
Association Between B-Cell Marker Expression and RUNX1 Lesions in Acute Myeloid Leukemia, Beyond RUNX1::RUNX1T1 Fusion: Diagnostic Pitfalls with Mixed-Phenotype Acute Leukemia—B/Myeloid

 , , , , , ,
, , , , , , 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Case Selection
2.2. Flow Cytometry and Immunohistochemistry
2.3. Chromosomal Giemsa Banding and Fluorescence In Situ Hybridization
2.4. Next-Generation Sequencing
3. Results
3.1. Clinical Findings
3.2. Blast Cytomorphology
3.3. Immunophenotypic Evaluation
3.4. Cytogenetic and Molecular Evaluation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alaggio, R.A.C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; Chadburn, A.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef] [PubMed]
- Sood, R.; Kamikubo, Y.; Liu, P. Role of RUNX1 in hematological malignancies. Blood 2017, 129, 2070–2082. [Google Scholar] [CrossRef] [PubMed]
- Gaidzik, V.I.; Bullinger, L.; Schlenk, R.F.; Zimmermann, A.S.; Rock, J.; Paschka, P.; Corbacioglu, A.; Krauter, J.; Schlegelberger, B.; Ganser, A.; et al. RUNX1 mutations in acute myeloid leukemia: Results from a comprehensive genetic and clinical analysis from the AML study group. J. Clin. Oncol. 2011, 29, 1364–1372. [Google Scholar] [CrossRef]
- Rungjirajittranon, T.; Siriwannangkul, T.; Kungwankiattichai, S.; Leelakanok, N.; Rotchanapanya, W.; Vittayawacharin, P.; Mekrakseree, B.; Kulchutisin, K.; Owattanapanich, W. Clinical Outcomes of Acute Myeloid Leukemia Patients Harboring the RUNX1 Mutation: Is It Still an Unfavorable Prognosis? A Cohort Study and Meta-Analysis. Cancers 2022, 14, 5239. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Menter, T.; Lundberg, P.; Wenzel, F.; Dirks, J.; Fernandez, P.; Friess, D.; Dirnhofer, S.; Tzankov, A. RUNX1 Mutations Can Lead to Aberrant Expression of CD79a and PAX5 in Acute Myelogenous Leukemias: A Potential Diagnostic Pitfall. Pathobiology 2019, 86, 162–166. [Google Scholar] [CrossRef]
- George, G.V.J.A.; Burack, W.R.; Evans, A.G.; Bennett, J.M.; El Hussein, S. Association Between B-Cell Marker Expression and RUNX1 Lesions in Acute Myeloid Leukemia, Beyond RUNX1::RUNX1T1 Fusion: Diagnostic Pitfalls with Mixed-Phenotype Acute Leukemia—B/Myeloid. J. Mol. Diagn. 2024, 26, S18. [Google Scholar]
- Xiao, W.; Chan, A.; Waarts, M.R.; Mishra, T.; Liu, Y.; Cai, S.F.; Yao, J.; Gao, Q.; Bowman, R.L.; Koche, R.P.; et al. Plasmacytoid dendritic cell expansion defines a distinct subset of RUNX1-mutated acute myeloid leukemia. Blood 2021, 137, 1377–1391. [Google Scholar] [CrossRef]
