addressing this issue may require innovative approaches, including combination therapies and precision medicine strategies, tailored to the molecular profile of each patient. Therefore, a comprehensive understanding of the pathophysiologic mechanisms driving breast cancer progression and resistance is crucial for the development of advanced targeted therapies with greater precision and efficacy. This review aims to explore recent advancements in molecular research related to breast cancer subtypes and provide a critical analysis of current therapeutic approaches within the framework of precision medicine.
1. Introduction
Breast cancer remains the most common malignancy among women worldwide, affecting 2.3 million individuals and causing 670,000 deaths in 2022 alone. With an incidence rate of 8954 per 100,000 women in Portugal, as shown in Figure 1, this disease continues to pose a major public health [1,2,3]. Despite depending on the gender, both incidence and mortality rates depend on age, ethnicity and geographics. Typically, women with African descent have lower incidence but higher mortality rate in comparison to Caucasian women, which can be attributed to differences in the access of healthcare treatments [4]. Comparing countries with high, moderate and low income, the first have a higher incidence of breast cancer than low-income countries, possibly due to better screening programs that allow cancer to be detected earlier. On the contrary, countries with low income have a higher mortality rate due to the lack of screening programs and access to healthcare [5].
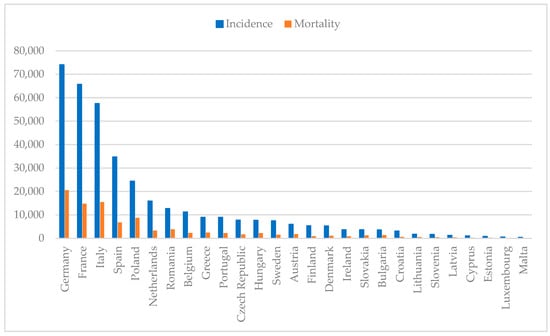
Figure 1.
Estimated breast cancer numbers of incidence and mortality in European countries per 100,000 population in 2022 (developed from 1–3).
The spectrum of risk factors associated with this disease is wide and diverse, going from genetic to environmental factors. Although gender and ethnicity are major risk factors for breast cancer, age is considered as one of the most important. Studies indicate a higher incidence of breast cancer in older women ranging from 55 to 64 years [4,6,7]. Respectively, new guidelines were approved to start the screening programs earlier than 50 years [8,9]. Younger women present a glandular-fibrous tissue which have a radiopaque appearance on a mammogram making more difficult to detect microcalcifications, while older women have more fatty tissue, which facilitates detection [7,10]. Studies demonstrated that as women grow older, tissue morphology changes and becomes fattier, and denser breast tissue poses a higher risk of developing breast cancer [11,12]. For these women, the guidelines are changing, and they are now eligible for magnetic resonance imaging (MRI) as strong data have emerged supporting a reduction in breast cancer mortality [13,14].
Furthermore, the personal and family history of breast cancer poses an increased predisposition for developing breast cancer. The personal history of breast cancer represents an increased risk of 2- to 6-fold of having breast cancer recurrence and the second malignancy may appear in a different breast as the first one or in the same [10,15]. Family history of breast cancer is responsible for 5% to 10% of breast cancer cases and studies have shown that women with blood relatives with breast cancer have a higher risk of developing breast cancer [16]. The same study demonstrated that women with two or more relatives diagnosed with breast cancer under the age of 45 are more susceptible to develop breast cancer [16,17]. In line with this, genetic factors play an important role in the predisposition of breast cancer. This is often related to inherited genetic variations that besides increasing the risk of cancer on their own, contribute to an increased familial risk [18]. The most common mutations studied and associated with breast carcinogenesis are BRCA1 and BRCA2 mutations, which increase the risk of cancer up to 40% [19]. These genes are known to regulate important mechanisms concerning cell growth and DNA damage repair; thus, a mutation in BRCA1 and BRCA2 is often related to genomic instability, as cells continue to proliferate despite having DNA damage, which promotes breast cancer [20]. Other mutations, such as TP53 and PTEN genes, also contribute to cell cycle irregularities and DNA repair dysregulation [21].
During a women’s reproductive years, she undergoes estrogen and progesterone fluctuations, increasing the risk of breast cancer. The more variations, the higher the risk, and this is largely explained by an early menarche and a late menopause [22]. As estrogen increases cell growth and proliferation, there is a higher probability of DNA replication errors and a reduction in DNA repair that result in cumulative alterations passed down into daughter cells that facilitate breast cancer development [23]. On the contrary, pregnancy and breastfeeding are considered protective factors of breast cancer; however, later pregnancy after the age of 35 is associated with increased risk [24,25,26]. Moreover, exogenous estrogen derived from hormonal replacement therapies or from oral contraceptives has been associated with an increased risk of breast cancer. Regarding the administration of hormonal replacement therapies, there is still some contradictory findings on whether it has a protective effect or if contributes to a higher risk of breast cancer [23,27]. A recent study demonstrated that women who were administered menopausal hormonal therapy have a higher risk of developing cancer in comparison to those that never used this therapy and revealed that the combination of estrogen with other molecules presents a higher risk than the administration of estrogen alone [28]. In addition, the usage of oral contraceptives also prolongs the exposure to estrogen, increasing the susceptibility for breast cancer development. Studies indicate that women that use oral contraceptives before the first full-term pregnancy and for more than 5 years have a significant increased risk of breast cancer [29,30].
Some environmental and lifestyle risk factors, including obesity, alcohol consumption, tobacco smoke and physical inactivity, enhance the risk of breast cancer. After menopause, the primary source of estrogen production is adipose tissue; therefore, obesity (body mass index > 30 [31]) represents a major risk factor for breast cancer [32]. The elevated levels of this hormone promote rapid cell proliferation and the overcoming of damage checkpoints, enabling mutations and DNA damage to be transmitted to daughter cells as cells have less time to correct the errors [33]. An increased alcohol consumption is associated with an increase in breast cancer risk [34]. It elevates estrogen levels in the blood, which triggers estrogen-related pathways and potentiates estrogen receptors’ production [35]. On the other hand, tobacco smoking is a major cause of several cancers [36], but there is still a controversial debate regarding its carcinogenic impact on breast cancer [37]. Recently, some studies indicated that tobacco smoking promotes breast cancer on premenopausal women, whereas it has no impact on postmenopausal women [38,39]. This may be explained by the increased estrogen levels in premenopausal women that potentiate the carcinogenic activity, while in postmenopausal women, estrogen levels decrease, thereby favoring anticarcinogenic effects [40]. Alongside these modifiable factors, physical inactivity is also associated with increased breast cancer risk [41]. A sedentary lifestyle together with a diet rich in fats contributes to obesity, which promotes breast cancer [42].
Recently, work shifts have been receiving new attention due to recent findings on increasing the risk of breast cancer development [43,44]. Working shift rotations disrupt the circadian rhythm and cause daily activities alterations [45]. The circadian rhythm works as a biological clock in the recognition of the dark–light cycle through the secretion of the melatonin hormone, helping to control the sleep cycle [46]. Findings suggest that melatonin exerts an inhibitory effect on estrogen levels; thus, an exposure to light during nighttime causes women who work night shifts to produce more estrogen than women who do not, increasing the breast cancer risk [47,48,49]. In addition, some studies point out that intensity, frequency and type of shift rotation may increase the risk [45,46,50].
Breast cancer classification is based on both morphological and molecular characteristics. Morphologically, tumors are categorized by their degree of invasiveness and site of origin within the breast tissue. Non-invasive breast cancer is confined to the epithelial structures of the mammary ducts or lobules, without invasion of the surrounding stroma [51]. This category includes ductal carcinoma in situ (DCIS) and lobular carcinoma in situ (LCIS), with DCIS representing neoplastic proliferation within the ductal system and LCIS involving lobular epithelium without stromal invasion [52,53].
In contrast, invasive breast carcinomas exhibit stromal infiltration and have the potential for regional and distant dissemination [54]. The most prevalent histotype, invasive ductal carcinoma (IDC), arises within the ducts and infiltrates adjacent breast parenchyma, often progressing to lymphovascular or systemic metastases [55]. Other subtypes include invasive lobular carcinoma (ILC), characterized by a diffuse growth pattern due to loss of E-cadherin expression [56], and inflammatory breast cancer (IBC), an aggressive variant in which dermal lymphatics are obstructed by neoplastic emboli, leading to erythema, peau d’orange and rapid progression [57]. It is noteworthy that 99% of breast carcinomas arise from ductal–lobular transition and the difference between IDC and ILC is mainly morphological and genetic. In addition, there are other types of breast cancer that are less common, such as metaplastic carcinoma, apocrine carcinoma, mucinous carcinoma, cribriform carcinoma, tubular carcinoma and neuroendocrine carcinoma [58,59].
Beyond morphological classification, molecular subtyping—based on hormone receptor status and human epidermal growth factor receptor 2 (HER2) expression—has become fundamental in guiding treatment strategies and prognostication. Advancements in molecular techniques have allowed researchers to subgroup different breast cancers, giving rise to luminal A, luminal B, HER2-positive and triple-negative breast cancer (TNBC) [60]. Luminal A refers to the presence of both hormone receptors, such as estrogen receptors (ER) and progesterone receptors (PR) in tumors; thus, this subtype is often called ER-positive. On the other hand, luminal B presents ER and may express PR, in addition to possibly expressing HER2 receptors and has a higher proliferation index than luminal A. HER2-positive breast cancer is characterized by the absence of hormone receptors and the presence of only HER2 receptors, and TNBC does not express any of these receptors, being the most aggressive subtype. This new categorization provides a new insight into the disease phenotype and prognosis, as well as guiding physicians in treatment options [59,61].
After breast cancer classification, tumor characteristics and biological behavior are assessed, helping physicians select the best treatment option among the existing ones including surgery, chemotherapy, radiotherapy, immunotherapy, hormone therapy and targeted therapies [62,63,64]. Nevertheless, the different characteristics of the tumor within the same patient (intra-heterogeneity) and between patients (inter-heterogeneity) along time and space (temporal and spatial heterogeneity) represent one of the biggest pitfalls in breast cancer treatment [65]. Intra-tumor heterogeneity refers to the presence of diverse cancer cell populations within the same tumor with distinct genetic, molecular and phenotypic aspects and it is often seen between the primary tumor and the metastatic lesion [65,66], while inter-tumor heterogeneity pertains to the difference between tumors in different patients driven by genetic mutations, molecular subtypes and hormone status [65]. In addition, genetic and molecular variations within the primary tumor or between the primary tumor and metastatic lesions across time and depending on the presence of therapy contribute to spatial and temporal heterogeneity to the tumor [67]. The manipulation of tumor genetics and epigenetics contributes to different heterogeneities, causing phenotypic alterations that may lead to therapy resistance [68,69].
Indeed, drug resistance poses a major challenge for breast cancer therapy, and it may lead to patient relapses. It can be acquired, and it is only detected after several failed treatment administrations, or it can be intrinsic and the therapy does not improve the patient’s survival [70]. Even though therapy resistance has been a problem in traditional therapies, it has been an emerging challenge in targeted therapies that target specific molecules and key signaling pathways due to epigenetic alterations, tumor heterogeneity and microenvironment, enhanced DNA repair and drug efflux [71,72]. Sometimes, this leads to treatment persistence or overtreatment resulting in patient toxicity and multiple side effects. Chemotherapy and radiotherapy usually lead to neurotoxicity, cardiotoxicity and nephrotoxicity besides encompassing side effects on the gastrointestinal and genitourinary tracts, lungs and skin, while targeted therapies possess a selective toxic profile [73,74,75].
To address these challenges, recent findings demonstrate that personalized and combinatorial approaches are the best methods to overcome it [76,77,78]. Therefore, the aim of this work is to provide an insight into the molecular mechanisms of breast cancer molecular subtypes and to enlighten the efficacy of novel personalized treatments of this disease over traditional therapy.
2. Types of Cancer Based on Molecular Classification
Recent advances in molecular technologies have facilitated breast cancer research, allowing researchers and physicians to use biomarkers expressed by cancerous cells to categorize this pathology into four distinct groups—luminal A, luminal B, HER2-positive and TNBC. Thereby, the main molecular markers used are ER, PR and HER2, and they are detected using immunohistochemical techniques. In addition to these markers, Ki-67 is a widely used biomarker of tumor proliferation that helps complement and understand the pathology as demonstrated in Table 1. Together, they provide insights on diagnosis, prognosis and treatment [79].
The hormone receptors ER and PR are associated with the female reproductive system. Tumor cells that are ER-positive (≥1%) are defined as luminal A breast cancer. In addition, this type of cancer expresses high levels of PR (≥20%) and presents a low expression of Ki-67 (<20%), indicative of low tumor proliferation. Consequently, among the several types of cancer, this is the least harmful and the most common, being responsible for 50–60% of cases [59,80,81]. Furthermore, breast cancers categorized as IDC and ILC, mucinous, cribriform and tubular carcinomas fall under the subtype of luminal A breast cancer [59]. Luminal B breast cancer expresses the hormone receptor ER (≥1%) and it can be HER2-negative (≤10%) or HER2-positive (>10%). Usually, if luminal B is HER2-negative, the expression of PR is low to none (<20%) and the biomarker Ki-67 is highly expressed (≥20%), but if the luminal B cancer is HER2-positive, the levels of PR and Ki-67 varies [59,81]. However, it often presents higher levels of Ki-67 than luminal A, accelerating tumor growth and being associated with worse prognosis [80,81].
HER2 is an onco-protein that upon a strong stimulus starts to divide uncontrollably and, as a response, the mammary cells produce an excessive amount of HER2 receptors. This overexpression of HER2 gives rise to HER2-positive breast cancer, which is characterized by the absence of hormone receptors (ER < 1% and PR < 20%) and the presence of high levels of HER2 (>10%) [59]. This subtype of cancer is further subdivided into three subgroups, namely, HER2-positive if the score of immunohistochemistry is 3+ or if the score is 2+ and in situ hybridization is positive, HER2-low if the score of immunohistochemistry is 2+ and the in situ hybridization assay is negative or if the score of immunohistochemistry is 1+, and HER2-negative if the score of immunohistochemistry is 0 [82]. The HER2-enriched breast cancer is typically associated with ILC, and it is responsible for 10–15% of all breast cancers, presenting a high expression of Ki-67 (>20%), which makes this cancer the one with worse prognosis and with the fastest pace of growth in comparison to luminal breast cancer [79,80,81].
TNBC is defined by the absence of hormone receptors (ER < 1% and PR < 20%) and HER2 (≤10%) and represents 10% of all breast cancer cases. It is characterized by DNA alterations and genetic mutations, being mostly associated with mutations in the BRCA1 gene [59]. It is the most aggressive among the four subtypes of breast cancer with the highest proliferation index of Ki-67 (>30%) and poorer prognosis, and it is usually related to IDC and variants of apocrine and metaplastic carcinomas [59,80,81]. Furthermore, several studies of gene expression revealed that TNBC can be divided into seven different subtypes, including basal-like 1 (BL1), basal-like 2 (BL2), mesenchymal-like (M), mesenchymal stem-like (MSL), immunomodulatory (IM), luminal androgen receptor (LAR) and claudin-low type (CL) [59,83].

Table 1.
Molecular characteristics and diagnostic guidelines of molecular subgroups of breast cancer.
Table 1.
Molecular characteristics and diagnostic guidelines of molecular subgroups of breast cancer.
| Types of Cancer | Subgroups | Biomarkers | Other Markers | Diagnostic Guidelines | Reference |
|---|---|---|---|---|---|
| Luminal | Luminal A | ER (≥1%), PR (≥20%), Ki-67 (<20%) | BCL2, CK8/18, ESR1, GATA3, TRPS1, GPR160, TMEM45B, BAG1 | [84] | [85,86,87] |
| Luminal B (HER2-negative) | ER (≥1%), PR (<20%), HER2 (≤10%) Ki-67 (≥20%) | CK8/18, GATA3, TRPS1, ESR1, FOXA1, CXXC5, BIRC5, BLVRA, UBE2T | [86,87] | ||
| Luminal B (HER2-positive) | ER (≥1%), PR (<20%), HER2 (>10%), Ki-67 (15–30%) | CK8/18, GATA3, TRPS1, MKI67, cyclin B1 | [85,86] | ||
| HER2-enriched | HER2-positive | ER (<1%), PR (<20%), HER2 (>10%), Ki-67 (>30%) | ERBB2, MYC, MAPK, TP53, VHL, ATM | [88] | [89,90] |
| HER2-low | ER (≥1%), HER2 (>10%), Ki-67 (≤30%) | BCL2, BAG1, FOXA1, ESR1, CCNE1, CCNB1, MYBL2, MKI67, MELK | [91,92] | ||
| TNBC | BL1 | ER (<1%), PR (<20%), HER2 (≤10%), Ki-67 (>30%) | EGFR, CK5/6, CK14, CK17, MYC, PIK3CA, CDK6, AKT2, KRAS, FGFR1, IGF1R, CCNE1, CDKN2A/B, BRCA, PTEN, MDM2, RB1, TP53 | [84,88,93] | [83,94] |
| BL2 | EGFR, MET, NGF, IGF-1R, Wnt-β-catenin | [83] | |||
| M | Wnt, TGF-β, DNMT3A, TP53, PDGFRA, MAP3K1 | [83,94] | |||
| MSL | ABCA8, PROCR, ENG, ALDHA1, PER1, ABCB1, TERT2IP, BCL2, BMP2, THY, HOXA5, HOXA10, MEIS1, MEIS2, MEOX1, MEOX2, MSX1, ITGAV, KDR, NGFR, NT5E, PDGFR, THY1, VCAM1 | [83] | |||
| IM | Th1/Th2, IL-12, IL-7, TP53 | [83,95] | |||
| CL | ALDH1A1, PROCR, ZEB1, VIM, CDH2, EPCAM, CDH1, IL-13, ALDH1A1, PROCR, ZEB1, VIM, CDH2, EPCAM, CDH1, IL-13, IL6, CXCL8, VEGF-C, NRF1, EREG | [96,97] | |||
| LAR | AR (≥10%), ER (<1%), PR (<20%), HER2 (≤10%), Ki-67 (>30%) | DHCR24, ALCAM, FASN, FKBP5, APOD, PIP, SPDEF, CLDN8, PIK3CA, AKT1, NF1, GATA3, CDH1, KMT2C | [83,94,95] |
3. Molecular Mechanisms of Breast Cancer
Genetic mutations and alterations in signaling pathways play pivotal roles in the onset and progression of breast cancer. These mutations can disrupt key regulatory processes, including cell growth, survival, proliferation, apoptosis and DNA repair, leading to tumorigenesis. In particular, the crosstalk between genetic alterations and dysregulated signaling pathways contributes to the heterogeneity observed across the different breast cancer subtypes [98]. Understanding the molecular mechanisms underlying these mutations is critical for identifying therapeutic targets and improving patient outcomes.
