What Have We Learnt from the Recent Multimodal Managements of Young Patients with ATRT?


Simple Summary
Abstract
1. Introduction/Historical Background
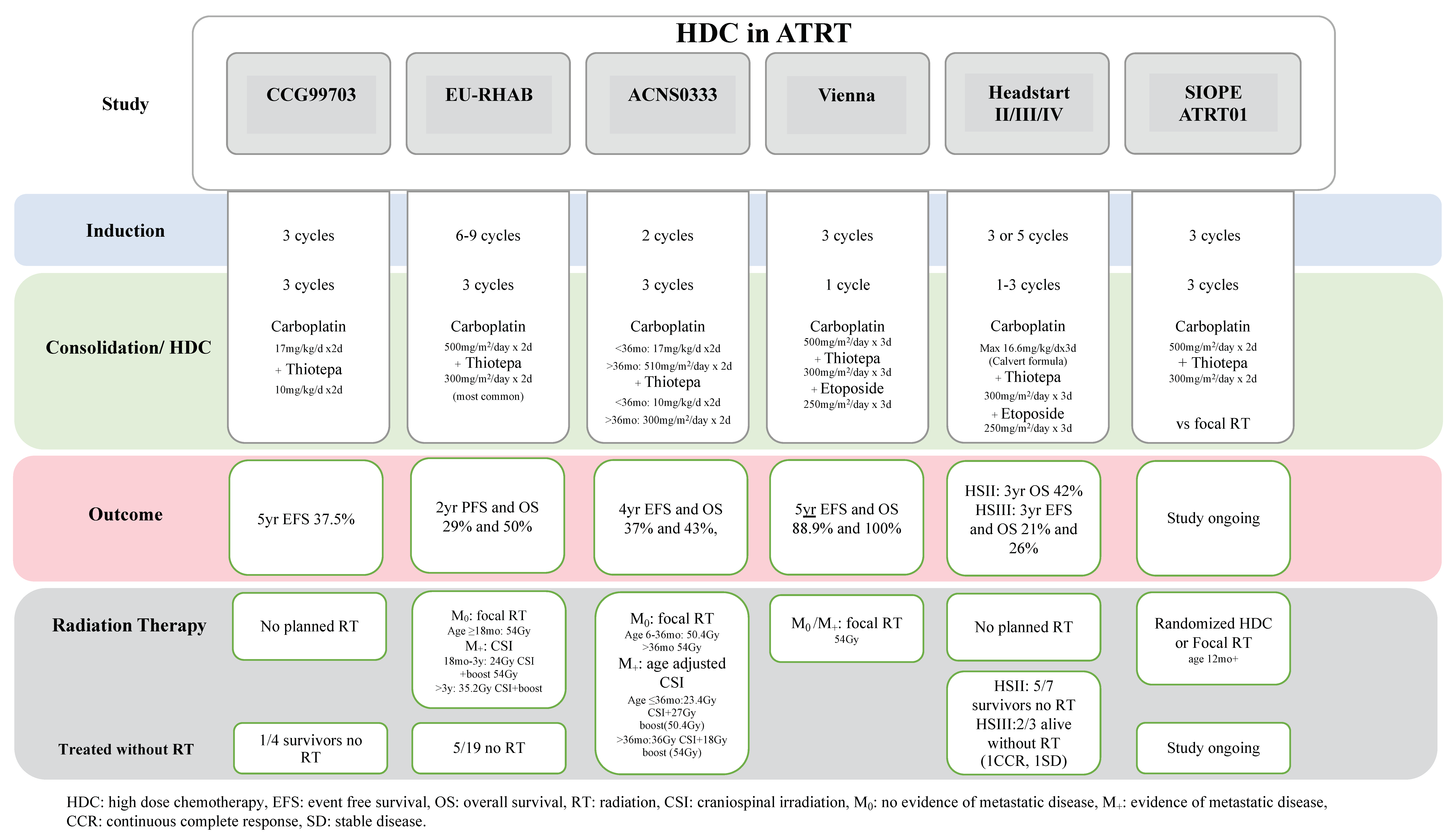
2. HDC for Atypical Teratoid Rhabdoid Tumors
3. Contribution of Molecular Characterization
3.1. Molecular Landscape of ATRT
3.2. Prognostic Value of ATRT Molecular Subgroups
4. Who Benefits from These Intensive Multimodal Therapies?
5. Approach to Relapse/Recurrent ATRT
6. Future Perspectives and Research
6.1. Exploring the Role of Intrathecal Therapy (It) in Upfront Treatment for ATRT
6.2. Exploring the Role of Maintenance Therapy in ATRT
6.3. Preclinical Drug Screening for Molecularly Informed Treatment
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Woehrer, A.; Slavc, I.; Waldhoer, T.; Heinzl, H.; Zielonke, N.; Czech, T.; Benesch, M.; Hainfellner, J.A.; Haberler, C.; Registry, A.B.T. Incidence of atypical teratoid/rhabdoid tumors in children: A population-based study by the Austrian Brain Tumor Registry, 1996–2006. Cancer 2010, 116, 5725–5732. [Google Scholar] [CrossRef]
- Fossey, M.; Li, H.; Afzal, S.; Carret, A.S.; Eisenstat, D.D.; Fleming, A.; Hukin, J.; Hawkins, C.; Jabado, N.; Johnston, D.; et al. Atypical teratoid rhabdoid tumor in the first year of life: The Canadian ATRT registry experience and review of the literature. J. Neuro-Oncol. 2017, 132, 155–162. [Google Scholar] [CrossRef]
- Squire, S.E.; Chan, M.D.; Marcus, K.J. Atypical teratoid/rhabdoid tumor: The controversy behind radiation therapy. J. Neurooncol. 2007, 81, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Sultan, I.; Qaddoumi, I.; Rodríguez-Galindo, C.; Nassan, A.A.; Ghandour, K.; Al-Hussaini, M. Age, stage, and radiotherapy, but not primary tumor site, affects the outcome of patients with malignant rhabdoid tumors. Pediatr. Blood Cancer 2010, 54, 35–40. [Google Scholar] [CrossRef]
- Isikay, I.; Hanalioglu, S.; Basar, I.; Narin, F.; Bilginer, B. Survival Benefit with Gross Total Resection and Adjuvant Radiotherapy in Childhood Atypical Teratoid/Rhabdoid Tumors: Results of a Single-Center Cohort of 27 Cases. Turk. Neurosurg. 2019, 29, 689–697. [Google Scholar] [CrossRef] [PubMed]
- von Hoff, K.; Hinkes, B.; Dannenmann-Stern, E.; von Bueren, A.O.; Warmuth-Metz, M.; Soerensen, N.; Emser, A.; Zwiener, I.; Schlegel, P.G.; Kuehl, J.; et al. Frequency, risk-factors and survival of children with atypical teratoid rhabdoid tumors (AT/RT) of the CNS diagnosed between 1988 and 2004, and registered to the German HIT database. Pediatr. Blood Cancer 2011, 57, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Strother, D.R.; Lafay-Cousin, L.; Boyett, J.M.; Burger, P.; Aronin, P.; Constine, L.; Duffner, P.; Kocak, M.; Kun, L.E.; Horowitz, M.E.; et al. Benefit from prolonged dose-intensive chemotherapy for infants with malignant brain tumors is restricted to patients with ependymoma: A report of the Pediatric Oncology Group randomized controlled trial 9233/34. Neuro-Oncol. 2014, 16, 457–465. [Google Scholar] [CrossRef]
