
Germline Testing in Breast Cancer: A Single-Center Analysis Comparing Strengths and Challenges of Different Approaches

 , ,
, ,  , ,
, ,  , ,
, ,  and
and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Molecular Analyses
- In 308 patients, genetic testing was carried out via an SGT approach using the Devyser BRCA NGS test (Devyser AB, Stockholm, Sweden), which allows for the analysis of BRCA genes.
- In the remaining 776 patients, analysis was performed through an MGPT approach with a custom Hereditary Cancer Solution panel (SOPHiA GENETICS, Geneva, Switzerland), which includes 28 cancer predisposition genes (ABRAXAS1, APC, ATM, BARD1, BRCA1, BRCA2, BRIP1, CDH1, CDK4, CDKN2A, CHEK2, EPCAM, MLH1, MRE11A, MSH2, MSH6, MUTYH, NBN, PALB2, PIK3CA, PMS2, PTEN, RAD50, RAD51C, RAD51D, STK11, TP53, XRCC2) and the pseudogene PMS2CL. This in-house validated panel initially covered 26 genes, but it was customized with two additional genes for familial melanoma predisposition (CDKN2A and CDK4).
2.3. Classification of Genetic Testing Results
- BRCA genes—BRCA1 and BRCA2;
- Other non-BRCA BC susceptibility genes—genes known to be associated with BC (i.e., ATM, BARD1, CDH1, CHEK2, PALB2, PTEN, RAD51C, RAD51D, STK11, TP53);
- Genes responsible for other CPSs not strictly known to be associated with BC risk (such as APC, CDK4, CDKN2A, MLH1, MSH2, MSH6, MUTYH, PMS2, EPCAM deletions of the 3′ region);
- Genes included in the panel and not clearly associated with BC risk (ABRAXAS1, BRIP1, MRE11A, NBN, PIK3CA, RAD50, XRCC2, point mutations in EPCAM).
2.4. Statistical Analyses
3. Results
3.1. Genetic Testing Results
3.1.1. SGT Results
3.1.2. MGPT Results
3.1.3. Clinical Interpretation of Genetic Testing Results
3.2. Comparison Between SGT and MGPT Approaches
3.3. Clinical Characterization of BC Patients Based on Genetic Testing Results
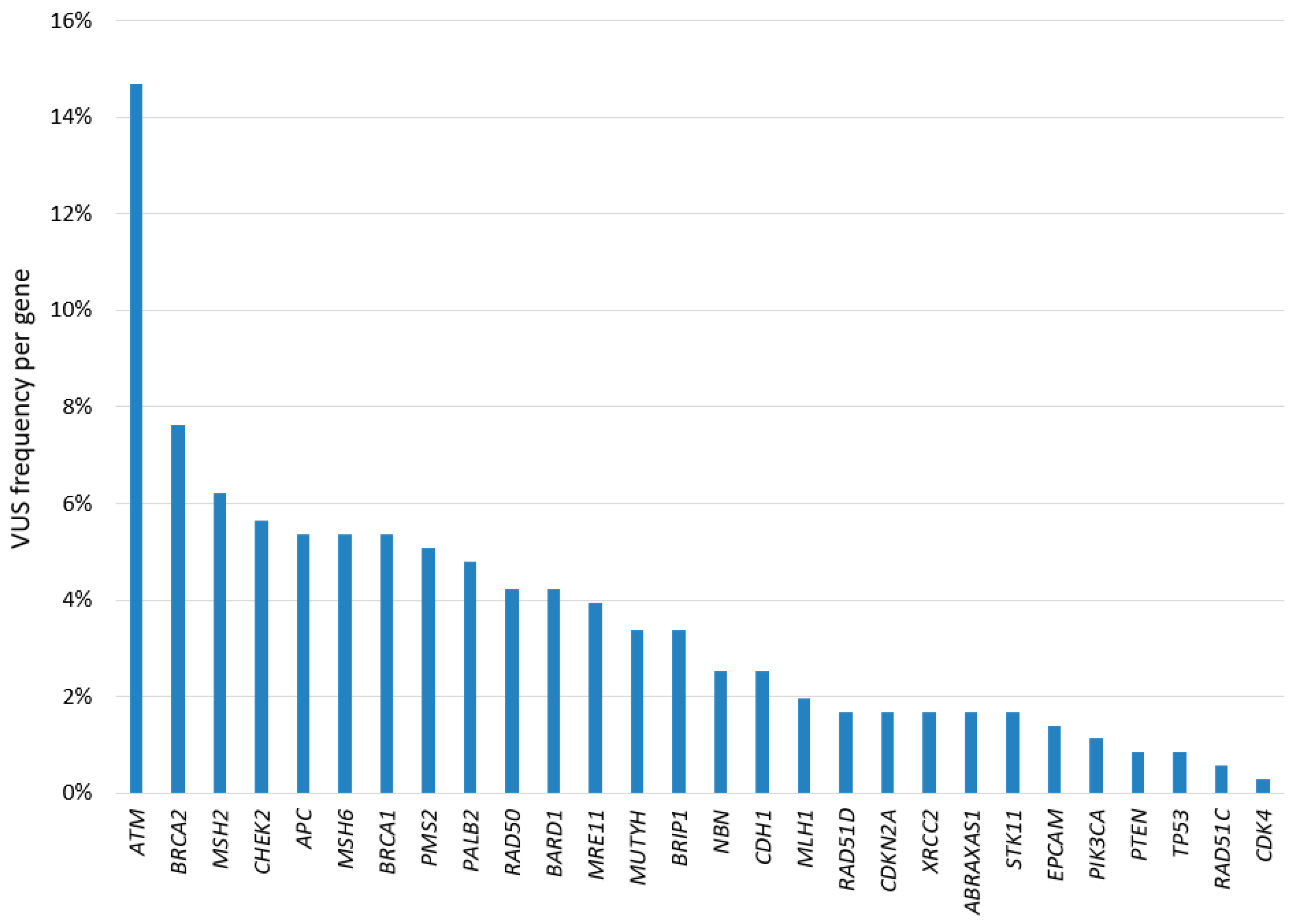
3.4. Overview of the Variants Identified in the Study
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACMG | American College of Medical Genetics and Genomics |
| AMP | Association for Molecular Pathology |
| BC | breast cancer |
| BRCA | BRCA1 and BRCA2 genes |
| CI | confidence interval |
| CNV | copy number variant |
| CPS | cancer predisposition syndrome |
| ER | estrogen receptor |
| FDR | false discovery rate |