3.1. Luminal Breast Cancer
Estrogen belongs to a group of steroid hormones, and it is associated with the female reproductive system. There are four distinct forms of estrogen including estrone (E1), estradiol (E2), estriol (E3) and estetrol (E4) [99]. E1 levels increase after menopause and have a pro-inflammatory effect in breast and adipose tissue, suggesting it is responsible for poor outcomes of ER-positive breast cancer in obesity [100]. The E2 form of estrogen is the most common in human blood circulation and it is produced by granulosa cells present in the ovaries [99,101]. E3 and E4 are both produced during pregnancy. Whereas E3 is synthesized by the placenta, E4 is a byproduct secreted by the fetus’ liver and they are both transported into the mother’s circulation through the placenta [101,102].
Estrogen, primarily E2, exerts its function by binding to two different ER, namely, ERα and ERβ, which are nuclear transcription factors involved in the regulation of many biological processes [101]. Mutations in these receptors have a great impact in cancer cells proliferation [103].
The receptor ERα plays a crucial role in cancer development through the regulation of target genes related to cell proliferation, apoptosis and cell cycle [80]. This receptor is activated upon E2 binding, causing a change in its configuration into an active form, dimerization and subsequent translocation to the nucleus. Through this mechanism, estrogen can regulate the expression of many estrogen-responsive elements (ERE) by binding to their gene promotor region instead of binding directly to DNA [80,104]. Once the estrogen is bound to EREs, ER recruits co-activators to assist the transcription of target genes. However, a dysregulation in ERα expression causes an unbalance between Ap1 and SP1 transcription factors and coregulators of target genes, as SRC1, AIB1, NCOR and MTA1, playing a major role in the development of luminal breast cancers [80,105,106].
Additionally, E2 can also bind to ERs present in the plasma membrane, activating the plasmatic ERα which attaches to caveolin-1, thus inducing downstream signaling pathways including PI3K/AKT/mTOR, a major pathway typically involved in cell growth, metabolism, proliferation, apoptosis and angiogenesis [80,107].
Under normal conditions, after the activation of ERα, E2 directly interacts with the p85 subunit of PI3K, activating the p110 catalytic subunit that promotes the phosphorylation of PIP2 into PIP3 which in turn recruits AKT. Following this, the AKT is phosphorylated by mTOR complex 2, changing its conformation into an active form that promotes the phosphorylation of target proteins present in the cell membrane, cytosol and in the nucleus. However, in breast cancer, PI3K suffers many changes due to genetic alterations and amplifications that are responsible for key products that ensure the normal signaling pathway. The catalytic subunit p110α regulated by the PIK3CA gene of PI3K is mutated, causing an hyperactivation of this gene, while there is a loss of PTEN, resulting in tumor growth. This overactivation of the signaling pathway PI3K/AKT/mTOR increases cell proliferation and inhibits apoptosis, enhancing breast cancer development [103,107].
On the other hand, ERβ is often co-expressed with ERα, expressing similar transcription factors and co-activators, but it plays an antagonistic role in comparison to ERα [80]. Studies find that ERβ acts as a tumor suppressor by inhibiting its proliferation, reduces inflammation and induces apoptosis of breast cancer cells, aiding in breast cancer therapy and being associated with better prognosis [108,109]. Recent publications support this mechanism, stating that ERβ has an anti-proliferative effect by downregulating the expression of ERα, diminishing epithelial–mesenchymal transition, which reduces cell invasion, and induces the entering in G2 phase arrest that promotes the inhibition of tumor growth [110,111].
Progesterone belongs to the same group of steroid hormones of estrogen and is responsible for the regulation of female pregnancy, menstrual cycle and embryogenesis. In addition, it promotes mammary epithelial cells proliferation and mammary gland expansion [112].
In breast cancer, progesterone signaling provokes a switch from paracrine regulation to autocrine regulation, being a hallmark of tumor progression. A progesterone-derived hormone named hormone medroxyprogesterone acetate turns epithelial cells more susceptible to breast tumors by triggering the nuclear factor-κB (NF-κB). Therefore, the progesterone binds to PR, activating this receptor, which results in an upregulation of RANKL, thereby increasing the expression of RANK receptor [112,113]. Other studies support this finding, as an increase of RANKL correlates with breast cancer development, which is also associated with poor prognosis in patients with luminal B breast cancer [114]. Hence, this pathway plays a major role in mammary tumorigenesis in response to progesterone. Nevertheless, PR has two isoforms, PRA and PRB, and the ratio between these two significantly influences breast cancer progression. It has been demonstrated that an increase in PRA is related to breast cancer development and metastasis, while an increase in PRB correlates with tumor proliferation [114]. The role of progesterone in breast carcinogenesis has not yet been clarified; therefore, further research needs to be conducted to enlighten the mechanism of PR in breast cancer.
3.2. HER2-Positive Breast Cancer
HER2 is a tyrosine kinase receptor and is a part of the epidermal growth factor receptor (EGFR) family. Commonly, its structure presents an intracellular tyrosine kinase that regulates genes involved in several cellular functions, a membrane domain that transmits the signal to the nucleus and an extracellular component where ligands, namely, HER1, HER3 and HER4, bind to their respective receptors and trigger a specific signal [115]. However, unlike the hormone receptors ER and PR, HER2 does not have ligand-dependent activity and thus depend on other family members to heterodimerize or homodimerize with itself to be activated. This phosphorylation of tyrosine kinases induces a conformational change on the intracellular component that promotes the activation of secondary signaling pathways, such as PI3K/AKT and MAPK [115,116].
Typically, phosphorylated HER2 and dephosphorylated HER2 are at an equilibrium, which is responsible for normal breast development. Upon a strong oncogenic stimulus, phosphorylated HER2 becomes upregulated, disrupting the balance that causes cell invasion, differentiation and migration, which are common features of tumor development and progression [117]. Thus, the amplification of HER2 is a hallmark of HER2-positive breast cancer that provokes hyperactivation of the signaling pathways, contributing to a more aggressive biological behavior and worse clinical outcomes [115,116]. However, recent findings have suggested a new subtype of HER2-positive breast cancer called HER2-low breast cancer [82]. Several studies were performed to understand the molecular biology of HER2-low subgroup of breast cancer, and it was predominantly associated with luminal A and luminal B cancer [118]. Nevertheless, a clear definition of this condition has not yet been formulated; thus, further investigations are needed to better understand it and optimize treatments for eligible patients [119].
When HER2 heterodimers with HER3, it interacts with the p85 subunit of PI3K activating the catalytic subunit p110. In HER2-positive breast cancer, the HER2 overexpression causes cellular signaling stress and increases DNA replication errors, resulting in mutations in the PIK3CA and AKT genes of the PI3K/AKT signaling pathway. Moreover, PTEN acts as a negative regulator of PI3K/AKT, maintaining it inactive and keeping the balance between cell survival and apoptosis. However, in cancerous conditions, this regulator is dysfunctional and thus promotes the activation of this pathway. Even though PTEN deletion is more correlated to poor outcomes in ER-positive breast cancer, its conjugation with PIK3CA and AKT gene mutations is associated with mammary tumor initiation and proliferation [115,117]. In addition, findings assessing the molecular landscape of HER2-positive breast cancer patients reported an elevated frequency of PIK3CA mutations (61.5%), but the difference to PIK3CA wild-type was not significant [120]. This indicates that this pathway may not be the main player in the development of this breast cancer subtype.
Moreover, the MAPK signaling cascade is also a major player in breast cancer. The aberrant overexpression of HER2 recruits more growth factors receptor-bound proteins 2 (GRB2) to be activated, which, together with the SOS, facilitates the exchange of guanosine diphosphate (GDP) into guanosine triphosphate (GTP) that activates RAS [121]. This hyperactivation of RAS causes a dysregulation in the activation of downstream protein kinases by promoting the phosphorylation and dimerization of RAF, which is strongly associated with carcinogenic conditions. Furthermore, the kinases RAF and RAS form a heterodimer that activates RAF, which phosphorylates MEK1 and MEK2 that, in turn, triggers the activation of ERK1 and ERK2. These activated ERK translocate to the nucleus, where they regulate several transcription factors including ETS1/2, ELK-1 and JUN involved in tumor proliferation, resistance to apoptosis and enhanced metastasis that contribute to cancer progression [121,122,123].
3.3. Triple-Negative Breast Cancer
TNBC does not express any hormone receptor or HER2 receptors and often arises from genetic alterations and mutations, causing an extended genomic heterogeneity. The BRCA1 and BRCA2 genes play a critical role in DNA repair by homologous recombination pathways or by influencing the cell to enter in non-homologous end joining pathways; they activate important cell cycle checkpoints to assess DNA damage and ensure that it does not present any abnormality; and they participate in cytoplasmic division and induce cell apoptosis and ubiquitination when the damage on the DNA cannot be repaired by inhibiting oncogenes amplification [124,125]. Typically, mutations in these genes are inherited from a relative that has this genetic mutation in their germinative cells and are related to the appearance of cancerous indicators at a young age, but mutations can also be acquired from several environmental factors that cause DNA damage. Mutations in somatic cells are not passed on to the offspring [124,126,127].
BRCA1 mutation inhibits the homologous recombination pathway, leading to the activation of alternative DNA damage repair pathways, while BRCA2 mutations compromise cytoplasmic division, increasing the number of binucleated cells and provoking chromosomic alterations which leads to aneuploid cells [127,128]. Taken together, they contribute to genomic instability and subsequent tumor initiation and progression. It has been stated that patients with BRCA1 gene mutations also present TP53 mutations [129] and several cohorts that studied the genetic profile of patients with TNBC proved this to be true. A study conducted with 95 female patients that have been submitted to primary surgery or neoadjuvant chemotherapy demonstrated that among the patients that suffered from TNBC, TP53 was the most frequent mutation (30%), followed by BRCA1 gene mutation (13%) and BRCA2 mutation (7%). Beyond this, they verified that BRCA2 mutations (16%) were more prevalent in luminal breast cancers in comparison to the other two genes and that TP53 mutations (18%) were also more common in HER2-positive breast cancer [130]. After assessing the prognostic value of each gene mutation, they concluded that TP53 and BRCA1 are the best prognostic biomarkers in TNBC being associated with worse outcomes. Another cohort with 52 patients with TNBC studied the mutation of genes related to DNA damage repair pathways, such as BRCA1, BRCA2 and TP53, among others. It was found that 75% of the patients presented BRCA1 mutations against 25% that presented BRCA2 mutations. When assessing the non-BRCA mutations landscape, they reported TP53 mutations in almost all the patients, and although BRCA1 was more prevalent than BRCA2 mutations, it was noteworthy that PIK3CA was more commonly co-mutated with the latter [131]. This last finding suggests that in TNBC, the PI3K/AKT/mTOR signaling pathway also suffers alterations affecting key cellular regulatory processes. An older study from 2022 with a larger sample composed of 42,680 female patients with diagnosed breast cancer from several countries associated TP53 mutations with HER2-positive breast cancer, but not with TNBC, while BRCA1 and BRCA2 variants were strongly present in this latter condition [132]. This is in line with what was recently found when comparing genetic mutations between the different subtypes of breast cancer.
Alterations in the PI3K/AKT/mTOR signaling pathway result in a disruption of regulatory cellular processes; therefore, PIK3CA mutations increase tumor cells proliferation, migration and invasion, as well as resistance to apoptosis [133]. Numerous studies comparing PIK3CA mutations in hormone receptor-positive breast cancer against TNBC have demonstrated that this mutation is less prevalent in the most aggressive subtype [133]. Mosele F. et al. found that 10% of TNBC cases reported PIK3CA mutations against 28% in hormone receptor-positive breast cancer [134]. Consistent with this finding, a study with 1270 female patients with breast cancer showed that PIK3CA mutations are more prevalent in hormone receptor-positive breast cancer, especially with HER2-negative status (31.4%) in comparison to TNBC (11.7%) [135]. Therefore, TNBC is more affected by mutations in genes involved in DNA repair than by PIK3CA mutations. Impairment of the machinery involved in DNA reparation promotes the propagation of mutations throughout the cell division, which contributes to uncontrollable tumor cell growth and genetic instability conferring the aggressive nature of this cancer subtype.
4. Therapeutic Approaches
Breast cancer treatment has evolved over the past century, transitioning from surgeries to personalized medicine. Technological advances have allowed for a better understanding of medical science and a deeper knowledge of cancer biology, which paved the way for a multidisciplinary approach that integrates surgery, chemotherapy and targeted therapies. Today, molecular profiling and disease subtyping guide treatment strategies, ensuring that therapies are tailored to each patient to improve survival rates. Table 2 summarizes the main findings and conclusions of the studies mentioned in each therapy.
4.1. Endocrine Therapy
Endocrine therapy is the treatment of choice to manage luminal breast cancer, specifically ER-positive breast cancer. This strategy directly targets ERs to prevent ligands from binding to it, and inhibits ovarian estrogen production, which promotes a reduction in estrogen levels [136,137]. To this end, various drugs are used including tamoxifen (TAM) and aromatase inhibitors (AI), such as anastrozole, letrozole and exemestane [138,139]. TAM is a selective estrogen response modulator (SERM) and is mostly used in patients with breast cancer who cannot endure AI [140]. This drug acts as an ER blocker by binding to ERα of breast cancer cells, which prevents estrogen from binding to the receptor. This mechanism of action provokes estrogen inactivity by suppressing transcriptional action or by reducing its effect, thereby stopping the onset and progression of breast cancer as illustrated in Figure 2 [140,141]. AI works by blocking the aromatase enzyme, preventing the production of E2 from androgen’s conversion or the synthesis of E1 from testosterone’s conversion, which inhibits its action on ER, thus decreasing estrogen levels [142,143].
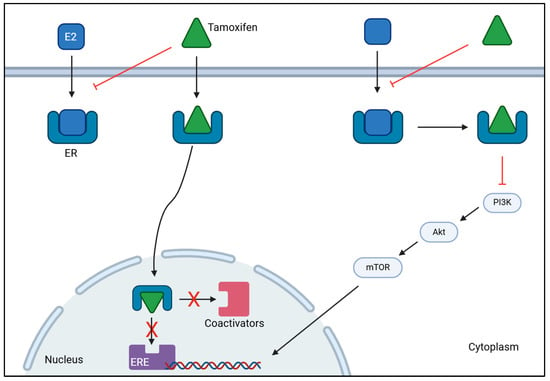
Figure 2.
Mechanism of action of tamoxifen on ER-positive breast cancer. Tamoxifen binds to estrogen receptors (ER), preventing estrogen (E2) from binding. This inhibits the activation of estrogen-responsive elements (ERE) and the subsequent recruitment of coactivators, stopping gene transcription. Tamoxifen can also bind to estrogen receptors to inhibit the activation of PI3K/AKT/mTOR signaling cascade to decrease the transcription of genes involved in tumor progression. Together, they hamper cancer proliferation and progression. [BioRender, 2025].
Since ER-positive breast cancer development depends on estrogen, these inhibitors are particularly effective in treating breast cancer in postmenopausal women [144]. A study with 31,920 women with ER-positive breast cancer was conducted to determine the effect of TAM treatment versus AI treatment on recurrence risk and mortality rate in a 5-year period. Although no significant differences were found between the group under TAM plus AI therapy and the group under AI monotherapy, women treated with AI had a 30% recurrence risk reduction and a 15% mortality rate reduction when treatment differed [145]. Consistent with this finding, Rosie B. et al. performed a meta-analysis in female premenopausal patients with ER-positive breast cancer receiving ovarian suppression to assess if they would benefit from AI treatment in a 5-year period. Results showed that patients treated with AI had lower recurrence risk (3.2%) of breast cancer than women who underwent TAM treatment (6.9%). Regarding mortality, they observed that patients who received AI treatment had higher mortality rates in the 0–4 period of follow-up, but the same patients had a mortality rate reduction in the following years (5–9 years) [146]. This suggests that AI is more effective in reducing breast cancer, serving as a more effective treatment than TAM by having a long-term effect on ER-positive breast cancer.
4.2. Anti-HER2 Therapy
Anti-HER2 therapy is the recommended treatment for patients with HER2-positive breast cancer and typically relies on pertuzumab and trastuzumab as primary treatment [138,147]. This strategy works by blocking the HER2 signaling pathway by monoclonal antibodies, such as trastuzumab, binding to HER2 receptor which prevents HER2 activation and dimerization, thereby inhibiting HER2 overexpression [148]. Likewise, pertuzumab blocks HER2 overexpression by preventing HER2-HER3 dimerization resulting in HER2 signaling pathway inhibition as it is demonstrated in Figure 3 [149]. A study conducted by Hamid M. and Zhixiang W. on the effect of trastuzumab on the development of HER2-positive breast cancer cells revealed that this drug inhibits cancer cells proliferation by promoting the entering in G1 phase arrest, but did not have any effect on HER2 dimerization [150]. Despite having less HER2, the remaining HER2 molecules continue to heterodimerize with HER3 or homodimerize with itself; thus, research has arisen combining trastuzumab and pertuzumab to completely block the proliferation of tumorous cells and improve the outcome of patients with this disease [151]. Li-Chung T. et al. tested the combination of trastuzumab and pertuzumab both in vitro and in vivo and highlighted that dual therapy maximized the therapeutic effect in comparison to monotherapy of either drug promoting tumor recognition by the immune system to destroy it, facilitating cancerous cell lysis and subsequent phagocytosis [152]. Multiple research reenforced that the usage of these two drugs together with chemotherapy would significantly improve patient outcomes and overall survival (OS) rates [153,154]. A domestic study performed in China evaluated the efficacy of dual anti-HER2 therapy using trastuzumab with pertuzumab in combination with different types of chemotherapy. After treatment administration, the overall pathological complete response (pCR) rate was 60.9% and the expression of HER2 was reduced in four patients, becoming HER2-negative and confirming the efficacy of this treatment [155]. Consistently, a muti-center study confirmed the efficacy of trastuzumab and pertuzumab with chemotherapy in reducing the recurrence risk (7.4 years versus 6.7 years) and mortality rate (7.6 years versus 7.2 years) in patients who achieved pCR [156]. Additionally, these two studies also demonstrated the effect of combinational therapy, proving that the adverse effects are tolerable and acceptable.