- Chi, S.N.; Zimmerman, M.A.; Yao, X.; Cohen, K.J.; Burger, P.; Biegel, J.A.; Rorke-Adams, L.B.; Fisher, M.J.; Janss, A.; Mazewski, C.; et al. Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J. Clin. Oncol. 2009, 27, 385–389. [Google Scholar] [CrossRef]
- Bartelheim, K.; Nemes, K.; Seeringer, A.; Kerl, K.; Buechner, J.; Boos, J.; Graf, N.; Durken, M.; Gerss, J.; Hasselblatt, M.; et al. Improved 6-year overall survival in AT/RT—Results of the registry study Rhabdoid 2007. Cancer Med. 2016, 5, 1765–1775. [Google Scholar] [CrossRef]
- Upadhyaya, S.A.; Robinson, G.W.; Onar-Thomas, A.; Orr, B.A.; Johann, P.; Wu, G.; Billups, C.A.; Tatevossian, R.G.; Dhanda, S.K.; Srinivasan, A.; et al. Relevance of Molecular Groups in Children with Newly Diagnosed Atypical Teratoid Rhabdoid Tumor: Results from Prospective St. Jude Multi-institutional Trials. Clin. Cancer Res. 2021, 27, 2879–2889. [Google Scholar] [CrossRef]
- Cohen, B.H.; Geyer, J.R.; Miller, D.C.; Curran, J.G.; Zhou, T.; Holmes, E.; Ingles, S.A.; Dunkel, I.J.; Hilden, J.; Packer, R.J.; et al. Pilot Study of Intensive Chemotherapy With Peripheral Hematopoietic Cell Support for Children Less Than 3 Years of Age With Malignant Brain Tumors, the CCG-99703 Phase I/II Study. A Report From the Children’s Oncology Group. Pediatr. Neurol. 2015, 53, 31–46. [Google Scholar] [CrossRef]
- Gardner, S.L.; Asgharzadeh, S.; Green, A.; Horn, B.; McCowage, G.; Finlay, J. Intensive induction chemotherapy followed by high dose chemotherapy with autologous hematopoietic progenitor cell rescue in young children newly diagnosed with central nervous system atypical teratoid rhabdoid tumors. Pediatr. Blood Cancer 2008, 51, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.; Bartelheim, K.; Fleischhack, G.; Gruhn, B.; Schlegel, P.G.; Witt, O.; Stachel, K.D.; Hauch, H.; Urban, C.; Quehenberger, F.; et al. High-dose chemotherapy (HDCT) with auto-SCT in children with atypical teratoid/rhabdoid tumors (AT/RT): A report from the European Rhabdoid Registry (EU-RHAB). Bone Marrow Transpl. 2014, 49, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Slavc, I.; Chocholous, M.; Leiss, U.; Haberler, C.; Peyrl, A.; Azizi, A.A.; Dieckmann, K.; Woehrer, A.; Peters, C.; Widhalm, G.; et al. Atypical teratoid rhabdoid tumor: Improved long-term survival with an intensive multimodal therapy and delayed radiotherapy. The Medical University of Vienna Experience 1992–2012. Cancer Med. 2014, 3, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.T.; Strother, D.R.; Judkins, A.R.; Burger, P.C.; Pollack, I.F.; Krailo, M.D.; Buxton, A.B.; Williams-Hughes, C.; Fouladi, M.; Mahajan, A.; et al. Efficacy of High-Dose Chemotherapy and Three-Dimensional Conformal Radiation for Atypical Teratoid/Rhabdoid Tumor: A Report From the Children’s Oncology Group Trial ACNS0333. J. Clin. Oncol. 2020, 38, 1175–1185. [Google Scholar] [CrossRef]