- Wang, W.; Xu, J.; Khoury, J.D.; Pemmaraju, N.; Fang, H.; Miranda, R.N.; Yin, C.C.; Hussein, S.E.; Jia, F.; Tang, Z.; et al. Immunophenotypic and Molecular Features of Acute Myeloid Leukemia with Plasmacytoid Dendritic Cell Differentiation Are Distinct from Blastic Plasmacytoid Dendritic Cell Neoplasm. Cancers 2022, 14, 3375. [Google Scholar] [CrossRef]
- Ray, D.; Kwon, S.Y.; Tagoh, H.; Heidenreich, O.; Ptasinska, A.; Bonifer, C. Lineage-inappropriate PAX5 expression in t(8;21) acute myeloid leukemia requires signaling-mediated abrogation of polycomb repression. Blood 2013, 122, 759–769. [Google Scholar] [CrossRef]
- Valbuena, J.R.; Medeiros, L.J.; Rassidakis, G.Z.; Hao, S.; Wu, C.D.; Chen, L.; Lin, P. Expression of B cell-specific activator protein/PAX5 in acute myeloid leukemia with t(8;21)(q22;q22). Am. J. Clin. Pathol. 2006, 126, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Kozmik, Z.; Wang, S.; Dorfler, P.; Adams, B.; Busslinger, M. The promoter of the CD19 gene is a target for the B-cell-specific transcription factor BSAP. Mol. Cell Biol. 1992, 12, 2662–2672. [Google Scholar] [CrossRef] [PubMed]
- Nutt, S.L.; Morrison, A.M.; Dorfler, P.; Rolink, A.; Busslinger, M. Identification of BSAP (Pax-5) target genes in early B-cell development by loss- and gain-of-function experiments. EMBO J. 1998, 17, 2319–2333. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimmons, D.; Hodsdon, W.; Wheat, W.; Maira, S.M.; Wasylyk, B.; Hagman, J. Pax-5 (BSAP) recruits Ets proto-oncogene family proteins to form functional ternary complexes on a B-cell-specific promoter. Genes. Dev. 1996, 10, 2198–2211. [Google Scholar] [CrossRef]
- Tiacci, E.; Pileri, S.; Orleth, A.; Pacini, R.; Tabarrini, A.; Frenguelli, F.; Liso, A.; Diverio, D.; Lo-Coco, F.; Falini, B. PAX5 expression in acute leukemias: Higher B-lineage specificity than CD79a and selective association with t(8;21)-acute myelogenous leukemia. Cancer Res. 2004, 64, 7399–7404. [Google Scholar] [CrossRef]
- Revilla, I.D.R.; Bilic, I.; Vilagos, B.; Tagoh, H.; Ebert, A.; Tamir, I.M.; Smeenk, L.; Trupke, J.; Sommer, A.; Jaritz, M.; et al. The B-cell identity factor Pax5 regulates distinct transcriptional programmes in early and late B lymphopoiesis. Embo J. 2012, 31, 3130–3146. [Google Scholar] [CrossRef]
- El Hussein, S.; Wang, W. Flow Cytometry Profiling of Plasmacytoid Dendritic Cell Neoplasms. Cancers 2024, 16, 2118. [Google Scholar] [CrossRef]
- El Hussein, S.; Wang, W. Plasmacytoid dendritic cells in the setting of myeloid neoplasms: Diagnostic guide to challenging pathologic presentations. Br. J. Haematol. 2023, 200, 545–555. [Google Scholar] [CrossRef]
- Orfao, A.; Matarraz, S.; Perez-Andres, M.; Almeida, J.; Teodosio, C.; Berkowska, M.A.; van Dongen, J.J.M.; EuroFlow. Immunophenotypic dissection of normal hematopoiesis. J. Immunol. Methods 2019, 475, 112684. [Google Scholar] [CrossRef]