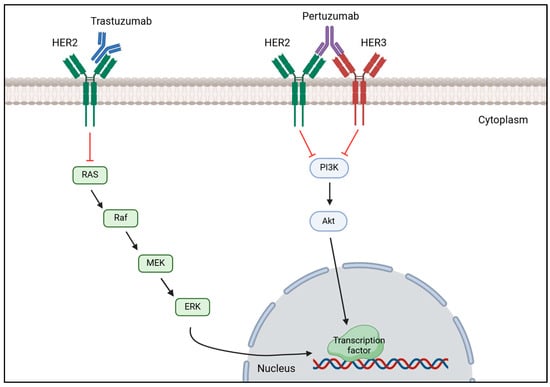
Figure 3.
Mechanism of action of trastuzumab and pertuzumab on HER2-positive breast cancer. Trastuzumab binds to the extracellular domain of HER2 receptor to prevent it from being activated, decreasing the expression of the MAPK signaling cascade that results in a reduction of transcription factors’ transcription important for tumor proliferation. Pertuzumab binds to HER3, preventing HER2-HER3 heterodimerization, which inhibits the phosphorylation of PI3K subunits, decreasing the activation of the PI3K/AKT signaling pathway, which in turn does not activate the transcription of transcription factors important for cell proliferation. [Biorender, 2025].
Moreover, HER2-positive cancer is also treated with antibody-drug conjugates (ADC) that are targeted therapies that combine cytotoxic agents with monoclonal antibodies with the purpose of minimizing the toxic side effects typically associated with chemotherapy while maximizing the efficacy of the therapy [76]. Trastuzumab emtansine (T-DM1) and trastuzumab deruxtecan (T-DXd) are the most used ADC to treat this condition. When trastuzumab binds to HER2 in the cell surface, it is internalized by endocytosis, and when emtansine is released inside the cell, it inhibits microtubule assembly and causes cell death, while deruxtecan inhibits DNA replication and repair, induces cell cycle arrest and promotes apoptosis [157,158]. Phase 3 of the DESTINY-Breast03 randomized trial compared the efficacy of T-DM1 to T-DXd in HER2-postive breast cancer patients, and individuals treated with T-DXd had higher progression-free survival (PFS) and OS rate [159]. A subgroup analysis of the patients enrolled in this study confirmed these results that patients treated with T-DXd had longer PFS and higher OS rate in comparison to individuals treated with T-DM1 [160]. Together, they highlight the benefit of using T-DXd as a secondary treatment to treat HER2-positive breast cancer.
4.3. TNBC Therapy
The standard treatment of care for TNBC is chemotherapy, but immunotherapy and radiotherapy have been attracting a lot of attention as different ways to treat this condition [161]. Systemic chemotherapy is the most common and effective treatment for TNBC, and it can be used as a neoadjuvant or adjuvant therapy. Drugs such as anthracyclines, platinum, taxanes and capecitabine are used to target and kill cancerous cells throughout the body, as seen in Figure 4, but treatment dosage should take tumor size and aggressive morphological features into consideration, as well as age and cancer stage [138,161,162]. A meta-analysis of 100,000 women from 86 randomized trials showed that adding anthracycline to a taxane-based chemotherapy regimen reduces the risk of recurrence by 14% and the annual mortality rate by about 12% in comparison to treatment with taxane monotherapy [163]. Another study assessed the effectiveness of platinum-based chemotherapy on patient’s survivability. Results showed that this treatment, both in the neoadjuvant and adjuvant setting, improved disease-free survival (DFS) and OS rates obtaining hazard ratios of 0.63 and 0.69, respectively, as a neoadjuvant therapy, and hazard ratios of 0.69 and 0.70, respectively, as an adjuvant modality. Moreover, it was reported that patients under platinum-based neoadjuvant chemotherapy had a 44% increase in achieving pCR, which is suggestive of better long-term outcomes [164]. Xi W. et al. compared the efficacy of adding capecitabine after standard adjuvant chemotherapy in early-stage TNBC between a placebo group and a capecitabine group. Research showed that maintaining capecitabine after chemotherapy improved DFS and OS, as well as locoregional recurrence-free survival by 10%, 4% and 4%, respectively. This proves that combining capecitabine with chemotherapy provides a survival benefit with tolerable adverse effects, but after a year of treatment, side effects worsen and may promote hand–foot syndrome [165].
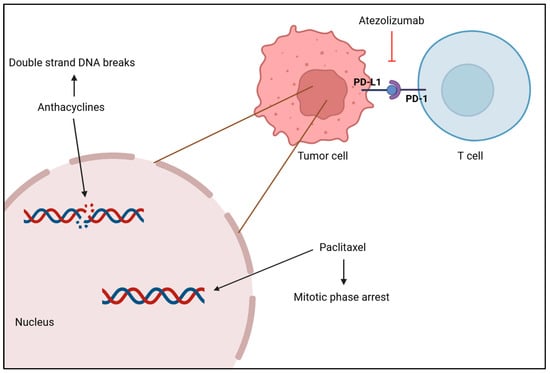
Figure 4.
Anthracycline intercalates with DNA base pairs, causing the strand to break and leading to cell apoptosis. Paclitaxel-based chemotherapy increases the microtubule stabilization, causing the loss of their dynamic instability, important for chromosome alignment and segregation, which leads to M phase arrest and cell death. Atezolizumab inhibits PD-L1 from binding to PD-1 by binding itself to PD-L1, which promotes the T cell to recognize tumor cells again and leads to tumor cell death. [Biorender, 2025].
Radiotherapy is typically used after a mastectomy to remove possible remaining tumor cells in the breast and lymph nodes [161]. A study with the aim of clarifying the role of radiotherapy in TNBC’s survival after surgery demonstrated that patients who received radiation had higher OS and better breast cancer-specific survival than patients who did not receive it, plus it reports that although it was efficient in several breast cancer stages, it was more efficient in later stages [166]. Moreover, immunotherapy has been emerging as a promising therapy for TNBC in which tumor cells are killed by the immune system, namely, by immune checkpoint inhibitors (ICI), which include cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), programmed death receptor-ligand 1 (PD-L1) and programmed death receptor-1 (PD-1) as shown in Figure 4 [167]. Atezolizumab is a monoclonal antibody that targets PD-L1 and inhibits its activity, enhancing the ability of T cells to recognize and destroy tumor cells. Research has shown that patients with PD-L1-positive tumors presented an OS higher when treated with atezolizumab plus nab-paclitaxel than patients treated with placebo plus nab-paclitaxel (25 months versus 18 months) and a longer PFS (7.5 months versus 5.3 months) [168], which highlights the clinical relevance of atezolizumab in treating patients with PD-L1-positive tumors with TNBC. Phase II of the KEYNOTE-086 study evaluated the efficacy of pembrolizumab monotherapy in patients with PD-L1-positive TNBC and observed a higher PFS within a disease-free interval higher than 12 months (2.2 months versus 2 months). Furthermore, results showed that this monotherapy improved the OS, especially when the disease-free interval was higher than 12 months (23 months versus 9.4 months), supporting the effectiveness of pembrolizumab treatment in these patients [169].
The heterogeneous nature of this disease and the lack of biomarkers poses an obstacle for the development of targeted therapies that could be tailored to each patient and would be beneficial to improve clinical outcomes. Consequently, clinicians treat every case of TNBC similarly with neoadjuvant therapy [170]. However, advances in the field show immunotherapy as a promising approach by leveraging the immune system to target tumor cells. Further research shows the beginning of the development of targeted therapies to treat TNBC and offers new therapeutic strategies for the creation of patient-tailored treatments [171,172].
Table 2.
Main findings and conclusion of the studies involving different therapies to treat breast cancer.
Table 2.
Main findings and conclusion of the studies involving different therapies to treat breast cancer.
| Study Objective | Methodology | Main Findings | Conclusions | Reference |
|---|---|---|---|---|
| Endocrine Therapy | ||||
| Understand the benefits of using AI and TAM and the effectiveness and impact of different treatment schedules over a 5-year period | Meta-analysis of individual patient data |
| Benefits were clearer when the treatment was different | [145] |
| Investigate if premenopausal women receiving ovarian suppression therapy benefit from AI in comparison to TAM to reduce breast cancer recurrence and improve patient survival over a 5-year period | Meta-analysis of individual patient data |
| Main benefit from taking AI was best seen when patients were treated differently | [146] |
| Anti-HER2 Therapies | ||||
| Investigate the mechanisms by which the combination of trastuzumab and pertuzumab enhance antitumor activity in HER2-positive breast cancer | In vitro (BT-474, KPL-4, SKBR3, Au565, SKOV3 and NIH/3T3 cells) In vivo (SCID-beige mice and C1qa-KO mice) |
| Dual therapy of pertuzumab plus trastuzumab promotes tumor cell lysis and tumor phagocytosis by macrophages | [152] |
| ||||
| Investigate if adding pertuzumab to trastuzumab and chemotherapy improves disease recurrence and reduces mortality in HER2-positive early breast cancer patients | Randomized, multicenter, multinational clinical trial |
| The addition of pertuzumab to trastuzumab and chemotherapy increases the treatment efficacy by reducing patient relapses and mortality risk | [153] |
| Evaluate OS and PFS in HER2-positive metastatic breast cancer patients receiving trastuzumab plus pertuzumab or trastuzumab plus pertuzumab with chemotherapy | Multicenter randomized clinical trial |
| Dual anti-HER2 therapy with chemotherapy increases OS and significantly enhances PFS | [154] |
| Assess the efficacy and safety of dual anti-HER2 therapy in combination with chemotherapy in HER2-positive breast cancer patients in China | Retrospective study in China |
| Elevated pCR demonstrates the efficacy of trastuzumab plus pertuzumab with chemotherapy and the side effects are tolerable | [155] |
| Assess the combinatory treatment of pertuzumab plus trastuzumab and chemotherapy in HER2-positive breast cancer patients | Retrospective, multicenter study |
| This combinatory therapy promotes higher pCR, which is linked to decreases disease recurrence and high survivability | [156] |
| Compare the efficacy and safety between T-DXd and T-DM1 in patients with metastatic HER2-positive breast cancer | Randomized, multicenter clinical trial |
| T-DXd significantly improved the OS and PFS and reduced the risk of mortality | [159] |
| Evaluate the efficacy and safety of trastuzumab deruxtecan compared to trastuzumab emtansine in Asian patients with HER2-positive metastatic breast cancer who were previously treated with trastuzumab and taxane | Subgroup analysis of a group of patients enrolled in the Breast03 study |
| T-DXd increased PFS, improved OS and reduced the risk of death | [160] |
| TNBC Therapy | ||||
| Understand the benefits and risks of including anthracyclines, and the comparative benefits of different anthracycline–taxane regimens | Patient-level meta-analysis |
| Adding anthracyclines to a taxane regimen increases the benefit by reducing the recurrence and the risk of mortality | [163] |
| Assess the benefits and harms of platinum-based chemotherapy as adjuvant and neoadjuvant treatment in people with early triple-negative breast cancer | Systematic review |
| Platinum-based chemotherapy improved DFS and OS in both treatment modalities | [164] |
| Investigate the efficacy and adverse effects of low-dose capecitabine maintenance after standard adjuvant chemotherapy in early-stage TNBC | Randomized clinical trial in China |
| Adding capecitabine after standard chemotherapy improved DFS, OS and locoregional recurrence-free survival | [165] |
| Evaluate the efficacy and safety of atezolizumab plus nab-paclitaxel in patients with unresectable, locally advanced or metastatic TNBC | IMpassion130 randomized, double-blind clinical trial |
| Atezolizumab benefits OS and PFS, although its efficacy is confined to PD-L1-possitive tumors | [168] |
| Evaluate the efficacy of pembrolizumab as first-line therapy for patients with PD-L1-positive metastatic TNBC | International, open-label, multicohort clinical trial |
| Higher response rate to pembrolizumab as first-line treatment benefits patients with metastatic TNBC, especially those with PD-L1-positive tumors | [169] |
4.4. Therapy Resistance
Breast cancer therapies, despite their effectiveness, often face significant challenges due to tumor heterogeneity, treatment resistance and toxicity that may reduce therapy efficacy and enhance disease recurrence [68,71]. As tumor heterogeneity pertains to a wide range of genetic and epigenetic modifications, it may difficult disease diagnostic, lead to treatment resistance and reduce prediction of prognosis resulting in treatment inefficacy, metastasis and disease recurrence [173]. Technology advancements allowed physicians to better understand this variability with single cell transcriptomics, also known as single-cell RNA sequencing. This method constructs the genetic profile of each individual cell within each tumor of the patient, providing a clear characterization of the tumor heterogeneity mechanisms, interaction between cells and signaling cascades related to poor therapeutic response or prognosis [174]. Although single-cell RNA sequencing proved to be a thorough molecular technique in explaining the oncologic mechanisms involved in tumor heterogeneity, the processes that lead to intratumor heterogeneity are still far from being understood and further research is needed as it represents an obstacle to the efficacy of targeted therapies [175].
A major source of tumor heterogeneity is cancer metastasis that originates from genetic mutations and epigenetic modifications that promote cancer cells to migrate and colonize other organs, predominantly bones, liver, brain and lungs. Additionally, the immune cells surrounding the primary tumor exert a selective pressure that creates treatment resistance and stimulates cancer metastasis [176,177]. Unraveling these interactions and modifications with single-cell RNA sequencing helps pave the way for the development of patient-tailored therapies with great efficacy potential using a combinatory drug approach. Recent studies using single-cell RNA sequencing to explore immune cell clusters on tumor microenvironment (TME) that modulate tumor heterogeneity revealed that each cell has a unique molecular signature within the same tumor and that its presence or absence can regulate different responses to breast cancer therapies and aid physicians in treatment selection [178,179].
Endocrine therapy resistance involves ER alterations, gene mutations, dysregulated expression patterns of co-regulatory proteins and alterations in signaling pathways and cell cycle modulators [180,181,182]. The ER molecule is a key component in ER-positive breast cancer and the main target of endocrine therapies; thus, ER alterations or mutations are a major cause of therapy resistance. The loss of ER expression prevents therapeutic drugs from targeting cancer cells, and this is strongly attributed to methylation of ER gene promoter region [183]. Manouk K.B. et al. confirmed that, in fact, ER expression can be regulated by DNA methylation, decreasing its levels of expression [184]. A small cohort of patients with ESR1-positive methylation and ESR1-negative methylation revealed that this alteration is related to reduced survival and increased therapy resistance [185]. Additionally, mutations in ESR1 result in ligand-independent constitutive activation and in the activation of multiple sets of target genes, increasing metastasis [186]. The hot spot mutations of this gene are D538G (18.6%), Y537S (13.1%), Y537N (8.8%) and E380Q (3.3%) [187]. In addition to therapy resistance caused by ESR1 mutations, evidence shows that PIK3CA gene mutations have a major role in the development of endocrine therapy resistance and that insulin-like growth factor 1 interacts with ER, using the PIK3/AKT/mTOR pathway to cause estrogen-independent activation of ER and therapy resistance [188,189].
Anti-HER2 therapy resistance can be attributed to changes in HER2 expression, loss of PTEN or overexpression of p95HER2. The upregulation of this truncated form of HER2 lacks an extracellular domain where drugs would bind, rendering therapy resistance [190]. Overexpression of HER2 is the main trigger for HER2-positive breast cancer, but the loss of HER2 is the cause of therapy resistance. This can be explained by a shedding mechanism that cleaves the extracellular and the intracellular juxtamembrane region that also induces p95HER2 production [191]. Recent findings report the existence of trastuzumab resistance whenever p95HER2 expression was elevated, and it was noteworthy that therapy resistance was significantly associated with lymphatic invasion, perinodal invasion, nodal involvement and metastasis [192]. Additionally, the upregulation of CMTM6 causes trastuzumab resistance and promotes breast cancer proliferation by increasing HER2 expression through the manipulation of downstream MAPK and PI3K/AKT/mTOR signaling pathways [193]. Furthermore, breast cancer cells may also be resistant to ADC due to changes in the routes of HER2 internalization, alterations in antibody–drug ratios may affect ADC efficacy, and mutations in PIK3CA gene and loss of PTEN may disrupt PI3K/AKT/mTOR signaling pathway, decreasing the effectiveness of ADC treatment [194,195].