- Lafay-Cousin, L.; Hawkins, C.; Carret, A.S.; Johnston, D.; Zelcer, S.; Wilson, B.; Jabado, N.; Scheinemann, K.; Eisenstat, D.; Fryer, C.; et al. Central nervous system atypical teratoid rhabdoid tumours: The Canadian Paediatric Brain Tumour Consortium experience. Eur. J. Cancer 2012, 48, 353–359. [Google Scholar] [CrossRef]
- Hilden, J.M.; Meerbaum, S.; Burger, P.; Finlay, J.; Janss, A.; Scheithauer, B.W.; Walter, A.W.; Rorke, L.B.; Biegel, J.A. Central nervous system atypical teratoid/rhabdoid tumor: Results of therapy in children enrolled in a registry. J. Clin. Oncol. 2004, 22, 2877–2884. [Google Scholar] [CrossRef]
- Lafay-Cousin, L.; Fay-McClymont, T.; Johnston, D.; Fryer, C.; Scheinemann, K.; Fleming, A.; Hukin, J.; Janzen, L.; Guger, S.; Strother, D.; et al. Neurocognitive evaluation of long term survivors of atypical teratoid rhabdoid tumors (ATRT): The Canadian registry experience. Pediatr. Blood Cancer 2015, 62, 1265–1269. [Google Scholar] [CrossRef]
- Torchia, J.; Golbourn, B.; Feng, S.; Ho, K.C.; Sin-Chan, P.; Vasiljevic, A.; Norman, J.D.; Guilhamon, P.; Garzia, L.; Agamez, N.R.; et al. Integrated (epi)-Genomic Analyses Identify Subgroup-Specific Therapeutic Targets in CNS Rhabdoid Tumors. Cancer Cell 2016, 30, 891–908. [Google Scholar] [CrossRef]
- Torchia, J.; Picard, D.; Lafay-Cousin, L.; Hawkins, C.E.; Kim, S.K.; Letourneau, L.; Ra, Y.S.; Ho, K.C.; Chan, T.S.; Sin-Chan, P.; et al. Molecular subgroups of atypical teratoid rhabdoid tumours in children: An integrated genomic and clinicopathological analysis. Lancet Oncol. 2015, 16, 569–582. [Google Scholar] [CrossRef]
- Johann, P.D.; Erkek, S.; Zapatka, M.; Kerl, K.; Buchhalter, I.; Hovestadt, V.; Jones, D.T.W.; Sturm, D.; Hermann, C.; Segura Wang, M.; et al. Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 2016, 29, 379–393. [Google Scholar] [CrossRef]
- Han, Z.Y.; Richer, W.; Fréneaux, P.; Chauvin, C.; Lucchesi, C.; Guillemot, D.; Grison, C.; Lequin, D.; Pierron, G.; Masliah-Planchon, J.; et al. The occurrence of intracranial rhabdoid tumours in mice depends on temporal control of Smarcb1 inactivation. Nat. Commun. 2016, 7, 10421. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.; Johann, P.D.; Grabovska, Y.; De Dieu Andrianteranagna, M.J.; Yao, F.; Frühwald, M.; Hasselblatt, M.; Bourdeaut, F.; Williamson, D.; Huang, A.; et al. Molecular subgrouping of atypical teratoid/rhabdoid tumors-a reinvestigation and current consensus. Neuro-Oncol. 2020, 22, 613–624. [Google Scholar] [CrossRef]