- Zhao, Q.; Ma, P.; Fu, P.; Wang, J.; Wang, K.; Chen, L.; Yang, Y. Myelodysplastic Syndrome/Acute Myeloid Leukemia Following the Use of Poly-ADP Ribose Polymerase (PARP) Inhibitors: A Real-World Analysis of Postmarketing Surveillance Data. Front. Pharmacol. 2022, 13, 912256. [Google Scholar] [CrossRef]
- Liu, H.; Wang, S.A.; Schlette, E.J.; Xu, J.; Jorgensen, J.L.; Cameron Yin, C.; Li, S.; Jeffrey Medeiros, L.; Tang, G. Myeloid neoplasms with t(16;21)(q24;q22)/RUNX1-RUNX1T3 mimics acute myeloid leukemia with RUNX1-RUNX1T1. Ann. Hematol. 2018, 97, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Koduru, P.; Cantu, M.; Fuda, F.; Chen, W. RUNX1::CBFA2T2 rearranged acute myeloid leukemia transformed from JAK2 V617F mutated primary myelofibrosis. EJHaem 2024, 5, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Loghavi, S. Acute myeloid leukemia with RUNX1::CBFA2T3 fusion. Blood 2025, 145, 1226. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Shallis, R.M.; Stempel, J.M.; Huntington, S.F.; Zeidan, A.M.; Gore, S.D.; Ma, X.; Podoltsev, N.A. Second malignancies among older patients with classical myeloproliferative neoplasms treated with hydroxyurea. Blood Adv. 2023, 7, 734–743. [Google Scholar] [CrossRef]
- Dohner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Hollein, A.; Nadarajah, N.; Meggendorfer, M.; Jeromin, S.; Kern, W.; Haferlach, C.; Haferlach, T. Molecular characterization of AML with RUNX1-RUNX1T1 at diagnosis and relapse reveals net loss of co-mutations. Hemasphere 2019, 3, e178. [Google Scholar] [CrossRef]
- Wolach, O.; Stone, R.M. How I treat mixed-phenotype acute leukemia. Blood 2015, 125, 2477–2485. [Google Scholar] [CrossRef]
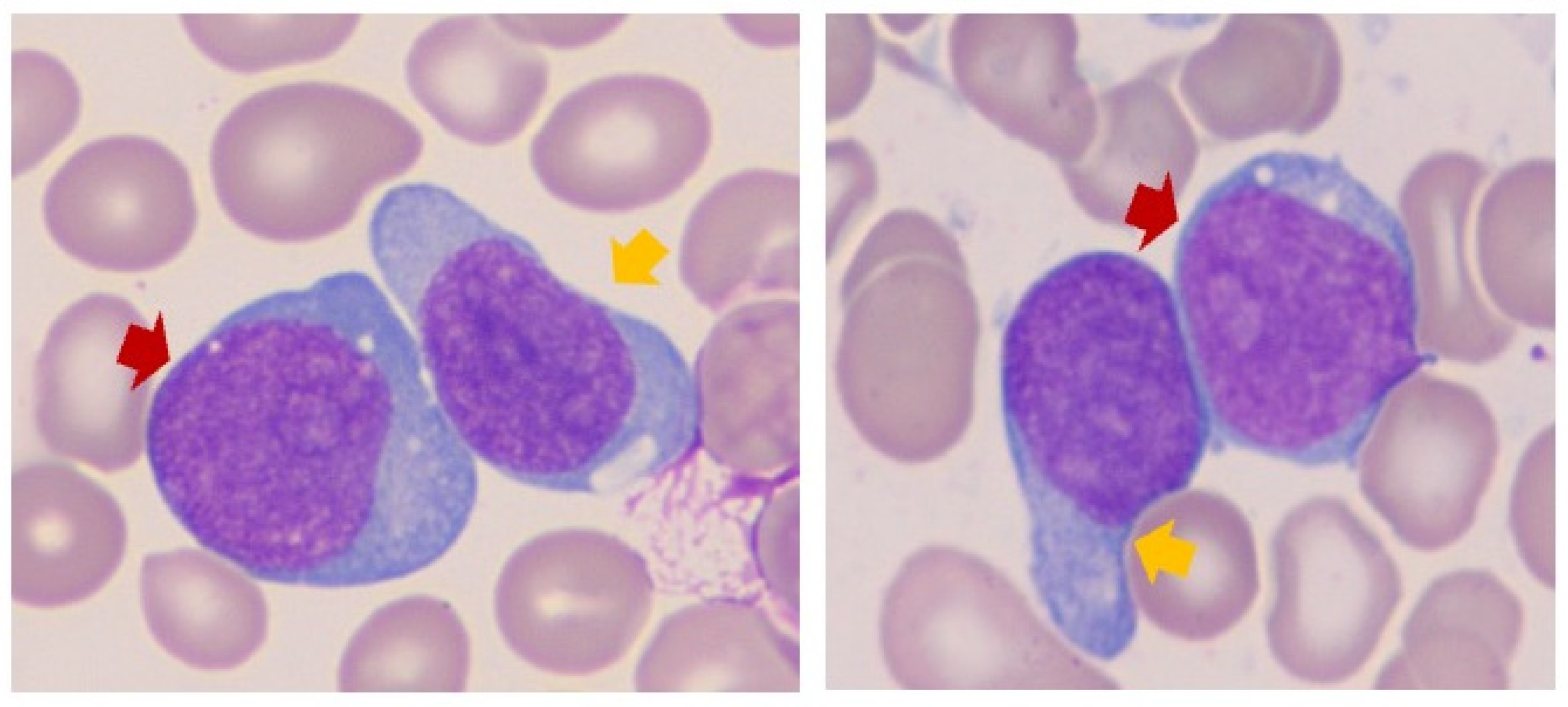
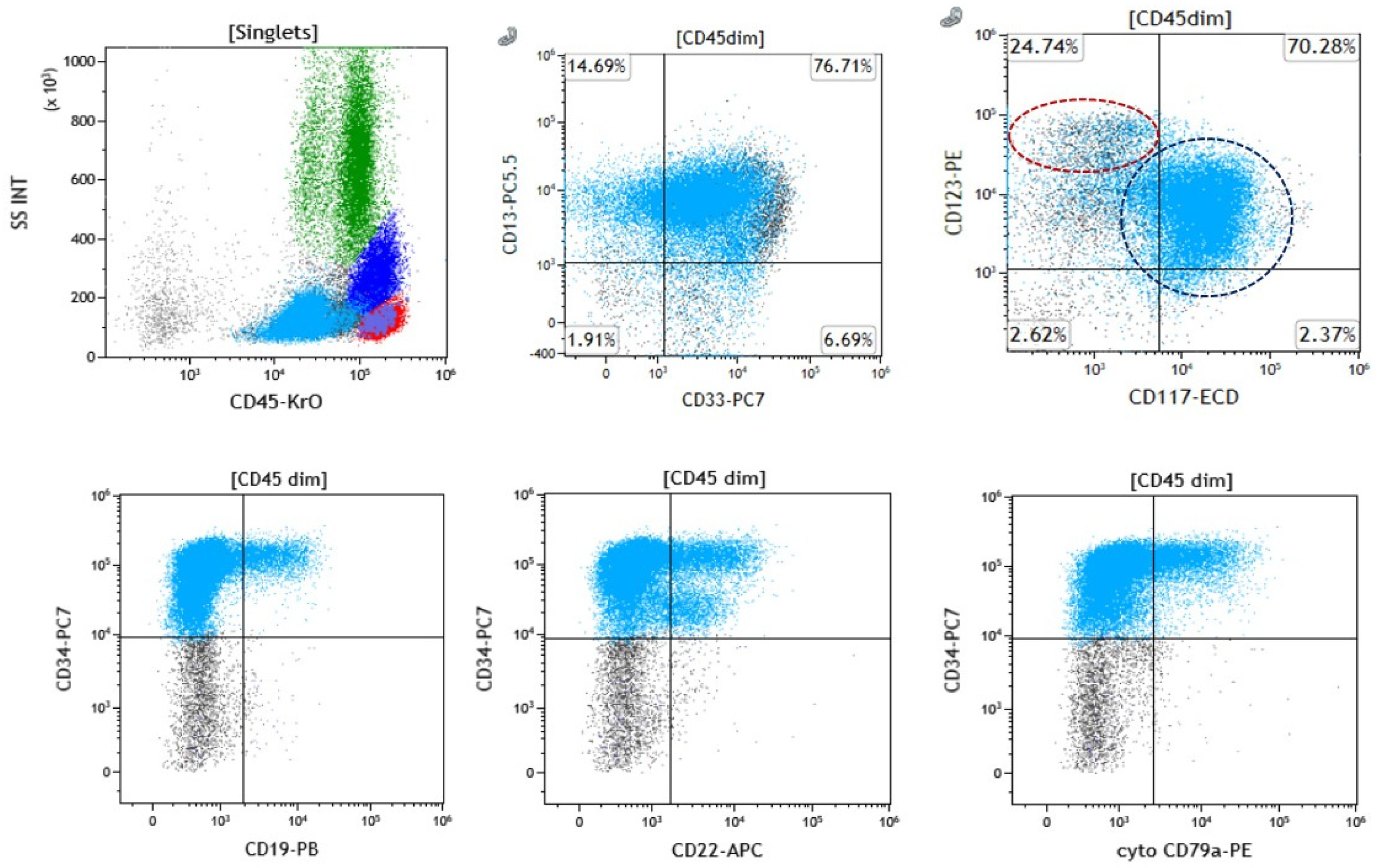


{kind=link}
{kind=link}
{kind=link}
| Case | Age/Sex | Diagnosis | Prior Malignancy (If Any), Details of Therapy, and Vital Status | Bone Marrow Aspirate or Peripheral Blood Blast Percentage | Fulfilling the Criteria for MPAL-B/Myeloid * by Flow Cytometry Analysis | pDC * Differentiation by Morphology | pDC Differentiation by Flow Cytometry Analysis |
|---|---|---|---|---|---|---|---|
| A | 64/F | AML-MR * | JAK-2 positive ET * treated with hydroxyurea; alive | 70% | Yes | Yes | Yes |
| B | 67/M | AML-MR | No history; alive | 80% | Yes | No | Yes |
| C | 76/M | AML post-cytotoxic therapy | Localized prostate adenocarcinoma and esophageal adenocarcinoma treated with neoadjuvant chemoradiotherapy (CROSS regimen) and esophagectomy; deceased | 60% | Yes | No | No |
| D | 59/F | AML post-cytotoxic therapy | Ovarian cancer treated with olaparib; deceased | 50% | Yes | No | No |