TME is a complex and dynamic network of cellular and non-cellular components, including mesenchymal cells, cancer-associated fibroblasts (CAF), tumor-associated macrophage and endothelial cells within a tumor. It is an active participant in tumor onset and progression and influences treatment resistance in TNBC [196]. Zhaoze G. et al. found that in vitro and in vivo models of breast CAFs promoted cancer growth and radioresistance due to high levels of IL-6 secretion from these cells by the modulation of STAT3 [197]. In addition, tumor-associated macrophages contribute to therapy resistance by secreting inflammatory molecules, such as IL-6, TNF-α and CCL18, which suppress the function of T cells and by reprogramming the metabolic system to induce tumorigenesis [198]. On another note, heat shock protein beta-1 (HSPB1) exerts antioxidant and anti-apoptotic effects, but the overexpression of this protein promotes chemotherapy resistance through the NF-κB signaling pathway and provokes epithelial–mesenchymal transition, thereby facilitating cancer cells migration and invasion [199]. ATP-binding cassettes (ABC) transporters use ATP to efflux molecules, such as therapeutic drugs, present within cell; thus, a great number of ABC decrease therapeutic effects and even confer therapy resistance [200]. Additionally, recent findings explored the impact of two different enzymes on breast cancer chemoresistance. The upregulation of paraoxanase-2 decreased TNBC cell’s susceptibility to doxorubicin, 5-Fluorouracil and cisplatin leading to cell proliferation and therapy resistance, while nicotinamide N-methyltransferase upregulation leads to SIRT1 stabilization by preventing its degradation, which increases its deacetylation activity promoting cell survival and therapy resistance [201,202].
Overcoming resistance remains a major challenge in breast cancer treatment, necessitating further research into combinatory therapies, novel drug development, and personalized treatment strategies to increase long-term patient outcomes. Recent studies using CDK4/6 inhibitors in combination with therapeutic drugs showed an improvement in endocrine resistance caused by ESR1 mutations, reducing treatment failure [203,204]. Moreover, targeting PI3K/AKT/mTOR signaling pathway with a combination therapy of PI3K inhibitors and trastuzumab to treat HER2-positive breast cancer can ameliorate resistance and restore apoptotic effects [205]. Additionally, a clinical trial using a tyrosine kinase inhibitor (tucatinib), together with trastuzumab and capecitabine, improved OS and PFS, suggesting overcoming therapy resistance [206]. A study on a combinatory strategy of pembrolizumab plus chemotherapy on TNBC revealed that the conjugation was more effective than using only chemotherapy to treat patients, significantly improving the OS of these patients [207]. Furthermore, a meta-analysis on PARP inhibitors on patients with germline BRCA1/2 mutations revelated that this targeted therapy increases the overall response to treatment overcoming chemoresistance [208]. Another study using PARP inhibitors (olaparib) and CDK4/6 inhibitors (palbociclib) demonstrated that this combinatory strategy was an effective approach to overcome cellular olaparib resistance in comparison to olaparib monotherapy, enhancing treatment response [209].
Despite clinical efforts for developing efficient treatment for primary tumors, metastatic breast cancer therapy does not always respond to treatment due to tumor heterogeneity that varies according to cancer subtype, metastatic site and patient, leading to therapy resistance [176]. However, recent research on using targeted therapies and combinatory approaches elucidates an efficient strategy to improve the survival of patients with metastatic disease and reduce resistance. A study comparing the use of fulvestrant plus capivasertib with fulvestrant plus placebo on patients with metastatic ER-positive and HER2-negative breast cancer patients indicates that capivasertib improves OS and PFS [210]. Another study using pyrotinib, trastuzumab and docetaxel found that this three-drug approach ameliorated PFS in comparison to a placebo group of patients with metastatic HER2-positive breast cancer [211]. A cohort of patients with metastatic TNBC treated with atezolizumb and nab-paclitaxel had higher PFS and higher OS in comparison to patients treated with placebo and nab-paclitaxel [212]. Contradictory, recent research on patients with metastatic TNBC did not find significant differences in OS between patients that received atezolizumb and chemotherapy in comparison to those that received a placebo plus chemotherapy [213]. This may be explained by the aggressive and heterogenous nature of TNBC that entails significant genetic, molecular and cellular variability in comparison to other breast cancer subtypes, making it more difficult to treat. Therefore, further research must be conducted to better understand how to respond to metastatic TNBC.
To this extent, numerous studies have positively correlated combinatory therapy strategies to treatment response over monotherapy. The employment of this approach offers several benefits, particularly in overcoming therapy resistance. By targeting multiple pathways simultaneously, combinatory approaches reduce the likelihood of cancer cells developing resistance to a single drug [214]. Additionally, combining targeted therapies with chemotherapy or immunotherapy can enhance tumor cell death while minimizing off-target effects [215]. This strategy also helps address tumor heterogeneity by targeting different subpopulations within a tumor, thereby reducing the chances of relapse [216].
5. Conclusions
Breast cancer remains a major public health challenge due to its molecular heterogeneity and therapy resistance. However, a comprehensive analysis of the molecular subtypes of breast cancer and their respective therapies helps physicians in treatment selection by reducing complicated side effects, therapeutic resistance and improving patient outcomes.
Advancements in technology have allowed the development of single-cell transcriptomic analysis to identify different cell populations within a tumor, providing a clear insight of tumor heterogeneity that often leads to therapy resistance. Nevertheless, despite significant efforts in managing advanced breast cancer, the heterogenous nature of the disease continues to be a major challenge, as it poses a key obstacle in promoting resistance to targeted therapies. Therefore, understanding tumor characteristics is crucial for the development of personalized therapeutic strategies and the continuous adaptation of therapeutic approaches.
Furthermore, to the best our knowledge, this is the first review that highlights the importance of combinatory therapies over monotherapies in overcoming therapy resistance in every breast cancer subtype, providing an extended explanation on resistance mechanisms and proposing the utilization of multi-targeted strategies to target multiple pathways involved in therapy resistance to reduce disease recurrence.
Addressing these limitations of tumor heterogeneity and treatment resistance by developing an integrative approach with molecular profiling, single-cell transcriptomic and combinatory therapies pave the way for the creation of multimodal treatments where cancer management is effective and patient-centered, contributing to great advancements in precision oncology. Therefore, future research should focus on validating these findings into clinical practice and explore novel drug combinations to target adaptive resistance mechanisms and technological enhancement to increase drug response prediction and personalized treatment selection, as well as to help optimize treatment regimen in real time.
Author Contributions
Conceptualization, E.C. and N.V.; methodology, E.C. and N.V.; validation, S.C., F.S. and N.V.; formal analysis, S.C., F.S. and N.V.; investigation, S.C., F.S. and N.V.; resources, N.V.; writing—original draft preparation, E.C.; writing—review and editing, S.C., F.S. and N.V.; visualization S.C., F.S. and N.V.; supervision, N.V.; project administration, F.S. and N.V.; funding acquisition, F.S. and N.V. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financed by FEDER—Fundo Europeu de Desenvolvimento Regional through the COMPETE 2020—Operational Programme for Competitiveness and Internationalization (POCI), Portugal 2020, and by Portuguese funds through FCT—Fundação para a Ciência e a Tecnologia, in a framework of the projects in CINTESIS, R&D Unit (reference UIDB/4255/2020), and within the scope of the project RISE—LA/P/0053/2020. N.V. would also like to gratefully acknowledge the support from FCT and FEDER (European Union), award number IF/00092/2014/CP1255/CT0004 and CHAIR in Onco-Innovation at FMUP.
Acknowledgments
E.C. thanks FMUP for her PhD grant.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ABC | ATP-Binding Cassette |
| ADC | Antibody-Drug Conjugate |
| AI | Aromatase Inhibitors |
| AIB1 | Amplified in Breast Cancer 1 |
| AKT | Protein Kinase B |
| Ap1 | Activator Protein 1 |
| BL1 | Basal-Like 1 |
| BL2 | Basal-Like 2 |
| BMI | Body Mass Index |
| BRCA1 | Breast Cancer Associated Gene 1 |
| BRCA2 | Breast Cancer Associated Gene 2 |
| CAF | Cancer-Associated Fibroblasts |
| CCL18 | Chemokine (C-C motif) Ligand 18 |
| CL | Claudin-Low Type |
| CMTM6 | CKLF-like MARVEL Transmembrane Domain-Containing 6 |
| CTLA-4 | Cytotoxic T-Lymphocyte-Associated Antigen 4 |
| DCIS | Ductal Carcinoma In Situ |
| DFS | Disease-Free Survival |
| E1 | Estrone |
| E2 | Estradiol |
| E3 | Estriol |
| E4 | Estetrol |
| EGFR | Epidermal Growth Factor Receptor |
| ER | Estrogen Receptors |
| ERE | Estrogen-Responsive Elements |
| ERK1 | Extracellular Signal-Regulated Kinase 1 |
| ERK2 | Extracellular Signal-Regulated Kinase 2 |
| GDP | Guanosine Diphosphate |
| GRB2 | Growth Factors Receptor-Bound Proteins 2 |
| GTP | Guanosine Triphosphate |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| HSPB1 | Heat Shock Protein Beta-1 |
| IBC | Inflammatory Breast Cancer |
| ICI | Immune Checkpoint Inhibitors |
| IDC | Invasive Ductal Carcinoma |
| IL-6 | Interleukin-6 |
| ILC | Invasive Lobular Carcinoma |
| IM | Immunomodulatory |
| LAR | Luminal Androgen Receptor |
| LCIS | Lobular Carcinoma In Situ |
| M | Mesenchymal-Like |
| MAPK | Mitogen-Activated Protein Kinase |
| MEK 2 | Mitogen-Activated Protein Kinase Kinase 2 |
| MEK1 | Mitogen-Activated Protein Kinase Kinase 1 |
| MRI | Magnetic Resonance Imaging |
| MSL | Mesenchymal Stem-Like |
| MTA1 | Metastasis-Associated Protein 1 |
| mTOR | Mammalian Target of Rapamycin |
| NCOR | Nuclear Receptor Corepressor |
| NFκB | Nuclear Factor-κB |
| OS | Overall Survival |
| pCR | Pathological Complete Response |
| PD-1 | Programmed Death Receptor-1 |
| PD-L1 | Programmed Death Receptor-Ligand 1 |
| PFS | Progression-Free Survival |
| PI3K | Phosphatidylinositol 3-Kinase |
| PR | Progesterone Receptors |
| PTEN | Phosphatase and Tensin Homolog |
| RANK | Receptor Activator of Nuclear Factor κB |
| RANKL | Receptor Activator of Nuclear Factor κB Ligand |
| RAS | Rat Sarcoma |
| SERM | Selective Estrogen Response Modulator |
| SOS | Son Of Sevenless |
| SP1 | Specificity Protein 1 |
| SRC 1 | Steroid Receptor Coactivor 1 |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| TAM | Tamoxifen |
| T-DM1 | Trastuzumab Emtansine |
| T-DXd | Trastuzumab Deruxtecan |
| TME | Tumor Microenvironment |
| TNBC | Triple-Negative Breast Cancer |
| TNF-α | Tumor Necrosis Factor Alpha |
References
- Breast Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer (accessed on 8 December 2024).
- Breast Cancer Statistics. Available online: https://www.wcrf.org/preventing-cancer/cancer-statistics/breast-cancer-statistics/ (accessed on 8 December 2024).
- Breast Cancer Facts. Available online: https://www.europadonna.org/breast-cancer/ (accessed on 29 January 2025).
- Grabinski, V.F.; Brawley, O.W. Disparities in Breast Cancer. Obstet. Gynecol. Clin. 2022, 49, 149–165. [Google Scholar]
- Xu, H.; Xu, B. Breast Cancer: Epidemiology, Risk Factors and Screening. Chin. J. Cancer Res. 2023, 35, 565–583. [Google Scholar]
- Benz, C.C. Impact of Aging on the Biology of Breast Cancer. Crit. Rev. Oncol. Hematol. 2007, 66, 65–74. [Google Scholar]
- Rojas, K.; Stuckey, A. Breast Cancer Epidemiology and Risk Factors. Clin. Obstet. Gynecol. 2016, 59, 651–672. [Google Scholar] [CrossRef] [PubMed]
- European Guidelines on Breast Cancer Screening and Diagnosis. Available online: https://cancer-screening-and-care.jrc.ec.europa.eu/en/ecibc/european-breast-cancer-guidelines?topic=63&usertype=60&filter_1=80&filter_2=79&updatef2=0 (accessed on 6 March 2025).
- Screening for Breast Cancer. Available online: https://www.cdc.gov/breast-cancer/screening/index.html (accessed on 6 March 2025).
- Obeagu, E.I.; Obeagu, G.U. Breast Cancer: A Review of Risk Factors and Diagnosis. Medicine 2024, 103, e36905. [Google Scholar] [PubMed]
- Bodewes, F.T.H.; van Asselt, A.A.; Dorrius, M.D.; Greuter, M.J.W.; de Bock, G.H. Mammographic Breast Density and the Risk of Breast Cancer: A Systematic Review and Meta-Analysis. Breast 2022, 66, 62–68. [Google Scholar]
- Mokhtary, A.; Karakatsanis, A.; Valachis, A. Mammographic Density Changes over Time and Breast Cancer Risk: A Systematic Review and Meta-Analysis. Cancers 2021, 13, 4805. [Google Scholar] [CrossRef]
- Mann, R.M.; Athanasiou, A.; Baltzer, P.A.T.; Camps-Herrero, J.; Clauser, P.; Fallenberg, E.M.; Forrai, G.; Fuchsjäger, M.H.; Helbich, T.H.; Killburn-Toppin, F.; et al. Breast Cancer Screening in Women with Extremely Dense Breasts Recommendations of the European Society of Breast Imaging (EUSOBI). Eur. Radiol. 2022, 32, 4036–4045. [Google Scholar]
- Lubinski, J.; Kotsopoulos, J.; Moller, P.; Pal, T.; Eisen, A.; Peck, L.; Karlan, B.Y.; Aeilts, A.; Eng, C.; Bordeleau, L.; et al. MRI Surveillance and Breast Cancer Mortality in Women With BRCA1 and BRCA2 Sequence Variations. JAMA Oncol. 2024, 10, 493–499. [Google Scholar]
- Trentham-Dietz, A.; Newcomb, P.A.; Nichols, H.B.; Hampton, J.M. Breast Cancer Risk Factors and Second Primary Malignancies among Women with Breast Cancer. Breast Cancer Res. Treat. 2007, 105, 195–207. [Google Scholar]
- Brewer, H.R.; Jones, M.E.; Schoemaker, M.J.; Ashworth, A.; Swerdlow, A.J. Family History and Risk of Breast Cancer: An Analysis Accounting for Family Structure. Breast Cancer Res. Treat. 2017, 165, 193–200. [Google Scholar] [PubMed]
- Liu, L.; Hao, X.; Song, Z.; Zhi, X.; Zhang, S.; Zhang, J. Correlation between Family History and Characteristics of Breast Cancer. Sci. Rep. 2021, 11, 6360. [Google Scholar]
- Collins, A.; Politopoulos, I. The Genetics of Breast Cancer: Risk Factors for Disease. Appl. Clin. Genet. 2011, 4, 11–19. [Google Scholar]
- Baretta, Z.; Mocellin, S.; Goldin, E.; Olopade, O.I.; Huo, D. Effect of BRCA Germline Mutations on Breast Cancer Prognosis: A Systematic Review and Meta-Analysis. Medicine 2016, 95, e4975. [Google Scholar] [PubMed]
- Gorodetska, I.; Kozeretska, I.; Dubrovska, A. BRCA Genes: The Role in Genome Stability, Cancer Stemness and Therapy Resistance. J. Cancer 2019, 10, 2109–2127. [Google Scholar]
- Naeem, M.; Hayat, M.; Ahmad Qamar, S.; Mehmood, T.; Munir, A.; Ahmad, G.; Rasool Azmir, U.; Adeel Faryad, M.; Zeeshan Talib, M.; Irfan, M.; et al. Risk Factors, Genetic Mutations and Prevention of Breast Cancer. Int. J. Biosci. 2019, 14, 492–496. [Google Scholar]
- Hamajima, N.; Hirose, K.; Tajima, K.; Rohan, T.; Friedenreich, C.M.; Calle, E.E.; Gapstur, S.M.; Patel, A.V.; Coates, R.J.; Liff, J.M.; et al. Menarche, Menopause, and Breast Cancer Risk: Individual Participant Meta-Analysis, Including 118 964 Women with Breast Cancer from 117 Epidemiological Studies. Lancet Oncol. 2012, 13, 1141–1151. [Google Scholar]
- Yue, W.; Yager, J.D.; Wang, J.P.; Jupe, E.R.; Santen, R.J. Estrogen Receptor-Dependent and Independent Mechanisms of Breast Cancer Carcinogenesis. Steroids 2013, 78, 161–170. [Google Scholar]
- Subramani, R.; Lakshmanaswamy, R. Pregnancy and Breast Cancer. Prog. Mol. Biol. Transl. Sci. 2017, 151, 81–111. [Google Scholar]
- Stordal, B. Breastfeeding Reduces the Risk of Breast Cancer: A Call for Action in High-income Countries with Low Rates of Breastfeeding. Cancer Med. 2022, 12, 4616–4625. [Google Scholar]
- Migliavacca Zucchetti, B.; Peccatori, F.A.; Codacci-Pisanelli, G. Pregnancy and Lactation: Risk or Protective Factors for Breast Cancer? Adv. Exp. Med. Biol. 2020, 1252, 195–197. [Google Scholar]
- Rozenberg, S.; Di Pietrantonio, V.; Vandromme, J.; Gilles, C. Menopausal Hormone Therapy and Breast Cancer Risk. Best. Pract. Res. Clin. Endocrinol. Metab. 2021, 35, 101577. [Google Scholar] [CrossRef]
- Yuk, J.-S.; Jin-Sung Yuk, C. Relationship between Menopausal Hormone Therapy and Breast Cancer: A Nationwide Population-Based Cohort Study. Int. J. Gynecol. Obstet. 2024, 166, 735–744. [Google Scholar] [CrossRef]
- Kanadys, W.; Barańska, A.; Malm, M.; Błaszczuk, A.; Polz-Dacewicz, M.; Janiszewska, M.; Jędrych, M. Use of Oral Contraceptives as a Potential Risk Factor for Breast Cancer: A Systematic Review and Meta-Analysis of Case-Control Studies up to 2010. Int. J. Environ. Res. Public Health 2021, 18, 4638. [Google Scholar] [CrossRef] [PubMed]
- Kamani, M.O.; Akgor, U.; Gültekin, M. Review of the Literature on Combined Oral Contraceptives and Cancer. Ecancermedicalscience 2022, 16, 1416. [Google Scholar] [CrossRef] [PubMed]
- Obesity. Available online: https://www.who.int/health-topics/obesity#tab=tab_1 (accessed on 6 March 2025).