- Federico, A.; Thomas, C.; Miskiewicz, K.; Woltering, N.; Zin, F.; Nemes, K.; Bison, B.; Johann, P.D.; Hawes, D.; Bens, S.; et al. ATRT-SHH comprises three molecular subgroups with characteristic clinical and histopathological features and prognostic significance. Acta Neuropathol. 2022, 143, 697–711. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Liu, A.P.Y.; Orr, B.A.; Northcott, P.A.; Robinson, G.W. Advances in the classification of pediatric brain tumors through DNA methylation profiling: From research tool to frontline diagnostic. Cancer 2018, 124, 4168–4180. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.E.; Johann, P.D.; Milne, K.; Zapatka, M.; Buellesbach, A.; Ishaque, N.; Iskar, M.; Erkek, S.; Wei, L.; Tessier-Cloutier, B.; et al. Identification and Analyses of Extra-Cranial and Cranial Rhabdoid Tumor Molecular Subgroups Reveal Tumors with Cytotoxic T Cell Infiltration. Cell Rep. 2019, 29, 2338–2354.e2337. [Google Scholar] [CrossRef]
- Frühwald, M.C.; Hasselblatt, M.; Nemes, K.; Bens, S.; Steinbügl, M.; Johann, P.D.; Kerl, K.; Hauser, P.; Quiroga, E.; Solano-Paez, P.; et al. Age and DNA methylation subgroup as potential independent risk factors for treatment stratification in children with atypical teratoid/rhabdoid tumors. Neuro-Oncol. 2020, 22, 1006–1017. [Google Scholar] [CrossRef]
- Hasselblatt, M.; Nagel, I.; Oyen, F.; Bartelheim, K.; Russell, R.B.; Schüller, U.; Junckerstorff, R.; Rosenblum, M.; Alassiri, A.H.; Rossi, S.; et al. SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol. 2014, 128, 453–456. [Google Scholar] [CrossRef]
- Holdhof, D.; Johann, P.D.; Spohn, M.; Bockmayr, M.; Safaei, S.; Joshi, P.; Masliah-Planchon, J.; Ho, B.; Andrianteranagna, M.; Bourdeaut, F.; et al. Atypical teratoid/rhabdoid tumors (ATRTs) with SMARCA4 mutation are molecularly distinct from SMARCB1-deficient cases. Acta Neuropathol. 2021, 141, 291–301. [Google Scholar] [CrossRef]
- Tran, Q.T.; Upadhyaya, S.A.; Billups, C.A.; Onar-Thomas, A.; Alom, M.Z.; Carey, S.S.; Robinson, G.W.; Ellison, D.W.; Gajjar, A.; Orr, B.A. DNA-methylation subgroups carry no prognostic significance in ATRT-SHH patients in clinical trial cohorts. Acta Neuropathol. 2023, 146, 543–545. [Google Scholar] [CrossRef]
- Zaky, W.; Dhall, G.; Ji, L.; Haley, K.; Allen, J.; Atlas, M.; Bertolone, S.; Cornelius, A.; Gardner, S.; Patel, R.; et al. Intensive induction chemotherapy followed by myeloablative chemotherapy with autologous hematopoietic progenitor cell rescue for young children newly-diagnosed with central nervous system atypical teratoid/rhabdoid tumors: The Head Start III experience. Pediatr. Blood Cancer 2014, 61, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Carey, S.S.; Huang, J.; Myers, J.R.; Mostafavi, R.; Orr, B.A.; Dhanda, S.K.; Michalik, L.H.; Tatevossian, R.G.; Klimo, P.; Boop, F.; et al. Outcomes for children with recurrent/refractory atypical teratoid rhabdoid tumor: A single-institution study with molecular correlation. Pediatr. Blood Cancer 2024, 71, e31208. [Google Scholar] [CrossRef] [PubMed]