| E | 91/F | AML-MR | No history; deceased | 85% | Yes | Yes | Yes |
| F | 82/F | AML-MR | ET treated with hydroxyurea; deceased | 80% | Yes | No | No |
| G | 56/M | AML-MR | No history; alive | 85% | Yes | No | No |
| H | 70/F | AML-MR | MDS/MPN * treated with hydroxyurea; deceased | 80% | Yes | No | No |
| I | 77/M | AML-MR | Prostate adenocarcinoma treated with a GnRH * antagonist; deceased | 33% | Yes | Yes | Yes |
| J | 79/M | AML-MR | No history; alive | 16% | No | Yes | Yes |
| K | 74/F | AML-MR | No history; deceased | 53% | Yes | No | No |
| L | 61/M | AML-MR | No history; deceased | 30% | Yes | No | No |
| M | 70/M | AML post-cytotoxic therapy | Stage IA CHL *, Stage IA NSCLC *, MDS * treated with VAD * (two cycles), FRT *, stereotactic radiosurgery, decitabine/venetoclax (12 cycles); deceased | 56% | Yes | No | Yes |
| N | 69/M | AML-MR | No history; deceased | 68% | Yes | No | Yes |
| O | 45/M | AML-MR | No history; deceased | 70% | Yes | No | Yes |
| P | 61/F | AML-MR | No history; alive | 18% | No | No | No |
| Case A | Case B | Case C | Case D | Case E | Case F | Case G | Case H | Case I | Case J | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PAX5 * | + (subset) | + (subset) | + (subset) | + (subset) | + (subset) | Not performed | Not performed | + (subset) | − | + (subset) | |
| CD2 | − | − | − | − | − | − | − | + (subset) | − | − | |
| Surface CD3 | − | − | − | − | − | − | − | − | − | − | |
| Cytoplasmic CD3 | − | − | − | − | − | − | − | − | − | − | |
| CD4 | + (subset) | + (subset) | − | − | + (subset) | N/A | + (subset) | + (subset) | − | + (subset) | |
| CD5 | + (subset) | − | − | − | − | N/A | − | − | + (subset) | − | |
| CD7 | + (subset) | − | + (subset) | − | + (subset) | − | + (subset) | − | + (subset) | + | |
| CD11b | − | + (subset) | − | − | − | N/A | + (subset) | − | + (subset) | − | |
| CD13 | + (subset) | + (subset) | + | − | + (subset) | + (subset) | + (subset) | + (subset) | + | + (subset) | |
| CD14 | − | − | + (subset) | − | + (subset) | − | − | − | − | − | |
| CD15 | − | − | + (subset) | + (subset) | − | + (subset) | − | − | − | − | |
| CD19 | + (subset) | + (subset) | + (subset) | + | + (subset) | + (subset) | + (subset) | + (subset) | − | + (subset) | |
| CD20 | − | − | − | − | + (subset) | − | − | − | − | − | |
| Cytoplasmic CD22 | + (subset) | + (subset) | + (subset) | − | + (subset) | N/A** | + (subset) | + (subset) | + (subset) | + (subset) | |
| CD33 | + | + (subset) | + (subset) | - | + (subset) | + (subset) | + (subset) | + (subset) | + | + (subset) | |
| CD34 | + | + | + | + (subset) | + | + | + | + | + | + | |
| CD56 | + (subset) | − | − | − | − | − | − | − | + | − | |
| CD64 | − | − | + (subset) | − | − | − | − | − | − | − | |