- Dehesh, T.; Fadaghi, S.; Seyedi, M.; Abolhadi, E.; Ilaghi, M.; Shams, P.; Ajam, F.; Mosleh-Shirazi, M.A.; Dehesh, P. The Relation between Obesity and Breast Cancer Risk in Women by Considering Menstruation Status and Geographical Variations: A Systematic Review and Meta-Analysis. BMC Womens Health 2023, 23, 392. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast Cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef]
- Starek-Świechowicz, B.; Budziszewska, B.; Starek, A. Alcohol and Breast Cancer. Pharmacol. Rep. 2022, 75, 69–84. [Google Scholar] [CrossRef]
- Lofterød, T.; Frydenberg, H.; Flote, V.; Eggen, A.E.; McTiernan, A.; Mortensen, E.S.; Akslen, L.A.; Reitan, J.B.; Wilsgaard, T.; Thune, I. Exploring the Effects of Lifestyle on Breast Cancer Risk, Age at Diagnosis, and Survival: The EBBA-Life Study. Breast Cancer Res. Treat. 2020, 182, 215–227. [Google Scholar] [CrossRef]
- National Cancer Policy Forum; Board on Health Care Services; Institute of Medicine. Tobacco use and cancer. In Reducing Tobacco-Related Cancer Incidence and Mortality: Workshop Summary; National Academies Press (US): Washington, DC, USA, 2013. [Google Scholar]
- Peñalver-Argüeso, B.; García-Esquinas, E.; Castelló, A.; de Larrea-Baz, N.F.; Castaño-Vinyals, G.; Amiano, P.; Fernández-Villa, T.; Guevara, M.; Fernández-Tardón, G.; Alguacil, J.; et al. Smoking History and Breast Cancer Risk by Pathological Subtype: MCC-Spain Study. Tob. Induc. Dis. 2023, 21, 157. [Google Scholar] [CrossRef]
- He, Y.; Si, Y.; Li, X.; Hong, J.; Yu, C.; He, N. The Relationship between Tobacco and Breast Cancer Incidence: A Systematic Review and Meta-Analysis of Observational Studies. Front. Oncol. 2022, 12, 961970. [Google Scholar]
- Rosenberg, L.; Boggs, D.A.; Bethea, T.N.; Wise, L.A.; Adams-Campbell, L.L.; Palmer, J.R. A Prospective Study of Smoking and Breast Cancer Risk among African American Women. Cancer Causes Control 2013, 24, 2207–2215. [Google Scholar]
- Sun, Y.S.; Zhao, Z.; Yang, Z.N.; Xu, F.; Lu, H.J.; Zhu, Z.Y.; Shi, W.; Jiang, J.; Yao, P.P.; Zhu, H.P. Risk Factors and Preventions of Breast Cancer. Int. J. Biol. Sci. 2017, 13, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Dixon-Suen, S.C.; Lewis, S.J.; Martin, R.M.; English, D.R.; Boyle, T.; Giles, G.G.; Michailidou, K.; Bolla, M.K.; Wang, Q.; Dennis, J.; et al. Physical Activity, Sedentary Time and Breast Cancer Risk: A Mendelian Randomization Study. Br. J. Sports Med. 2022, 56, 1157–1170. [Google Scholar] [CrossRef]
- Boraka, Ö.; Klintman, M.; Rosendahl, A.H. Physical Activity and Long-Term Risk of Breast Cancer, Associations with Time in Life and Body Composition in the Prospective Malmö Diet and Cancer Study. Cancers 2022, 14, 1960. [Google Scholar] [CrossRef]
- Stevens, R.G.; Hansen, J.; Costa, G.; Haus, E.; Kauppinen, T.; Aronson, K.J.; Castaño-Vinyals, G.; Davis, S.; Frings-Dresen, M.H.W.; Fritschi, L.; et al. Considerations of Circadian Impact for Defining “shift Work” in Cancer Studies: IARC Working Group Report. Occup. Environ. Med. 2011, 68, 154–162. [Google Scholar] [PubMed]
- Gómez-Salgado, J.; Fagundo-Rivera, J.; Ortega-Moreno, M.; Allande-Cussó, R.; Ayuso-Murillo, D.; Ruiz-Frutos, C. Night Work and Breast Cancer Risk in Nurses: Multifactorial Risk Analysis. Cancers 2021, 13, 1470. [Google Scholar] [CrossRef]
- Szkiela, M.; Kusideł, E.; Makowiec-Dabrowska, T.; Kaleta, D. How the Intensity of Night Shift Work Affects Breast Cancer Risk. Int. J. Environ. Res. Public Health 2021, 18, 4570. [Google Scholar] [CrossRef]
- Hansen, J.; Pedersen, J.E. Night Shift Work and Breast Cancer Risk—2023 Update of Epidemiologic Evidence. J. Natl. Cancer Cent. 2025, 5, 94–103. [Google Scholar] [CrossRef]
- Sánchez-Barceló, E.J.; Cos, S.; Mediavilla, D.; Martínez-Campa, C.; González, A.; Alonso-González, C. Melatonin–Estrogen Interactions in Breast Cancer. J. Pineal Res. 2005, 38, 217–222. [Google Scholar]
- Kubatka, P.; Zubor, P.; Busselberg, D.; Kwon, T.K.; Adamek, M.; Petrovic, D.; Opatrilova, R.; Gazdikova, K.; Caprnda, M.; Rodrigo, L.; et al. Melatonin and Breast Cancer: Evidences from Preclinical and Human Studies. Crit. Rev. Oncol. Hematol. 2018, 122, 133–143. [Google Scholar] [PubMed]
- Samanta, S. Melatonin: A Potential Antineoplastic Agent in Breast Cancer. J. Environ. Pathol. Toxicol. Oncol. 2022, 41, 55–84. [Google Scholar] [PubMed]
- Wei, F.; Chen, W.; Lin, X. Night-Shift Work, Breast Cancer Incidence, and All-Cause Mortality: An Updated Meta-Analysis of Prospective Cohort Studies. Sleep Breath. 2022, 26, 1509–1526. [Google Scholar]
- Barletta, J.A. Surgical Pathology of Carcinomas. In Pathobiology of Human Disease: A Dynamic Encyclopedia of Disease Mechanisms; Elsevier: Amsterdam, The Netherlands, 2014; pp. 3526–3545. [Google Scholar]
- Tomlinson-Hansen, S.E.; Khan, M.; Cassaro, S. Breast Ductal Carcinoma in Situ. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Sokolova, A.; Lakhani, S.R. Lobular Carcinoma in Situ: Diagnostic Criteria and Molecular Correlates. Mod. Pathol. 2021, 34, 8–14. [Google Scholar]
- Sahin, A.; Zhang, H. Invasive Breast Carcinoma. In Pathobiology of Human Disease: A Dynamic Encyclopedia of Disease Mechanisms; Elsevier: Amsterdam, The Netherlands, 2014; pp. 934–951. [Google Scholar]
- Makki, J. Diversity of Breast Carcinoma: Histological Subtypes and Clinical Relevance. Clin. Med. Insights Pathol. 2015, 8, 23–31. [Google Scholar] [CrossRef]
- McCart Reed, A.E.; Kalinowski, L.; Simpson, P.T.; Lakhani, S.R. Invasive Lobular Carcinoma of the Breast: The Increasing Importance of This Special Subtype. Breast Cancer Res. 2021, 23, 6. [Google Scholar] [PubMed]
- Rosenbluth, J.M.; Overmoyer, B.A. Inflammatory Breast Cancer: A Separate Entity. Curr. Oncol. Rep. 2019, 21, 86. [Google Scholar]
- Sharma, G.N.; Dave, R.; Sanadya, J.; Sharma, P.; Sharma, K.K. Various Types and Management of Breast Cancer: An Overview. J. Adv. Pharm. Technol. Res. 2010, 1, 109–126. [Google Scholar]
- Gomes Do Nascimento, R.; Otoni, K.M. Histological and Molecular Classification of Breast Cancer: What Do We Know? Mastology 2020, 30, e20200024. [Google Scholar]
- Zhang, X. Molecular Classification of Breast Cancer: Relevance and Challenges. Arch. Pathol. Lab. Med. 2023, 147, 46–51. [Google Scholar]
- Roy, M.; Fowler, A.M.; Ulaner, G.A.; Mahajan, A. Molecular Classification of Breast Cancer. PET Clin. 2023, 18, 441–458. [Google Scholar] [PubMed]
- Métodos de Tratamento—Cancro da Mama. Available online: https://www.ligacontracancro.pt/cancro-da-mama-metodos-de-tratamento/ (accessed on 7 March 2025).
- Treatment for Breast Cancer. Available online: https://www.cancerresearchuk.org/about-cancer/breast-cancer/treatment (accessed on 7 March 2025).
- Treatment Options for Breast Cancer. Available online: https://www.cancer.org/cancer/types/breast-cancer/treatment.html (accessed on 7 March 2025).
- Fumagalli, C.; Barberis, M. Breast Cancer Heterogeneity. Diagnostics 2021, 11, 1555. [Google Scholar] [CrossRef] [PubMed]
- Gawin, M.; Kurczyk, A.; Niemiec, J.; Stanek-Widera, A.; Grela-Wojewoda, A.; Adamczyk, A.; Biskup-Frużyńska, M.; Polańska, J.; Widłak, P. Intra-Tumor Heterogeneity Revealed by Mass Spectrometry Imaging Is Associated with the Prognosis of Breast Cancer. Cancers 2021, 13, 4349. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour Heterogeneity and Resistance to Cancer Therapies. Nat. Rev. Clin. Oncol. 2017, 15, 81–94. [Google Scholar]
- Proietto, M.; Crippa, M.; Damiani, C.; Pasquale, V.; Sacco, E.; Vanoni, M.; Gilardi, M. Tumor Heterogeneity: Preclinical Models, Emerging Technologies, and Future Applications. Front. Oncol. 2023, 13, 1164535. [Google Scholar]
- Lovly, C.M.; Salama, A.K.S.; Salgia, R. Tumor Heterogeneity and Therapeutic Resistance. Am. Soc. Clin. Oncol. Educ. Book 2016, 36, 585–593. [Google Scholar]
- Tufail, M.; Cui, J.; Wu, C. Breast Cancer: Molecular Mechanisms of Underlying Resistance and Therapeutic Approaches. Am. J. Cancer Res. 2022, 12, 2920–2949. [Google Scholar]
- Kinnel, B.; Singh, S.K.; Oprea-Ilies, G.; Singh, R. Targeted Therapy and Mechanisms of Drug Resistance in Breast Cancer. Cancers 2023, 15, 1320. [Google Scholar] [CrossRef]
- Masoud, V.; Pagès, G. Targeted Therapies in Breast Cancer: New Challenges to Fight against Resistance. World J. Clin. Oncol. 2017, 8, 120–134. [Google Scholar]
- Wang, K.; Tepper, J.E. Radiation Therapy-Associated Toxicity: Etiology, Management, and Prevention. CA Cancer J. Clin. 2021, 71, 437–454. [Google Scholar]
- Langeh, U.; Kumar, V.; Ahuja, P.; Singh, C.; Singh, A. An Update on Breast Cancer Chemotherapy-Associated Toxicity and Their Management Approaches. Health Sci. Rev. 2023, 9, 100119. [Google Scholar]
- Anders, C.K.; LeBoeuf, N.R.; Bashoura, L.; Faiz, S.A.; Shariff, A.I.; Thomas, A. What’s the Price? Toxicities of Targeted Therapies in Breast Cancer Care. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, 55–70. [Google Scholar] [PubMed]
- Mark, C.; Lee, J.S.; Cui, X.; Yuan, Y. Antibody–Drug Conjugates in Breast Cancer: Current Status and Future Directions. Int. J. Mol. Sci. 2023, 24, 13726. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Xu, Y.Y.; Shao, Z.M.; Yu, K.D. Resistance to Antibody-drug Conjugates in Breast Cancer: Mechanisms and Solutions. Cancer Commun. 2022, 43, 297–337. [Google Scholar]
- Chang, H.L.; Schwettmann, B.; McArthur, H.L.; Chan, I.S. Antibody-Drug Conjugates in Breast Cancer: Overcoming Resistance and Boosting Immune Response. J. Clin. Investig. 2023, 133, e172156. [Google Scholar]
- Mohanty, S.S.; Sahoo, C.R.; Padhy, R.N. Role of Hormone Receptors and HER2 as Prospective Molecular Markers for Breast Cancer: An Update. Genes Dis. 2022, 9, 648–658. [Google Scholar]
- Clusan, L.; Ferrière, F.; Flouriot, G.; Pakdel, F. A Basic Review on Estrogen Receptor Signaling Pathways in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 6834. [Google Scholar] [CrossRef]
- Orrantia-Borunda, E.; Anchondo-Nuñez, P.; Acuña-Aguilar, L.E.; Gómez-Valles, F.O.; Ramírez-Valdespino, C.A. Subtypes of Breast Cancer. In Breast Cancer; Mayrovitz, H.N., Ed.; Exon Publications: Brisbane (AU), Australia, 2022; pp. 31–42. [Google Scholar]
- Tarantino, P.; Hamilton, E.; Tolaney, S.M.; Cortes, J.; Morganti, S.; Ferraro, E.; Marra, A.; Viale, G.; Trapani, D.; Cardoso, F.; et al. HER2-Low Breast Cancer: Pathological and Clinical Landscape. J. Clin. Oncol. 2020, 38, 1951–1962. [Google Scholar]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-Negative Breast Cancer Molecular Subtyping and Treatment Progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Allison, K.H.; Hammond, M.E.H.; Dowsett, M.; McKernin, S.E.; Carey, L.A.; Fitzgibbons, P.L.; Hayes, D.F.; Lakhani, S.R.; Chavez-MacGregor, M.; Perlmutter, J.; et al. Estrogen and Progesterone Receptor Testing in Breast Cancer: ASCO/CAP Guideline Update. J. Clin. Oncol. 2020, 38, 1346–1366. [Google Scholar]
- Eroles, P.; Bosch, A.; Alejandro Pérez-Fidalgo, J.; Lluch, A. Molecular Biology in Breast Cancer: Intrinsic Subtypes and Signaling Pathways. Cancer Treat. Rev. 2012, 38, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Breast Cancer—Molecular Subtypes. Available online: https://www.pathologyoutlines.com/topic/breastmolecularsubtypes.html (accessed on 12 March 2025).