- Peyrl, A.; Chocholous, M.; Kieran, M.W.; Azizi, A.A.; Prucker, C.; Czech, T.; Dieckmann, K.; Schmook, M.T.; Haberler, C.; Leiss, U.; et al. Antiangiogenic metronomic therapy for children with recurrent embryonal brain tumors. Pediatr. Blood Cancer 2012, 59, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.N.; Yi, J.S.; Williams, P.M.; Roy-Chowdhuri, S.; Patton, D.R.; Coffey, B.D.; Reid, J.M.; Piao, J.; Saguilig, L.; Alonzo, T.A.; et al. Tazemetostat for tumors harboring SMARCB1/SMARCA4 or EZH2 alterations: Results from NCI-COG pediatric MATCH APEC1621C. J. Natl. Cancer Inst. 2023, 115, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Vejmelkova, K.; Pokorna, P.; Noskova, K.; Faustmannova, A.; Drabova, K.; Pavelka, Z.; Bajciova, V.; Broz, M.; Tinka, P.; Jezova, M.; et al. Tazemetostat in the therapy of pediatric INI1-negative malignant rhabdoid tumors. Sci. Rep. 2023, 13, 21623. [Google Scholar] [CrossRef]
- Nemes, K.; Johann, P.D.; Steinbugl, M.; Gruhle, M.; Bens, S.; Kachanov, D.; Teleshova, M.; Hauser, P.; Simon, T.; Tippelt, S.; et al. Infants and Newborns with Atypical Teratoid Rhabdoid Tumors (ATRT) and Extracranial Malignant Rhabdoid Tumors (eMRT) in the EU-RHAB Registry: A Unique and Challenging Population. Cancers 2022, 14, 2185. [Google Scholar] [CrossRef]
- Upadhyaya, S.A.; Campagne, O.; Billups, C.A.; Orr, B.A.; Onar-Thomas, A.; Tatevossian, R.G.; Mostafavi, R.; Myers, J.R.; Vinitsky, A.; Moreira, D.C.; et al. Phase II study of alisertib as a single agent for treating recurrent or progressive atypical teratoid/rhabdoid tumor. Neuro Oncol. 2023, 25, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Loning, L.; Zimmermann, M.; Reiter, A.; Kaatsch, P.; Henze, G.; Riehm, H.; Schrappe, M. Secondary neoplasms subsequent to Berlin-Frankfurt-Munster therapy of acute lymphoblastic leukemia in childhood: Significantly lower risk without cranial radiotherapy. Blood 2000, 95, 2770–2775. [Google Scholar]
- Pullen, J.; Boyett, J.; Shuster, J.; Crist, W.; Land, V.; Frankel, L.; Iyer, R.; Backstrom, L.; van Eys, J.; Harris, M. Extended triple intrathecal chemotherapy trial for prevention of CNS relapse in good-risk and poor-risk patients with B-progenitor acute lymphoblastic leukemia: A Pediatric Oncology Group study. J. Clin. Oncol. 1993, 11, 839–849. [Google Scholar] [CrossRef]
- Rutkowski, S.; Bode, U.; Deinlein, F.; Ottensmeier, H.; Warmuth-Metz, M.; Soerensen, N.; Graf, N.; Emser, A.; Pietsch, T.; Wolff, J.E.; et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N. Engl. J. Med. 2005, 352, 978–986. [Google Scholar] [CrossRef]
- Underiner, R.M.; Eltobgy, M.; Stanek, J.R.; Finlay, J.L.; AbdelBaki, M.S. Meta-Analysis of Treatment Modalities in Metastatic Atypical Teratoid/Rhabdoid Tumors in Children. Pediatr. Neurol. 2020, 108, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein-Shechter, T.; Gassas, A.; Mabbott, D.; Huang, A.; Bartels, U.; Tabori, U.; Janzen, L.; Hawkins, C.; Taylor, M.; Bouffet, E. Atypical teratoid or rhabdoid tumors: Improved outcome with high-dose chemotherapy. J. Pediatr. Hematol Oncol. 2010, 32, e182–e186. [Google Scholar] [CrossRef] [PubMed]