| CD79a | + (subset) | + (subset) | + (subset) | + (subset) | + (subset) | + (subset) | + (subset) | + (subset) | − | − | |
| CD117 | + (subset) | − | + (subset) | + | + (subset) | + | + (subset) | + (subset) | + | + | |
| CD123 | + (subset) | + | + | + | + | N/A | + | + | + | + | |
| HLA-DR | + | + | + | + | + | + | + | + | + | + | |
| MPO | − | − | + (subset) | + | + (subset) | − | − | + (subset) | + (subset) | - | |
| TdT | + (subset) | + (subset) | + (subset) | + (subset) | + (subset) | + (subset) | + (subset) | + (subset) | − | + | |
| Case K | Case L | Case M | Case N | Case O | Case P | ||||||
| PAX5 * | Not performed | Not performed | Not available | Not performed | + (subset) | Not available | |||||
| CD2 | − | − | − | − | − | − | |||||
| Surface CD3 | − | − | − | − | − | − | |||||
| Cytoplasmic CD3 | N/A | − | − | − | − | − | |||||
| CD4 | N/A | + (subset) | + (subset) | − | + (subset) | − | |||||
| CD5 | N/A | − | − | − | − | − | |||||
| CD7 | + (subset) | + (subset) | − | − | + (subset) | + (subset) | |||||
| CD11b | N/A | + (subset) | + (subset) | − | + (subset) | − | |||||
| CD13 | + | + | - | + | + | + (subset) | |||||
| CD14 | − | - | - | − | − | − | |||||
| CD15 | − | + (subset) | + (subset) | − | + (subset) | − | |||||
| CD19 | − | − | + (subset) | − | + (subset) | + (subset) | |||||
| CD20 | − | − | − | − | − | − | |||||
| Cytoplasmic CD22 | N/A ** | + (subset) | + (subset) | + (subset) | − | − | |||||
| CD33 | + (subset) | + | + (subset) | + | + | − | |||||
| CD34 | + (subset) | + (subset) | + | + | + (subset) | + | |||||
| CD56 | + (subset) | − | − | + (subset) | − | − | |||||
| CD64 | + (subset) | + (subset) | − | − | + (subset) | - | |||||
| CD79a | N/A | - | + (subset) | − | + (subset) | + (subset) | |||||
| CD117 | + | + (subset) | − | + | + (subset) | - | |||||
| CD123 | N/A | + (subset) | + | + (subset) | + | + | |||||
| HLA-DR | + (subset) | + (subset) | + | + | + | + | |||||
| MPO | N/A | + (subset) | + (subset) | + (subset) | + | − | |||||
| TdT | N/A | − | + (subset) | + (subset) | − | + (subset) | |||||
| Case | Diagnosis | RUNX1 Lesion | Pathogenic Mutations (VAF * %) |
|---|---|---|---|
| A | AML-MR * | Mutation and copy number gain | RUNX1 c.318G>T, p.Trp106Cys (68%), SF3B1 c.2098A>G, p.Lys700Glu (46%), NRAS c.34G>A, p.Gly12Ser (42%), ASXL1 c.2338C>T, p.Gln780Ter (43%) |
| B | AML-MR | Mutations | RUNX1 c.611G > A, p.Arg204Gln (49%), RUNX1 c.259_277dup, p.Asp93GlyfsTer51 (28%) SRSF2 c.284C > G,p.Pro95Arg (47%) |
| C | AML post-cytotoxic therapy | Copy number gain | TP53 c.856G>A, p.Glu286Lys (87%) |
| D | AML post-cytotoxic therapy | Copy number gain and rearrangement | TP53 c.273G > A, p.Trp91Ter (26%) |