- Atallah, N.M.; Haque, M.; Quinn, C.; Toss, M.S.; Makhlouf, S.; Ibrahim, A.; Green, A.R.; Alsaleem, M.; Rutland, C.S.; Allegrucci, C.; et al. Characterisation of Luminal and Triple-Negative Breast Cancer with HER2 Low Protein Expression. Eur. J. Cancer 2023, 195, 113371. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.C.; Somerfield, M.R.; Dowsett, M.; Hammond, M.E.H.; Hayes, D.F.; Mcshane, L.M.; Saphner, T.J.; Spears, P.A.; Allison, K.H. Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: ASCO-College of American Pathologists Guideline Update. J. Clin. Oncol. 2023, 41, 3867–3872. [Google Scholar] [CrossRef]
- Rediti, M.; Venet, D.; Joaquin Garcia, A.; Maetens, M.; Vincent, D.; Majjaj, S.; El-Abed, S.; Di Cosimo, S.; Ueno, T.; Izquierdo, M.; et al. Identification of HER2-Positive Breast Cancer Molecular Subtypes with Potential Clinical Implications in the ALTTO Clinical Trial. Nat. Commun. 2024, 15, 10402. [Google Scholar] [CrossRef]
- Zakaria, N.H.; Hashad, D.; Saied, M.H.; Hegazy, N.; Elkayal, A.; Tayae, E. Genetic Mutations in HER2-Positive Breast Cancer: Possible Association with Response to Trastuzumab Therapy. Hum. Genom. 2023, 17, 43. [Google Scholar] [CrossRef]
- Chen, C.; Li, W.; Wu, W.; Lu, X. The Characteristics of HER2 Low-Expressing Breast Cancer: A China Single-Center Real-World Data Analysis. Breast Cancer Manag. 2023, 12, BMT71. [Google Scholar] [CrossRef]
- El Haddad, G.; Diab, E.; Hajjar, M.; Aoun, M.; Mallat, F.; Zalaquett, Z.; Kourie, H.R. Insights Into the Emerging Entity of HER2-Low Breast Cancer. Int. J. Breast Cancer 2024, 2024, 2853007. [Google Scholar] [CrossRef]
- Jayachandran, P.; Deshmukh, S.K.; Wu, S.; Ribeiro, J.; Kang, I.; Xiu, J.; Farrell, A.P.; Battaglin, F.; Spicer, D.V.; Soni, S.; et al. Molecular and Immunological Characterization of Androgen Receptor Expression in Different Breast Cancer Subtypes. J. Clin. Oncol. 2024, 42, 421114. [Google Scholar] [CrossRef]
- Derakhshan, F.; Reis-Filho, J.S. Pathogenesis of Triple-Negative Breast Cancer. Annu. Rev. Pathol. 2022, 17, 181–204. [Google Scholar] [CrossRef]
- Jiang, Y.Z.; Ma, D.; Suo, C.; Shi, J.; Xue, M.; Hu, X.; Xiao, Y.; Yu, K.D.; Liu, Y.R.; Yu, Y.; et al. Genomic and Transcriptomic Landscape of Triple-Negative Breast Cancers: Subtypes and Treatment Strategies. Cancer Cell 2019, 35, 428–440.e5. [Google Scholar] [CrossRef]
- Pommier, R.M.; Sanlaville, A.; Tonon, L.; Kielbassa, J.; Thomas, E.; Ferrari, A.; Sertier, A.S.; Hollande, F.; Martinez, P.; Tissier, A.; et al. Comprehensive Characterization of Claudin-Low Breast Tumors Reflects the Impact of the Cell-of-Origin on Cancer Evolution. Nat. Commun. 2020, 11, 3431. [Google Scholar] [PubMed]
- Pan, C.; Xu, A.; Ma, X.; Yao, Y.; Zhao, Y.; Wang, C.; Chen, C. Research Progress of Claudin-Low Breast Cancer. Front. Oncol. 2023, 13, 1226118. [Google Scholar]
- Ortega, M.A.; Fraile-Martínez, O.; Asúnsolo, Á.; Buján, J.; García-Honduvilla, N.; Coca, S. Signal Transduction Pathways in Breast Cancer: The Important Role of PI3K/Akt/MTOR. J. Oncol. 2020, 2020, 9258396. [Google Scholar] [CrossRef]
- Miziak, P.; Baran, M.; Błaszczak, E.; Przybyszewska-Podstawka, A.; Kałafut, J.; Smok-Kalwat, J.; Dmoszyńska-Graniczka, M.; Kiełbus, M.; Stepulak, A. Estrogen Receptor Signaling in Breast Cancer. Cancers 2023, 15, 4689. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, R.; Picon-Ruiz, M.; Aurrekoetxea-Rodriguez, I.; Nunes de Paiva, V.; D’Amico, M.; Yoon, H.; Radhakrishnan, R.; Morata-Tarifa, C.; Ince, T.; Lippman, M.E.; et al. The Major Pre- and Postmenopausal Estrogens Play Opposing Roles in Obesity-Driven Mammary Inflammation and Breast Cancer Development. Cell Metab. 2020, 31, 1154–1172. [Google Scholar] [PubMed]
- Fuentes, N.; Silveyra, P. Estrogen Receptor Signaling Mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar]
- Gallez, A.; Dias Da Silva, I.; Wuidar, V.; Foidart, J.M.; Péqueux, C. Estetrol and Mammary Gland: Friends or Foes? J. Mammary Gland. Biol. Neoplasia 2021, 26, 297–308. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, H.; Yao, J. ERα, A Key Target for Cancer Therapy: A Review. Onco Targets Ther. 2020, 13, 2183–2191. [Google Scholar] [CrossRef]
- Björnström, L.; Sjöberg, M. Mechanisms of Estrogen Receptor Signaling: Convergence of Genomic and Nongenomic Actions on Target Genes. Mol. Endocrinol. 2005, 19, 833–842. [Google Scholar]
- Altwegg, K.A.; Vadlamudi, R.K. Role of Estrogen Receptor Coregulators in Endocrine Resistant Breast Cancer. Explor. Target. Antitumor Ther. 2021, 2, 385–400. [Google Scholar]
- Mal, R.; Magner, A.; David, J.; Datta, J.; Vallabhaneni, M.; Kassem, M.; Manouchehri, J.; Willingham, N.; Stover, D.; Vandeusen, J.; et al. Estrogen Receptor Beta (ERβ): A Ligand Activated Tumor Suppressor. Front. Oncol. 2020, 10, 587386. [Google Scholar] [CrossRef]
- Miricescu, D.; Totan, A.; Stanescu-Spinu, I.I.; Badoiu, S.C.; Stefani, C.; Greabu, M. PI3K/AKT/MTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects. Int. J. Mol. Sci. 2020, 22, 173. [Google Scholar] [CrossRef]
- Nagandla, H.; Thomas, C. Estrogen Signals through ERβ in Breast Cancer; What We Have Learned since the Discovery of the Receptor. Receptors 2024, 3, 182–200. [Google Scholar] [CrossRef] [PubMed]
- Paruthiyil, S.; Parmar, H.; Kerekatte, V.; Cunha, G.R.; Firestone, G.L.; Leitmant, D.C. Estrogen Receptor β Inhibits Human Breast Cancer Cell Proliferation and Tumor Formation by Causing a G2 Cell Cycle Arrest. Cancer Res. 2004, 64, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, X. The Role of Estrogen Receptor Beta in Breast Cancer. Biomark. Res. 2020, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y. Estrogen Receptor β Expression and Its Clinical Implication in Breast Cancers: Favorable or Unfavorable? J. Breast Cancer 2022, 25, 75–93. [Google Scholar] [CrossRef]
- Li, Z.; Wei, H.; Li, S.; Wu, P.; Mao, X. The Role of Progesterone Receptors in Breast Cancer. Drug Des. Dev. Ther. 2022, 16, 305–314. [Google Scholar] [CrossRef]
- Trabert, B.; Sherman, M.E.; Kannan, N.; Stanczyk, F.Z. Progesterone and Breast Cancer. Endocr. Rev. 2020, 41, 320–344. [Google Scholar] [CrossRef]
- Azeez, J.M.; Susmi, R.; Remadevi, V.; Ravindran, V.; Sujatha, A.S.; Nair, R.; Ayswarya, S.; Sreeja, S. New Insights into the Functions of Progesterone Receptor (PR) Isoforms and Progesterone Signaling. Am. J. Cancer Res. 2021, 11, 5214–5232. [Google Scholar]
- Pan, L.; Li, J.; Xu, Q.; Gao, Z.; Yang, M.; Wu, X.; Li, X. HER2/PI3K/AKT Pathway in HER2-Positive Breast Cancer A Review. Medicine 2024, 103, e38508. [Google Scholar] [CrossRef]
- Gutierrez, C.; Schiff, R. HER 2: Biology, Detection, and Clinical Implications. Arch. Pathol. Lab. Med. 2011, 135, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Zhou, Z.; Wang, H.; Wang, R.; Shen, K.; Huang, R.; Zhong, H.; Zhou, Z.; Wang, H.; Wang, R.; et al. The Biological Roles and Clinical Applications of the PI3K/AKT Pathway in Targeted Therapy Resistance in HER2-Positive Breast Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2024, 25, 13376. [Google Scholar] [CrossRef]
- Shirman, Y.; Lubovsky, S.; Shai, A. HER2-Low Breast Cancer: Current Landscape and Future Prospects. Breast Cancer Targets Ther. 2023, 15, 605–616. [Google Scholar]
- Zhang, H.; Katerji, H.; Turner, B.M.; Hicks, D.G. HER2-Low Breast Cancers: New Opportunities and Challenges. Am. J. Clin. Pathol. 2022, 157, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.J.; Ma, D.; Xu, Y.Z.; Li, M.; Li, Y.W.; Xiao, Y.; Jin, X.; Wu, S.Y.; Zhao, Y.X.; Wang, H.; et al. Molecular Features and Clinical Implications of the Heterogeneity in Chinese Patients with HER2-Low Breast Cancer. Nat. Commun. 2023, 14, 5112. [Google Scholar] [CrossRef] [PubMed]
- Dittrich, A.; Gautrey, H.; Browell, D.; Tyson-Capper, A. The HER2 Signaling Network in Breast Cancer—Like a Spider in Its Web. J. Mammary Gland. Biol. Neoplasia 2014, 19, 253–270. [Google Scholar]
- Bahar, M.E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK Pathway for Cancer Therapy: From Mechanism to Clinical Studies. Signal Transduct. Target. Ther. 2023, 8, 455. [Google Scholar]
- Rocca, A.; Braga, L.; Volpe, M.C.; Maiocchi, S.; Generali, D. The Predictive and Prognostic Role of RAS–RAF–MEK–ERK Pathway Alterations in Breast Cancer: Revision of the Literature and Comparison with the Analysis of Cancer Genomic Datasets. Cancers 2022, 14, 5306. [Google Scholar] [CrossRef]
- Karim, A.M.; Eun Kwon, J.; Ali, T.; Jang, J.; Ullah, I.; Lee, Y.G.; Park, D.W.; Park, J.; Jeang, J.W.; Kang, S.C. Triple-Negative Breast Cancer: Epidemiology, Molecular Mechanisms, and Modern Vaccine-Based Treatment Strategies. Biochem. Pharmacol. 2023, 212, 115545. [Google Scholar]
- Choi, E.; Mun, G.I.; Lee, J.; Lee, H.; Cho, J.; Lee, Y.S. BRCA1 Deficiency in Triple-Negative Breast Cancer: Protein Stability as a Basis for Therapy. Biomed. Pharmacother. 2023, 158, 114090. [Google Scholar]
- Kumar, P.; Aggarwal, R. An Overview of Triple-Negative Breast Cancer. Arch. Gynecol. Obstet. 2016, 293, 247–269. [Google Scholar] [CrossRef]
- Xie, C.; Luo, J.; He, Y.; Jiang, L.; Zhong, L.; Shi, Y. BRCA2 Gene Mutation in Cancer. Medicine 2022, 101, e31705. [Google Scholar] [PubMed]
- Fu, X.; Tan, W.; Song, Q.; Pei, H.; Li, J. BRCA1 and Breast Cancer: Molecular Mechanisms and Therapeutic Strategies. Front. Cell Dev. Biol. 2022, 10, 813457. [Google Scholar] [CrossRef] [PubMed]
- Na, B.; Yu, X.; Withers, T.; Gilleran, J.; Yao, M.; Foo, T.K.; Chen, C.; Moore, D.; Lin, Y.; Kimball, S.D.; et al. Therapeutic Targeting of BRCA1 and TP53 Mutant Breast Cancer through Mutant P53 Reactivation. NPJ Breast Cancer 2019, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Arimura, A.; Sakai, K.; Kaneshiro, K.; Morisaki, T.; Hayashi, S.; Mizoguchi, K.; Yamada, M.; Kai, M.; Ono, M.; Nishio, K.; et al. TP53 and/or BRCA1 Mutations Based on CtDNA Analysis as Prognostic Biomarkers for Primary Triple-Negative Breast Cancer. Cancers 2024, 16, 1184. [Google Scholar] [CrossRef]
- Telli, M.L.; Litton, J.K.; Beck, J.T.; Jones, J.M.; Andersen, J.; Mina, L.A.; Brig, R.; Danso, M.; Yuan, Y.; Symmans, W.F.; et al. Neoadjuvant Talazoparib in Patients with Germline BRCA1/2 Mutation-Positive, Early-Stage Triple-Negative Breast Cancer: Exploration of Tumor BRCA Mutational Status. Breast Cancer 2024, 31, 886–897. [Google Scholar]
- Mavaddat, N.; Dorling, L.; Carvalho, S.; Allen, J.; González-Neira, A.; Keeman, R.; Bolla, M.K.; Dennis, J.; Wang, Q.; Ahearn, T.U.; et al. Pathology of Tumors Associated With Pathogenic Germline Variants in 9 Breast Cancer Susceptibility Genes. JAMA Oncol. 2022, 8, e216744. [Google Scholar]
- Mallick, S.; Duttaroy, A.K.; Dutta, S. The PIK3CA Gene and Its Pivotal Role in Tumor Tropism of Triple-Negative Breast Cancer. Transl. Oncol. 2024, 50, 102140. [Google Scholar]