- Athale, U.H.; Duckworth, J.; Odame, I.; Barr, R. Childhood atypical teratoid rhabdoid tumor of the central nervous system: A meta-analysis of observational studies. J. Pediatr. Hematol Oncol. 2009, 31, 651–663. [Google Scholar] [CrossRef]
- Alarcon-Vargas, D.; Zhang, Z.; Agarwal, B.; Challagulla, K.; Mani, S.; Kalpana, G.V. Targeting cyclin D1, a downstream effector of INI1/hSNF5, in rhabdoid tumors. Oncogene 2006, 25, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Cimica, V.; Smith, M.E.; Zhang, Z.; Mathur, D.; Mani, S.; Kalpana, G.V. Potent inhibition of rhabdoid tumor cells by combination of flavopiridol and 4OH-tamoxifen. BMC Cancer 2010, 10, 634. [Google Scholar] [CrossRef]
- Kolb, E.A.; Gorlick, R.; Houghton, P.J.; Morton, C.L.; Lock, R.B.; Tajbakhsh, M.; Reynolds, C.P.; Maris, J.M.; Keir, S.T.; Billups, C.A.; et al. Initial testing of dasatinib by the pediatric preclinical testing program. Pediatr. Blood Cancer 2008, 50, 1198–1206. [Google Scholar] [CrossRef]
- Leruste, A.; Tosello, J.; Ramos, R.N.; Tauziède-Espariat, A.; Brohard, S.; Han, Z.Y.; Beccaria, K.; Andrianteranagna, M.; Caudana, P.; Nikolic, J.; et al. Clonally Expanded T Cells Reveal Immunogenicity of Rhabdoid Tumors. Cancer Cell 2019, 36, 597–612.e598. [Google Scholar] [CrossRef]
- Theruvath, J.; Sotillo, E.; Mount, C.W.; Graef, C.M.; Delaidelli, A.; Heitzeneder, S.; Labanieh, L.; Dhingra, S.; Leruste, A.; Majzner, R.G.; et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat. Med. 2020, 26, 712–719. [Google Scholar] [CrossRef]
- Alva, E.; Rubens, J.; Chi, S.; Rosenberg, T.; Reddy, A.; Raabe, E.H.; Margol, A. Recent progress and novel approaches to treating atypical teratoid rhabdoid tumor. Neoplasia 2023, 37, 100880. [Google Scholar] [CrossRef]
- Pompe, R.S.; von Bueren, A.O.; Mynarek, M.; von Hoff, K.; Friedrich, C.; Kwiecien, R.; Treulieb, W.; Lindow, C.; Deinlein, F.; Fleischhack, G.; et al. Intraventricular methotrexate as part of primary therapy for children with infant and/or metastatic medulloblastoma: Feasibility, acute toxicity and evidence for efficacy. Eur. J. Cancer 2015, 51, 2634–2642. [Google Scholar]
- Blaney, S.M.; Boyett, J.; Friedman, H.; Gajjar, A.; Geyer, R.; Horowtiz, M.; Hunt, D.; Kieran, M.; Kun, L.; Packer, R.; et al. Phase I clinical trial of mafosfamide in infants and children aged 3 years or younger with newly diagnosed embryonal tumors: A pediatric brain tumor consortium study (PBTC-001). J. Clin. Oncol. 2005, 23, 525–531. [Google Scholar] [CrossRef]