| E | AML-MR | Mutation | IDH2 c.419G>A p.Arg140Gln (26%), SRSF2 c.284C>T p.Pro95Leu (23%), RUNX1 c.485G>A p.Arg162Lys (25%) |
| F | AML-MR | Mutations | DNMT3A c.2645G>A p.Arg882His (40%), PHF6 c.482_483insG p.Ser162LysfsTer10 (8%), PTPN11 c.211T>C p.Phe71Leu (12%), RUNX1 c.979delC p.Leu327Ter (18%), SRSF2 c.284C>A p.Pro95His (43%), RUNX1 c.508G>A p.Gly170Arg (39%) |
| G | AML-MR | Mutation and copy number gain | ASXL1 c.2056_2057dupAA, p.Cys687SerfsTer17 (12%), DNMT3A c.2645G>A, p.Arg882His (47%), FLT3 ITD (7%), FLT3 ITD (1%), IDH2 c.419G>A, p.Arg140Gln (48%), RUNX1 c.743dupA, p.Asn248LysfsTer13 (89%), SRSF2 c.284C>T, p.Pro95Leu (50%) |
| H | AML-MR | Mutations | DNMT3A c.2645G>A p.Arg882His (48%), JAK2 c.1849G>T p.Val617Phe (55%) RUNX1 c.425_426insCCGGC p.Glu143ArgfsTer4 (21%), TET2 c.4537+1G>A p.? (95%), RUNX1 c.484A>G p.Arg162Gly (31%) |
| I | AML-MR | Mutation | ASXL1 c.2077C>T, p.Arg693Ter (31%), CBL c.1192C>T, p.His398Tyr (18%), EZH2 c.1650delG, p.Lys550AsnfsTer125 (71%), NRAS c.35G>C, p.Gly12Ala (3%), NRAS c.176C>A, p.Ala59Asp (11%), RUNX1 c.485G>A, p.Arg162Lys (9%) |
| J | AML-MR | Mutations | BCOR c.472delA p.Ser158ValfsTer3 (24%), RUNX1 c.965C>G p.Ser322Ter (12%), RUNX1 c.618_619insAACC p.Arg207AsnfsTer7 (4%) |
| K | AML-MR | Copy number gain | TP53 c.401T>G p.Phe134Cys (84%) |
| L | AML-MR | Copy number gain | TP53 c.527G>T p.Cys176Phe (61%) |
| M | AML post-cytotoxic therapy | Copy number gain | PTPN11 c.227A>G p.Glu76Gly (14%), TP53 c.818G>A p.Arg273His (31%), TP53 c.752T>A p.Ile251Asn (29%) |
| N | AML-MR | Copy number gain | TP53 c.376-1_386del p.? (76%) |
| O | AML-MR | Copy number gain | NRAS c.182A>G, p.Gln61Arg (47%) |
| P | AML-MR | Copy number gain | IDH1 c.394C>A, p.Arg132Ser (32%) |
| Case | Diagnosis | RUNX1 lesion | Karyotype | FISH * |
|---|---|---|---|---|
| A | AML-MR * | Mutation and copy number gain | 46,XX,7,+13[15]./48,XX,+13,+21[1]./46,XX[4]. | Copy number gains of RUNX1 (21q22) (5%), monosomy 7 (73%) |
| B | AML-MR | Mutations | 46,XY,del(7)(q22)[12]./46,XY[7]. | Monosomy 7 (45.5%), TP53 (17p13.1) deletion (38.5%) |
| C | AML post-cytotoxic therapy | Copy number gain | 77<4n>,XXYY,-1,-3,-3,-4,-5,-5,+7,add(7)(q11.2) × 3,-8,+9,-10,-11,-11,-11,-11,-13,-13,-14,-16,add(17)(p11.2)× 2,-19,-20,-21,add(21)(p11.2) × 2, + marx2 [15]./44-45,XY,-5,del(5)(q12q33),-7,add(17)(p11.2)[cp5]. | Deletion 5q31 (16.5%), monosomy 7 (4.5%), copy number gains of RUNX1 (21q22) (70.5%), RUNX1T1 (8q22) (70.5%), ABL1 (9q34) (79%), BCR (22q11.2) (79%), and IgH (14q32) (71%) |
| D | AML post-cytotoxic therapy | Copy number gain and rearrangement | 46,XX,der(7)t(7;11)(q22;q13)[19]./46,XX[1]..ish t(16;21)(q24;q22) RUNX1::CBFA2T3 [8/9]. | Monosomy 7 (48.5%), copy number gains of RUNX1 (21q22) (65%) and MLL (11q23) (64%) |