- Mosele, F.; Stefanovska, B.; Lusque, A.; Tran Dien, A.; Garberis, I.; Droin, N.; Le Tourneau, C.; Sablin, M.P.; Lacroix, L.; Enrico, D.; et al. Outcome and Molecular Landscape of Patients with PIK3CA-Mutated Metastatic Breast Cancer. Ann. Oncol. 2020, 31, 377–386. [Google Scholar] [CrossRef]
- Reinhardt, K.; Stückrath, K.; Hartung, C.; Kaufhold, S.; Uleer, C.; Hanf, V.; Lantzsch, T.; Peschel, S.; John, J.; Pöhler, M.; et al. PIK3CA-Mutations in Breast Cancer. Breast Cancer Res. Treat. 2022, 196, 483–493. [Google Scholar]
- Spring, L.M.; Gupta, A.; Reynolds, K.L.; Gadd, M.A.; Ellisen, L.W.; Isakoff, S.J.; Moy, B.; Bardia, A. Neoadjuvant Endocrine Therapy for Estrogen Receptor-Positive Breast Cancer: A Systematic Review and Meta-Analysis. JAMA Oncol. 2016, 2, 1477–1486. [Google Scholar]
- Reinbolt, R.E.; Mangini, N.; Hill, J.L.; Levine, L.B.; Dempsey, J.L.; Singaravelu, J.; Koehler, K.A.; Talley, A.; Lustberg, M.B. Endocrine Therapy in Breast Cancer: The Neoadjuvant, Adjuvant, and Metastatic Approach. Semin. Oncol. Nurs. 2015, 31, 146–155. [Google Scholar]
- Kerr, A.J.; Dodwell, D.; McGale, P.; Holt, F.; Duane, F.; Mannu, G.; Darby, S.C.; Taylor, C.W. Adjuvant and Neoadjuvant Breast Cancer Treatments: A Systematic Review of Their Effects on Mortality. Cancer Treat. Rev. 2022, 105, 102375. [Google Scholar] [CrossRef] [PubMed]
- Barchiesi, G.; Mazzotta, M.; Krasniqi, E.; Pizzuti, L.; Marinelli, D.; Capomolla, E.; Sergi, D.; Amodio, A.; Natoli, C.; Gamucci, T.; et al. Neoadjuvant Endocrine Therapy in Breast Cancer: Current Knowledge and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 3528. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; He, Y.; Li, S.; Chen, H. Crosstalk of Methylation and Tamoxifen in Breast Cancer (Review). Mol. Med. Rep. 2024, 30, 180. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Ye, L.; Sun, S.; Yuan, J.; Huang, J.; Zeng, Z. Involvement of RFC3 in Tamoxifen Resistance in ER-Positive Breast Cancer through the Cell Cycle. Aging 2023, 15, 13738–13752. [Google Scholar] [CrossRef]
- Generali, D.; Berardi, R.; Caruso, M.; Cazzaniga, M.; Garrone, O.; Minchella, I.; Paris, I.; Pinto, C.; De Placido, S. Aromatase Inhibitors: The Journey from the State of the Art to Clinical Open Questions. Front. Oncol. 2023, 13, 1249160. [Google Scholar] [CrossRef]
- Cairns, J.; Ingle, J.N.; Dudenkov, T.M.; Kalari, K.R.; Carlson, E.E.; Na, J.; Buzdar, A.U.; Robson, M.E.; Ellis, M.J.; Goss, P.E.; et al. Pharmacogenomics of Aromatase Inhibitors in Postmenopausal Breast Cancer and Additional Mechanisms of Anastrozole Action. JCI Insight 2020, 5, e137571. [Google Scholar] [CrossRef]
- Kharb, R.; Haider, K.; Neha, K.; Yar, M.S. Aromatase Inhibitors: Role in Postmenopausal Breast Cancer. Arch. Pharm. 2020, 353, e2000081. [Google Scholar] [CrossRef]
- Bradley, R.; Burrett, J.; Clarke, M.; Davies, C.; Duane, F.; Evans, V.; Gettins, L.; Godwin, J.; Gray, R.; Liu, H.; et al. Aromatase Inhibitors versus Tamoxifen in Early Breast Cancer: Patient-Level Meta-Analysis of the Randomised Trials. Lancet 2015, 386, 1341–1352. [Google Scholar]
- Bradley, R.; Braybrooke, J.; Gray, R.; Hills, R.K.; Liu, Z.; Pan, H.; Peto, R.; Dodwell, D.; McGale, P.; Taylor, C.; et al. Aromatase Inhibitors versus Tamoxifen in Premenopausal Women with Oestrogen Receptor-Positive Early-Stage Breast Cancer Treated with Ovarian Suppression: A Patient-Level Meta-Analysis of 7030 Women from Four Randomised Trials. Lancet Oncol. 2022, 23, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Stanowicka-Grada, M.; Senkus, E. Anti-HER2 Drugs for the Treatment of Advanced HER2 Positive Breast Cancer. Curr. Treat. Options Oncol. 2023, 24, 1633–1650. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Shastry, M.; Hamilton, E. Targeting HER2-Positive Breast Cancer: Advances and Future Directions. Nat. Rev. Drug Discov. 2022, 22, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Mehanna, J.; Haddad, F.G.H.; Eid, R.; Lambertini, M.; Kourié, H.R. What Is the Role of HER2-Specific Antibody Immunity in Patients with HER2-Positive Early Breast Cancer Receiving Chemotherapy plus Trastuzumab? Transl. Cancer Res. 2018, 7, 1354–1356. [Google Scholar] [CrossRef]
- Maadi, H.; Wang, Z. A Novel Mechanism Underlying the Inhibitory Effects of Trastuzumab on the Growth of HER2-Positive Breast Cancer Cells. Cells 2022, 11, 4093. [Google Scholar] [CrossRef]
- Liu, X.; Fang, Y.; Li, Y.; Li, Y.; Qi, L.; Wang, X. Pertuzumab Combined with Trastuzumab Compared to Trastuzumab in the Treatment of HER2-Positive Breast Cancer: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Front. Oncol. 2022, 12, 894861. [Google Scholar] [CrossRef]
- Tsao, L.C.; Crosby, E.J.; Trotter, T.N.; Wei, J.; Wang, T.; Yang, X.; Summers, A.N.; Lei, G.; Rabiola, C.A.; Chodosh, L.A.; et al. Trastuzumab/Pertuzumab Combination Therapy Stimulates Antitumor Responses through Complement-Dependent Cytotoxicity and Phagocytosis. JCI Insight 2022, 7, e155636. [Google Scholar] [CrossRef]
- von Minckwitz, G.; Procter, M.; de Azambuja, E.; Zardavas, D.; Benyunes, M.; Viale, G.; Suter, T.; Arahmani, A.; Rouchet, N.; Clark, E.; et al. Adjuvant Pertuzumab and Trastuzumab in Early HER2-Positive Breast Cancer. N. Eng. J. Med. 2017, 377, 122–131. [Google Scholar] [CrossRef]
- Huober, J.; Weder, P.; Ribi, K.; Thürlimann, B.; Thery, J.-C.; Li, Q.; Vanlemmens, L.; Guiu, S.; Brain, E.; Grenier, J.; et al. Pertuzumab Plus Trastuzumab With or Without Chemotherapy Followed by Emtansine in ERBB2-Positive Metastatic Breast Cancer: A Secondary Analysis of a Randomized Clinical Trial. JAMA Oncol. 2023, 9, 1381–1389. [Google Scholar] [CrossRef]
- Peng, D.M.; Li, J.; Qiu, J.X.; Zhao, L. Neoadjuvant Pertuzumab plus Trastuzumab in Combination with Chemotherapy for Human Epidermal Growth Factor Receptor 2 Positive Breast Cancer: A Real-World Retrospective Single-Institutional Study in China. World J. Surg. Oncol. 2024, 22, 88. [Google Scholar] [CrossRef]
- González, A.F.; Jurado, J.C.; Valenti, E.L.; Cubero, R.U.; de la Gala, M.C.Á.; Guisado, M.A.M.; Ambite, R.Á.; González, C.J.R.; Góngora, M.A.; Pérez, L.R.; et al. Real-World Experience with Pertuzumab and Trastuzumab Combined with Chemotherapy in Neoadjuvant Treatment for Patients with Early-Stage HER2-Positive Breast Cancer: The NEOPERSUR Study. Clin. Transl. Oncol. 2024, 26, 2217–2226. [Google Scholar] [CrossRef]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab Emtansine: Mechanisms of Action and Drug Resistance. Breast Cancer Res. 2014, 16, 209. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.; Pandiella, A.; Vargas-Castrillón, E.; Díaz-Rodríguez, E.; Iglesias-Hernangómez, T.; Martínez Cano, C.; Fernández-Cuesta, I.; Winkow, E.; Perelló, M.F. Trastuzumab Deruxtecan in Breast Cancer. Crit. Rev. Oncol. Hematol. 2024, 198, 104355. [Google Scholar] [PubMed]
- Hurvitz, S.A.; Hegg, R.; Chung, W.P.; Im, S.A.; Jacot, W.; Ganju, V.; Chiu, J.W.Y.; Xu, B.; Hamilton, E.; Madhusudan, S.; et al. Trastuzumab Deruxtecan versus Trastuzumab Emtansine in Patients with HER2-Positive Metastatic Breast Cancer: Updated Results from DESTINY-Breast03, a Randomised, Open-Label, Phase 3 Trial. Lancet 2023, 401, 105–117. [Google Scholar] [CrossRef]
- Iwata, H.; Xu, B.; Kim, S.B.; Chung, W.P.; Park, Y.H.; Kim, M.H.; Tseng, L.M.; Chung, C.F.; Huang, C.S.; Kim, J.H.; et al. Trastuzumab Deruxtecan versus Trastuzumab Emtansine in Asian Patients with HER2-positive Metastatic Breast Cancer. Cancer Sci. 2024, 115, 3079–3088. [Google Scholar] [CrossRef] [PubMed]
- Sarno, F.; Dell’aversana, C.; Cappetta, D.; Megchelenbrink, W.L.; Obidiro, O.; Battogtokh, G.; Akala, E.O. Triple Negative Breast Cancer Treatment Options and Limitations: Future Outlook. Pharmaceutics 2023, 15, 1796. [Google Scholar] [CrossRef]
- van den Ende, N.S.; Nguyen, A.H.; Jager, A.; Kok, M.; Debets, R.; van Deurzen, C.H.M. Triple-Negative Breast Cancer and Predictive Markers of Response to Neoadjuvant Chemotherapy: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 2969. [Google Scholar] [CrossRef]
- Braybrooke, J.; Bradley, R.; Gray, R.; Hills, R.K.; Pan, H.; Peto, R.; Dodwell, D.; McGale, P.; Taylor, C.; Aihara, T.; et al. Anthracycline-Containing and Taxane-Containing Chemotherapy for Early-Stage Operable Breast Cancer: A Patient-Level Meta-Analysis of 100 000 Women from 86 Randomised Trials. Lancet 2023, 401, 1277–1292. [Google Scholar]
- Mason, S.R.E.; Willson, M.L.; Egger, S.J.; Beith, J.; Dear, R.F.; Goodwin, A. Platinum-Based Chemotherapy for Early Triple-Negative Breast Cancer. Cochrane Database Syst. Rev. 2023, 9, CD014805. [Google Scholar]
- Wang, X.; Wang, S.S.; Huang, H.; Cai, L.; Zhao, L.; Peng, R.J.; Lin, Y.; Tang, J.; Zeng, J.; Zhang, L.H.; et al. Effect of Capecitabine Maintenance Therapy Using Lower Dosage and Higher Frequency vs Observation on Disease-Free Survival Among Patients With Early-Stage Triple-Negative Breast Cancer Who Had Received Standard Treatment: The SYSUCC-001 Randomized Clinical Trial. JAMA 2020, 325, 50–58. [Google Scholar]
- Yao, Y.; Chu, Y.; Xu, B.; Hu, Q.; Song, Q. Radiotherapy after Surgery Has Significant Survival Benefits for Patients with Triple-negative Breast Cancer. Cancer Med. 2019, 8, 554–563. [Google Scholar]
- Liu, Y.; Hu, Y.; Xue, J.; Li, J.; Yi, J.; Bu, J.; Zhang, Z.; Qiu, P.; Gu, X. Advances in Immunotherapy for Triple-Negative Breast Cancer. Mol. Cancer 2023, 22, 145. [Google Scholar]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus Nab-Paclitaxel as First-Line Treatment for Unresectable, Locally Advanced or Metastatic Triple-Negative Breast Cancer (IMpassion130): Updated Efficacy Results from a Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2020, 21, 44–59. [Google Scholar]
- Adams, S.; Loi, S.; Toppmeyer, D.; Cescon, D.W.; De Laurentiis, M.; Nanda, R.; Winer, E.P.; Mukai, H.; Tamura, K.; Armstrong, A.; et al. Pembrolizumab Monotherapy for Previously Untreated, PD-L1-Positive, Metastatic Triple-Negative Breast Cancer: Cohort B of the Phase II KEYNOTE-086 Study. Ann. Oncol. 2019, 30, 405–411. [Google Scholar]
- Leon-Ferre, R.A.; Goetz, M.P. Advances in Systemic Therapies for Triple Negative Breast Cancer. BMJ 2023, 381, e071674. [Google Scholar]
- Li, Y.; Zhang, H.; Merkher, Y.; Chen, L.; Liu, N.; Leonov, S.; Chen, Y. Recent Advances in Therapeutic Strategies for Triple-Negative Breast Cancer. J. Hematol. Oncol. 2022, 15, 121. [Google Scholar] [CrossRef]
- Zhu, S.; Wu, Y.; Song, B.; Yi, M.; Yan, Y.; Mei, Q.; Wu, K. Recent Advances in Targeted Strategies for Triple-Negative Breast Cancer. J. Hematol. Oncol. 2023, 16, 100. [Google Scholar] [CrossRef]
- Guo, L.; Kong, D.; Liu, J.; Zhan, L.; Luo, L.; Zheng, W.; Zheng, Q.; Chen, C.; Sun, S. Breast Cancer Heterogeneity and Its Implication in Personalized Precision Therapy. Exp. Hematol. Oncol. 2023, 12, 3. [Google Scholar]