- Blaney, S.M.; Kocak, M.; Gajjar, A.; Chintagumpala, M.; Merchant, T.; Kieran, M.; Pollack, I.F.; Gururangan, S.; Geyer, R.; Phillips, P.; et al. Pilot study of systemic and intrathecal mafosfamide followed by conformal radiation for infants with intracranial central nervous system tumors: A pediatric brain tumor consortium study (PBTC-001). J. Neurooncol. 2012, 109, 565–571. [Google Scholar] [CrossRef]
- Benesch, M.; Sovinz, P.; Krammer, B.; Lackner, H.; Mann, G.; Schwinger, W.; Gadner, H.; Urban, C. Feasibility and toxicity of intrathecal liposomal cytarabine in 5 children and young adults with refractory neoplastic meningitis. J. Pediatr. Hematol. Oncol. 2007, 29, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, J.; Nishiyama, K.; Mori, H.; Takahashi, H.; Fujii, Y. Intrathecal chemotherapy for refractory disseminated medulloblastoma. Childs Nerv. Syst. 2008, 24, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Clayton, J.; Vloeberghs, M.; Jaspan, T.; Walker, D.; MacArthur, D.; Grundy, R. Intrathecal chemotherapy delivered by a lumbar-thecal catheter in metastatic medulloblastoma: A case illustration. Acta Neurochir. 2008, 150, 709–712. [Google Scholar] [CrossRef]
- Mastronuzzi, A.; Del Bufalo, F.; Iacono, A.; Secco, D.E.; Serra, A.; Colafati, G.S.; De Ioris, M.A.; Corsetti, T. Intrathecal liposomal cytarabine and leptomeningeal medulloblastoma relapse: A valuable therapeutic option. Anticancer Res. 2013, 33, 3515–3518. [Google Scholar] [PubMed]
- Yamasaki, K.; Okada, K.; Soejima, T.; Sakamoto, H.; Hara, J. Strategy to minimize radiation burden in infants and high-risk medulloblastoma using intrathecal methotrexate and high-dose chemotherapy: A prospective registry study in Japan. Pediatr. Blood Cancer 2020, 67, e28012. [Google Scholar] [CrossRef]
- Okada, K.; Soejima, T.; Sakamoto, H.; Hirato, J.; Hara, J. Phase II study of reduced-dose craniospinal irradiation and combination chemotherapy for children with newly diagnosed medulloblastoma: A report from the Japanese Pediatric Brain Tumor Consortium. Pediatr. Blood Cancer 2020, 67, e28572. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, S.; Cacciotti, C.; Yan, C.L.S.; Lafay-Cousin, L. What Have We Learnt from the Recent Multimodal Managements of Young Patients with ATRT? Cancers 2025, 17, 1116. https://doi.org/10.3390/cancers17071116
Cheng S, Cacciotti C, Yan CLS, Lafay-Cousin L. What Have We Learnt from the Recent Multimodal Managements of Young Patients with ATRT? Cancers. 2025; 17(7):1116. https://doi.org/10.3390/cancers17071116
Chicago/Turabian StyleCheng, Sylvia, Chantel Cacciotti, Carol L. S. Yan, and Lucie Lafay-Cousin. 2025. "What Have We Learnt from the Recent Multimodal Managements of Young Patients with ATRT?" Cancers 17, no. 7: 1116. https://doi.org/10.3390/cancers17071116
APA StyleCheng, S., Cacciotti, C., Yan, C. L. S., & Lafay-Cousin, L. (2025). What Have We Learnt from the Recent Multimodal Managements of Young Patients with ATRT? Cancers, 17(7), 1116. https://doi.org/10.3390/cancers17071116