| E | AML-MR | Mutation | 46, XX | Negative for all tested rearrangements |
| F | AML-MR | Mutations | 46,XX[19]., Non-clonal: 46,XX,del(6)(q10)[1]. | Negative for all tested rearrangements |
| G | AML-MR | Mutation and copy number gain | 47,XY,+13[10]./46,XY[10]. | Copy number gains of all probes, suspected genomic doubling |
| H | AML-MR | Mutations | 46,XX,+6,inv(6)(p25q13)x2,-20[20]., Non-clonal: add(2)(q32), questionable add(14)(q21) | Copy number loss of IgH (14q32) (16%) |
| I | AML-MR | Mutation | 46,XY,del(11)(q13q23)[19]./46,XY[1]. | Copy number loss of MLL (11q23) (84%) |
| J | AML-MR | Mutations | 46, XY | Negative for all tested rearrangements |
| K | AML-MR | Copy number gain | 54-58<2n>,XX,+X,+1,+2,+4,del(5)(q13q34), +del(5),+6,dic(7;11)(q11.2;q11.2),+9,+10,+11,del(11)(p11.1), +13,+15,-19,+21,+22,+der(?)t(?;13)(?;q14), +der(?)t(?;14)(?;q13),+1-2mar[cp20]. | Three RUNX1 signals (73.5%), three MLL signals (72.5%), and three to four PML signals (36.5%) |
| L | AML-MR | Copy number gain | 46,XY,del(5)(q22q35),+8,del(17)(p12),-18[1]./46,XY,del(5),del(16)(q21q22),del(17),+mar[1]./ 45-46,Y,del(X)(q21),add(3)(p21),del(5),i(8)(q10),del(9)(q21), der(10)ins(10:?)(q21;?),der(12)t(9;12)(q22;p13),del(16),del(17),-18,+19,-21,i(21)(q10),-22,i(22)(q10),+1-2mar[cp16]./46,XY[2]., Non-clonal: add(6)(q25-27), del(22)(q13) | EGR1 (5q31) deletion (87.5%), copy number gains of RUNX1T1 (8q22) (19–49.5%), copy number gains of RUNX1 (21q22) (19%), and one CBFB (16q22) fusion signal (75%) |
| M | AML post-cytotoxic therapy | Copy number gain | 66-69,XXY,del(1)(q41),-3,+5,del(5)(q33)x2,- 7,del(7)(q22),+8,der(9;14)(q1 0;q10),+10,+11,add(11)(p11.2),+12,-15,-16,-17,+22,+1-2mar[cp8]./ 46,XY[2]., Non-clonal: +2markers | Hyperdiploidy or near triploidy, three CRLF2 signals or copy number gains of Xp22.33/Yp11.32 (83.5%), three to four RUNX1T1 (8q22) signals (85%), three ABL1 (9q34) signals (89%), four MLL (11q23) signals (80%), four ETV6 (12p13) signals (83.5%), three IgH (14q32) signals (80%), three RUNX1 (21q22) signals (74.5%), four BCR (22q11.2) signals (89%) |
| N | AML-MR | Copy number gain | 46,XY,del(16)(q11.2q23)[2]./45,sl,del(5)(q22q35),add(17)(q23),-20,-21,+22[2]./49-50,sdl1,-7,add(11)(q23),-13,+22,+5-6mar[cp7]./50-51,sdl2,+del(5)[cp3]./50-51,sdl2,+X,+9,+14,-del(16)[cp4]./51-52,sdl4,+8[cp2]., Non-clonal:t(2;8)(p13;p13) | EGR1 (5q) deletion (80%), D7S486 (7q31) deletion (62.5%), monosomy 7 (9.5%), copy number gains of D5S721/D5S23 (5p15.2) (25%), copy number gains of RUNX1T1 (8q22) (39.5%), copy number gains of RUNX1T1 (8q22) and RUNX1 (21q22) (19.5%), three fused MLL (11q23) signals (10%) |