- Wu, F.; Fan, J.; He, Y.; Xiong, A.; Yu, J.; Li, Y.; Zhang, Y.; Zhao, W.; Zhou, F.; Li, W.; et al. Single-Cell Profiling of Tumor Heterogeneity and the Microenvironment in Advanced Non-Small Cell Lung Cancer. Nat. Commun. 2021, 12, 2540. [Google Scholar]
- Ding, S.; Chen, X.; Shen, K. Single-cell RNA Sequencing in Breast Cancer: Understanding Tumor Heterogeneity and Paving Roads to Individualized Therapy. Cancer Commun. 2020, 40, 329–344. [Google Scholar]
- Liang, Y.; Zhang, H.; Song, X.; Yang, Q. Metastatic Heterogeneity of Breast Cancer: Molecular Mechanism and Potential Therapeutic Targets. Semin. Cancer Biol. 2020, 60, 14–27. [Google Scholar] [CrossRef]
- Kundu, M.; Butti, R.; Panda, V.K.; Malhotra, D.; Das, S.; Mitra, T.; Kapse, P.; Gosavi, S.W.; Kundu, G.C. Modulation of the Tumor Microenvironment and Mechanism of Immunotherapy-Based Drug Resistance in Breast Cancer. Mol. Cancer 2024, 23, 92. [Google Scholar] [CrossRef]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2019, 174, 1293–1308.e36. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.; Mo, H.; Hu, X.; Gao, R.; Zhao, Y.; Liu, B.; Niu, L.; Sun, X.; Yu, X.; et al. Single-Cell Analyses Reveal Key Immune Cell Subsets Associated with Response to PD-L1 Blockade in Triple-Negative Breast Cancer. Cancer Cell 2021, 39, 1578–1593.e8. [Google Scholar] [CrossRef]
- Belachew, E.B.; Sewasew, D.T. Molecular Mechanisms of Endocrine Resistance in Estrogen-Positive Breast Cancer. Front Endocrinol. 2021, 12, 599586. [Google Scholar] [CrossRef]
- Yao, J.; Deng, K.; Huang, J.; Zeng, R.; Zuo, J. Progress in the Understanding of the Mechanism of Tamoxifen Resistance in Breast Cancer. Front. Pharmacol. 2020, 11, 592912. [Google Scholar] [CrossRef]
- Saatci, O.; Huynh-Dam, K.T.; Sahin, O. Endocrine Resistance in Breast Cancer: From Molecular Mechanisms to Therapeutic Strategies. J. Mol. Med. 2021, 99, 1691–1710. [Google Scholar] [CrossRef]
- Fan, W.; Chang, J.; Fu, P. Endocrine Therapy Resistance in Breast Cancer: Current Status, Possible Mechanisms and Overcoming Strategies. Future Med. Chem. 2015, 7, 1511–1519. [Google Scholar] [CrossRef]
- Bos, M.K.; Deger, T.; Sleijfer, S.; Martens, J.W.M.; Wilting, S.M. ESR1 Methylation Measured in Cell-Free DNA to Evaluate Endocrine Resistance in Metastatic Breast Cancer Patients. Int. J. Mol. Sci. 2022, 23, 5631. [Google Scholar] [CrossRef]
- Kirn, V.; Strake, L.; Thangarajah, F.; Richters, L.; Eischeid, H.; Koitzsch, U.; Odenthal, M.; Fries, J. ESR1-Promoter-Methylation Status in Primary Breast Cancer and Its Corresponding Metastases. Clin. Exp. Metastasis 2018, 35, 707–712. [Google Scholar] [CrossRef]
- Brett, J.O.; Spring, L.M.; Bardia, A.; Wander, S.A. ESR1 Mutation as an Emerging Clinical Biomarker in Metastatic Hormone Receptor-Positive Breast Cancer. Breast Cancer Res. 2021, 23, 85. [Google Scholar]
- André, F.; Solovieff, N.; Su, F.; Bardia, A.; Neven, P.; Yap, Y.S.; Tripathy, D.; Lu, Y.-S.; Slamon, D.; Chia, S.; et al. Acquired Gene Alterations in Patients Treated with Ribociclib plus Endocrine Therapy or Endocrine Therapy Alone Using Baseline and End-of-Treatment Circulating Tumor DNA Samples in the MONALEESA-2, -3, and -7 Trials. Ann. Oncol. 2025, 36, 54–64. [Google Scholar] [PubMed]
- Raheem, F.; Karikalan, S.A.; Batalini, F.; El Masry, A.; Mina, L. Metastatic ER+ Breast Cancer: Mechanisms of Resistance and Future Therapeutic Approaches. Int. J. Mol. Sci. 2023, 24, 16198. [Google Scholar] [CrossRef] [PubMed]
- Tolaney, S.M.; Toi, M.; Neven, P.; Sohn, J.; Grischke, E.M.; Llombart-Cussac, A.; Soliman, H.; Wang, H.; Wijayawardana, S.; Jansen, V.M.; et al. Clinical Significance of PIK3CA and ESR1 Mutations in Circulating Tumor DNA: Analysis from the MONARCH 2 Study of Abemaciclib plus Fulvestrant. Clin. Cancer Res. 2022, 28, 1500–1506. [Google Scholar] [PubMed]
- Wu, X.; Yang, H.; Yu, X.; Qin, J.J. Drug-Resistant HER2-Positive Breast Cancer: Molecular Mechanisms and Overcoming Strategies. Front. Pharmacol. 2022, 13, 1012552. [Google Scholar]
- Nami, B.; Ghanaeian, A.; Wang, Z. Epigenetic Downregulation of HER2 during EMT Leads to Tumor Resistance to HER2-Targeted Therapies in Breast Cancer. bioRxiv 2020, 1–38. [Google Scholar] [CrossRef]
- Eliyatkin, N.O.; Aktas, S.; Ozgur, H.; Ercetin, P.; Kupelioglu, A. The Role of P95HER2 in Trastuzumab Resistance in Breast Cancer. J. BUON 2016, 21, 382–389. [Google Scholar]
- Xing, F.; Gao, H.; Chen, G.; Sun, L.; Sun, J.; Qiao, X.; Xue, J.; Liu, C. CMTM6 Overexpression Confers Trastuzumab Resistance in HER2-Positive Breast Cancer. Mol. Cancer 2023, 22, 6. [Google Scholar]
- Zimmerman, B.S.; Esteva, F.J. Next-Generation HER2-Targeted Antibody–Drug Conjugates in Breast Cancer. Cancers 2024, 16, 800. [Google Scholar] [CrossRef]
- Hunter, F.W.; Barker, H.R.; Lipert, B.; Rothé, F.; Gebhart, G.; Piccart-Gebhart, M.J.; Sotiriou, C.; Jamieson, S.M.F. Mechanisms of Resistance to Trastuzumab Emtansine (T-DM1) in HER2-Positive Breast Cancer. Br. J. Cancer 2019, 122, 603–612. [Google Scholar]
- Mehraj, U.; Dar, A.H.; Wani, N.A.; Mir, M.A. Tumor Microenvironment Promotes Breast Cancer Chemoresistance. Cancer Chemother. Pharmacol. 2021, 87, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zhang, H.; Fu, Y.; Kuang, J.; Zhao, B.; Zhang, L.F.; Lin, J.; Lin, S.; Wu, D.; Xie, G. Cancer-Associated Fibroblasts Induce Growth and Radioresistance of Breast Cancer Cells through Paracrine IL-6. Cell Death Discov. 2023, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; He, J.; Yin, L.; Chen, X.; Zu, X.; Shen, Y. Tumor-Associated Macrophages: Critical Players in Drug Resistance of Breast Cancer. Front. Immunol. 2021, 12, 799428. [Google Scholar] [CrossRef]
- Liang, Y.; Wang, Y.; Zhang, Y.; Ye, F.; Luo, D.; Li, Y.; Jin, Y.; Han, D.; Wang, Z.; Chen, B.; et al. HSPB1 Facilitates Chemoresistance through Inhibiting Ferroptotic Cancer Cell Death and Regulating NF-ΚB Signaling Pathway in Breast Cancer. Cell Death Dis. 2023, 14, 434. [Google Scholar] [CrossRef] [PubMed]
- Nedeljković, M.; Damjanović, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer—How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef]
- Campagna, R.; Pozzi, V.; Giorgini, S.; Morichetti, D.; Goteri, G.; Sartini, D.; Serritelli, E.N.; Emanuelli, M. Paraoxonase-2 Is Upregulated in Triple Negative Breast Cancer and Contributes to Tumor Progression and Chemoresistance. Hum. Cell 2023, 36, 1108–1119. [Google Scholar] [CrossRef]
- Wang, Y.; Zeng, J.; Wu, W.; Xie, S.; Yu, H.; Li, G.; Zhu, T.; Li, F.; Lu, J.; Wang, G.Y.; et al. Nicotinamide N-Methyltransferase Enhances Chemoresistance in Breast Cancer through SIRT1 Protein Stabilization. Breast Cancer Res. 2019, 21, 64. [Google Scholar] [CrossRef]
- Lawson, M.; Cureton, N.; Ros, S.; Cheraghchi-Bashi, A.; Urosevic, J.; D’Arcy, S.; Delpuech, O.; DuPont, M.; Fisher, D.I.; Gangl, E.T.; et al. The Next-Generation Oral Selective Estrogen Receptor Degrader Camizestrant (AZD9833) Suppresses ER+ Breast Cancer Growth and Overcomes Endocrine and CDK4/6 Inhibitor Resistance. Cancer Res. 2023, 83, 3989–4004. [Google Scholar] [CrossRef]
- Crucitta, S.; Ruglioni, M.; Lorenzini, G.; Bargagna, I.; Luculli, G.I.; Albanese, I.; Bilancio, D.; Patanè, F.; Fontana, A.; Danesi, R.; et al. CDK4/6 Inhibitors Overcome Endocrine ESR1 Mutation-Related Resistance in Metastatic Breast Cancer Patients. Cancers 2023, 15, 1306. [Google Scholar] [CrossRef]
- Chung, W.-P.; Huang, W.-L.; Lee, C.-H.; Hsu, H.-P.; Huang, W.-L.; Liu, Y.-Y.; Su, W.-C. PI3K Inhibitors in Trastuzumab-Resistant HER2-Positive Breast Cancer Cells with PI3K Pathway Alterations. Am. J. Cancer Res. 2022, 12, 3067–3082. [Google Scholar]
- Murthy, R.K.; Loi, S.; Okines, A.; Paplomata, E.; Hamilton, E.; Hurvitz, S.A.; Lin, N.U.; Borges, V.; Abramson, V.; Anders, C.; et al. Tucatinib, Trastuzumab, and Capecitabine for HER2-Positive Metastatic Breast Cancer. N. Eng. J. Med. 2020, 382, 597–609. [Google Scholar]
- Cortes, J.; Rugo, H.S.; Cescon, D.W.; Im, S.-A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Perez-Garcia, J.; Iwata, H.; et al. Pembrolizumab plus Chemotherapy in Advanced Triple-Negative Breast Cancer. N. Eng. J. Med. 2022, 387, 217–226. [Google Scholar]
- Liu, X.; Wu, K.; Zheng, D.; Luo, C.; Fan, Y.; Zhong, X.; Zheng, H. Efficacy and Safety of PARP Inhibitors in Advanced or Metastatic Triple-Negative Breast Cancer: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 742139. [Google Scholar]
- Zhu, X.; Chen, L.; Huang, B.; Li, X.; Yang, L.; Hu, X.; Jiang, Y.; Shao, Z.; Wang, Z. Efficacy and Mechanism of the Combination of PARP and CDK4/6 Inhibitors in the Treatment of Triple-Negative Breast Cancer. J. Exp. Clin. Cancer Res. 2021, 40, 122. [Google Scholar] [CrossRef]
- Howell, S.J.; Casbard, A.; Carucci, M.; Ingarfield, K.; Butler, R.; Morgan, S.; Meissner, M.; Bale, C.; Bezecny, P.; Moon, S.; et al. Fulvestrant plus Capivasertib versus Placebo after Relapse or Progression on an Aromatase Inhibitor in Metastatic, Oestrogen Receptor-Positive, HER2-Negative Breast Cancer (FAKTION): Overall Survival, Updated Progression-Free Survival, and Expanded Biomarker Analysis from a Randomised, Phase 2 Trial. Lancet Oncol. 2022, 23, 851–864. [Google Scholar]
- Ma, F.; Yan, M.; Li, W.; Ouyang, Q.; Tong, Z.; Teng, Y.; Wang, Y.; Wang, S.; Geng, C.; Luo, T.; et al. Pyrotinib versus Placebo in Combination with Trastuzumab and Docetaxel as First Line Treatment in Patients with HER2 Positive Metastatic Breast Cancer (PHILA): Randomised, Double Blind, Multicentre, Phase 3 Trial. BMJ 2023, 383, e076065. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Eng. J. Med. 2018, 379, 2108–2121. [Google Scholar]
- Dent, R.; André, F.; Gonçalves, A.; Martin, M.; Schmid, P.; Schütz, F.; Kümmel, S.; Swain, S.M.; Bilici, A.; Loirat, D.; et al. IMpassion132 Double-Blind Randomised Phase III Trial of Chemotherapy with or without Atezolizumab for Early Relapsing Unresectable Locally Advanced or Metastatic Triple-Negative Breast Cancer. Ann. Oncol. 2024, 35, 630–642. [Google Scholar] [CrossRef]
- Correia, A.S.; Gärtner, F.; Vale, N. Drug Combination and Repurposing for Cancer Therapy: The Example of Breast Cancer. Heliyon 2021, 7, e05948. [Google Scholar]
- Wang, Y.; Minden, A. Current Molecular Combination Therapies Used for the Treatment of Breast Cancer. Int. J. Mol. Sci. 2022, 23, 11046. [Google Scholar] [CrossRef]
- Yeo, S.K.; Guan, J.L. Breast Cancer: Multiple Subtypes within a Tumor? Trends Cancer 2018, 3, 753–760. [Google Scholar] [CrossRef] [PubMed]
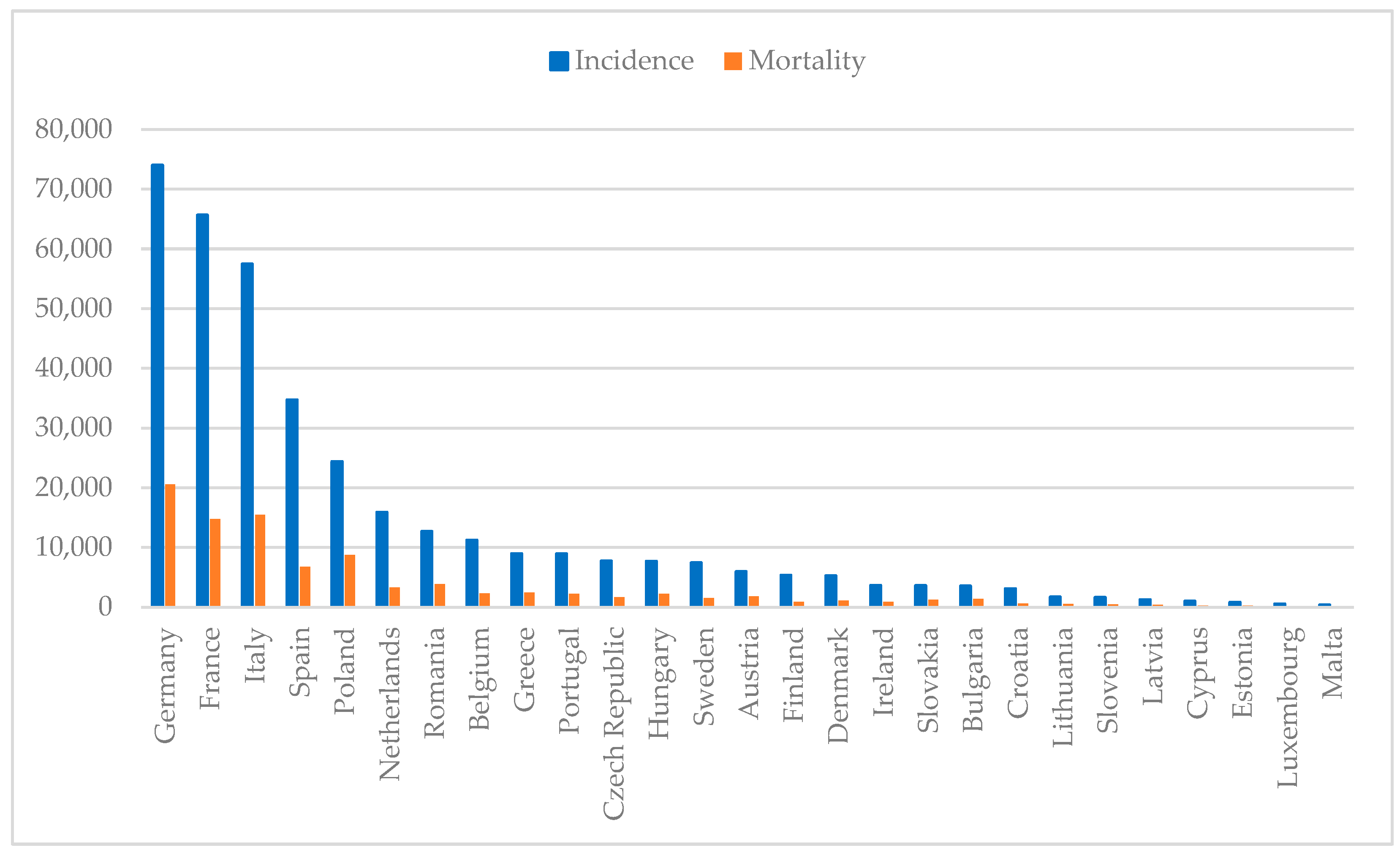
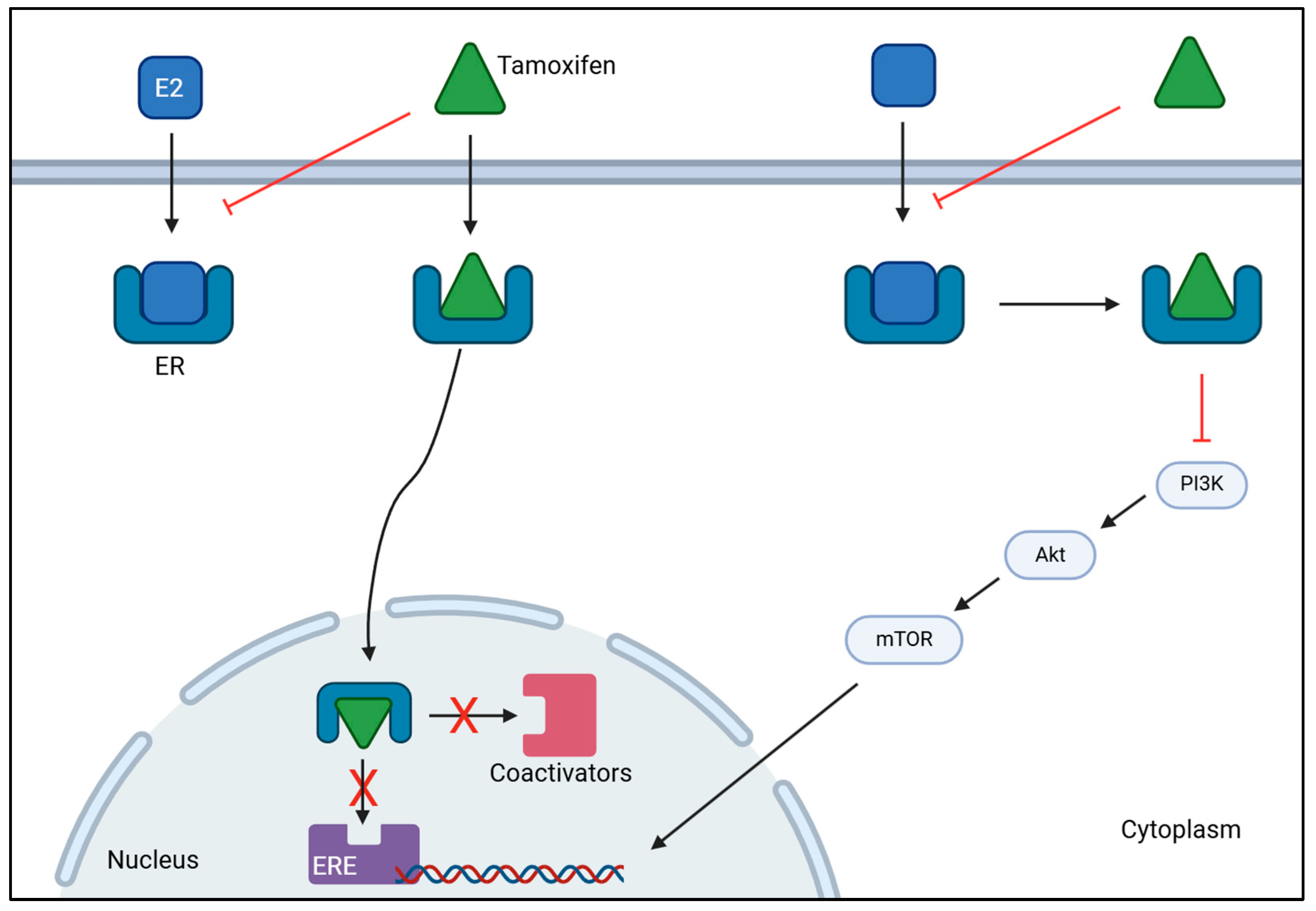
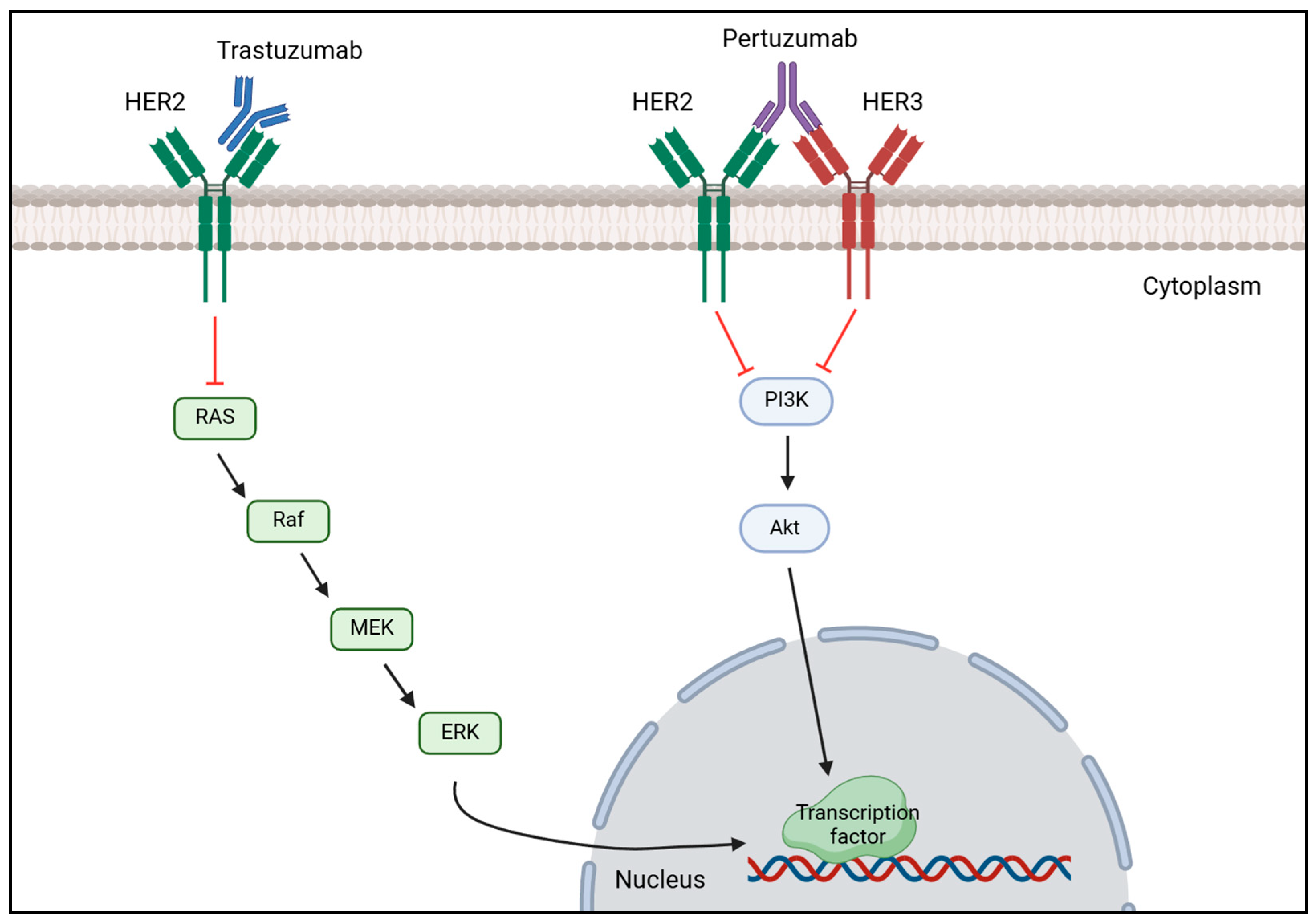
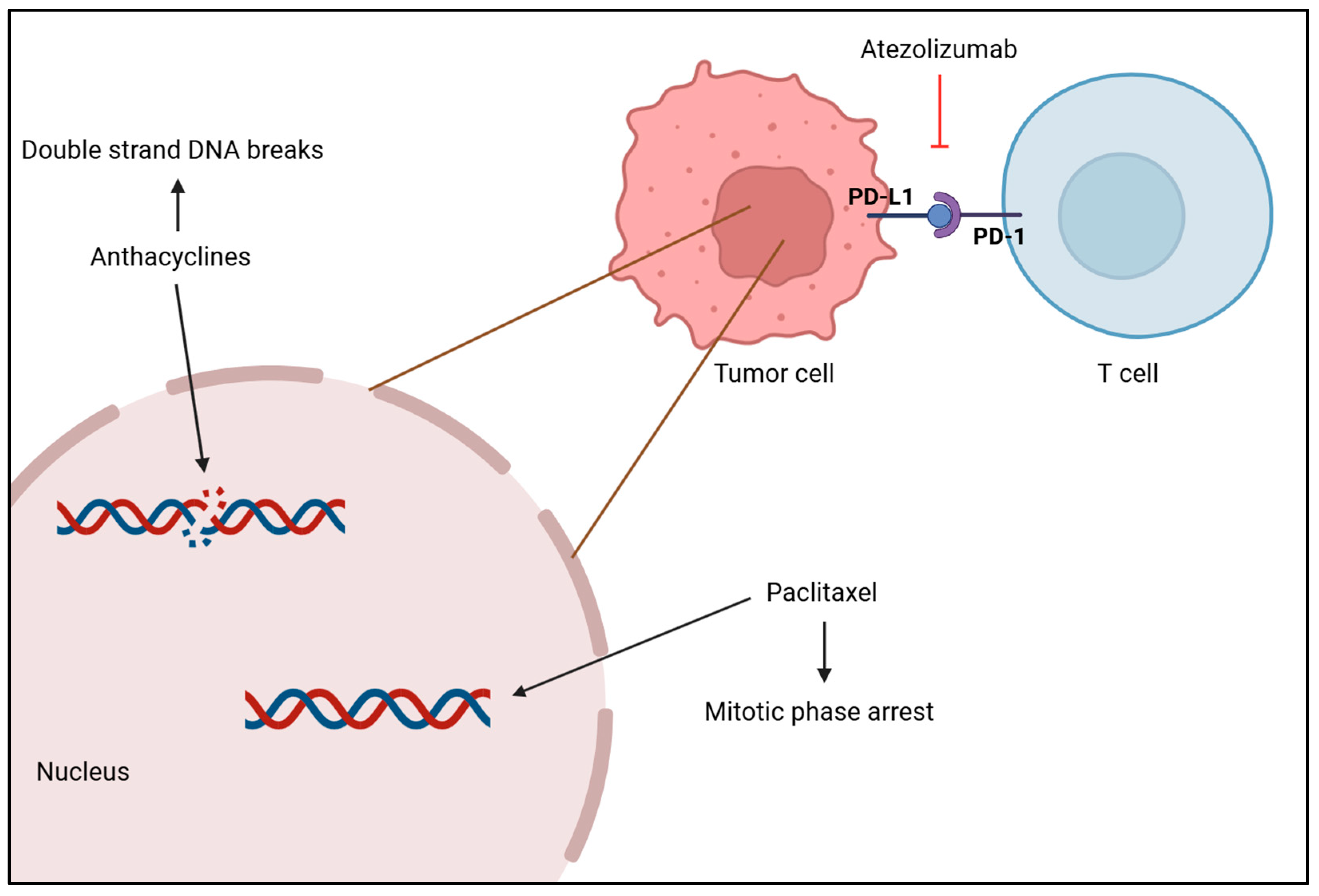
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).