| O | AML-MR | Copy number gain | 49,XY,+3,+8,?t(10;21)(q26;q21),+14[13]./46,XY[7]..nuc ish(RUNX1x3)[180/200]..ish ?t(10;21)(3′ or 5′RUNX1+;3′ or 5′RUNX1-)[2/2]. | Copy number gains of both RUNX1T1 (8q22) and RUNX1 (21q22) (95%) |
| P | AML-MR | Copy number gain | 46,XX,del(7)(q22q35)[4]./(46,idem)x2,-del(7),+8[16]. | D7S486 (7q31) deletion (19%), likely tetraploidy in ~50% of cells based on copy number gains of the following: tetrasomy 5 (53.5%), D7Z1 (CEP7) three copies, D7S486 (7q31) two copies (25.5%),loss of chromosome 7 relative to tetraploidy with additional loss of 7q, RUNX1T1 (8q21) four copies (10%), RUNX1T1 (8q21) five copies (40%),additional signal for 8q21 relative to tetraploidy, RUNX1 (21q22) four copies (50%), KMT2A (11q23) four copies (47%), KMT2A (11q23) three copies (6.5%), loss of 11q relative to tetraploidy, RB1 (13q14) four copies (49%), TP53 (17q13) four copies (49%), PML (15q22) four copies (46.5%), RARA (17q21) four copies, (46.5%), CBFB (16q22) four copies (44%), RARA (17q21) four copies (50%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
George, G.V.; Kajstura, M.; Jajosky, A.N.; Fang, H.; Jelloul, F.Z.; Evans, A.G.; Burack, W.R.; Bennett, J.M.; Medeiros, L.J.; Wang, W.; et al. Association Between B-Cell Marker Expression and RUNX1 Lesions in Acute Myeloid Leukemia, Beyond RUNX1::RUNX1T1 Fusion: Diagnostic Pitfalls with Mixed-Phenotype Acute Leukemia—B/Myeloid. Cancers 2025, 17, 1354. https://doi.org/10.3390/cancers17081354
George GV, Kajstura M, Jajosky AN, Fang H, Jelloul FZ, Evans AG, Burack WR, Bennett JM, Medeiros LJ, Wang W, et al. Association Between B-Cell Marker Expression and RUNX1 Lesions in Acute Myeloid Leukemia, Beyond RUNX1::RUNX1T1 Fusion: Diagnostic Pitfalls with Mixed-Phenotype Acute Leukemia—B/Myeloid. Cancers. 2025; 17(8):1354. https://doi.org/10.3390/cancers17081354
Chicago/Turabian StyleGeorge, Giby V., Malgorzata Kajstura, Audrey N. Jajosky, Hong Fang, Fatima Zahra Jelloul, Andrew G. Evans, W. Richard Burack, John M. Bennett, L. Jeffrey Medeiros, Wei Wang, and et al. 2025. "Association Between B-Cell Marker Expression and RUNX1 Lesions in Acute Myeloid Leukemia, Beyond RUNX1::RUNX1T1 Fusion: Diagnostic Pitfalls with Mixed-Phenotype Acute Leukemia—B/Myeloid" Cancers 17, no. 8: 1354. https://doi.org/10.3390/cancers17081354
APA StyleGeorge, G. V., Kajstura, M., Jajosky, A. N., Fang, H., Jelloul, F. Z., Evans, A. G., Burack, W. R., Bennett, J. M., Medeiros, L. J., Wang, W., & El Hussein, S. (2025). Association Between B-Cell Marker Expression and RUNX1 Lesions in Acute Myeloid Leukemia, Beyond RUNX1::RUNX1T1 Fusion: Diagnostic Pitfalls with Mixed-Phenotype Acute Leukemia—B/Myeloid. Cancers, 17(8), 1354. https://doi.org/10.3390/cancers17081354