| hetPV | heterozygous PV in genes associated with autosomal recessive conditions |
| HGVS | Human Genome Variation Society |
| IARC | International Agency for Research on Cancer |
| IEO | European Institute of Oncology |
| indel | small insertion/deletion |
| IQR | interquartile range |
| lowPV | low-penetrance PV |
| LS | Lynch syndrome |
| MGPT | multigene panel testing |
| MMR | mismatch repair |
| NGS | next generation sequencing |
| NST | no special type |
| OC | ovarian cancer |
| OR | odds ratio |
| PF | primary finding |
| PV | pathogenic/likely pathogenic variant |
| SF | secondary finding |
| SGT | single gene testing |
| SNV | single nucleotide variant |
| TNBC | triple-negative breast cancer |
| VUS | variant of uncertain significance |
| WT | wild-type |
References
- Bedrosian, I.; Somerfield, M.R.; Achatz, M.I.; Boughey, J.C.; Curigliano, G.; Friedman, S.; Kohlmann, W.K.; Kurian, A.W.; Laronga, C.; Lynce, F.; et al. Germline Testing in Patients with Breast Cancer: ASCO-Society of Surgical Oncology Guideline. J. Clin. Oncol. 2024, 42, 584–604. [Google Scholar] [CrossRef]
- Valencia, O.M.; Samuel, S.E.; Viscusi, R.K.; Riall, T.S.; Neumayer, L.A.; Aziz, H. The Role of Genetic Testing in Patients with Breast Cancer: A Review. JAMA Surg. 2017, 152, 589–594. [Google Scholar] [CrossRef]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Breast Cancer Association Consortium; Dorling, L.; Carvalho, S.; Allen, J.; González-Neira, A.; Luccarini, C.; Wahlström, C.; Pooley, K.A.; Parsons, M.T.; Fortuno, C.; et al. Breast Cancer Risk Genes—Association Analysis in More than 113,000 Women. N. Engl. J. Med. 2021, 384, 428–439. [Google Scholar] [CrossRef]
- Blair, V.R.; McLeod, M.; Carneiro, F.; Coit, D.G.; D’Addario, J.L.; van Dieren, J.M.; Harris, K.L.; Hoogerbrugge, N.; Oliveira, C.; van der Post, R.S.; et al. Hereditary diffuse gastric cancer: Updated clinical practice guidelines. Lancet Oncol. 2020, 21, e386–e397. [Google Scholar] [CrossRef] [PubMed]
- Hindorff, L.A.; Gillanders, E.M.; Manolio, T.A. Genetic architecture of cancer and other complex diseases: Lessons learned and future directions. Carcinogenesis 2011, 32, 945–954. [Google Scholar] [CrossRef] [PubMed]
- LaDuca, H.; Stuenkel, A.J.; Dolinsky, J.S.; Keiles, S.; Tandy, S.; Pesaran, T.; Chen, E.; Gau, C.L.; Palmaer, E.; Shoaepour, K.; et al. Utilization of multigene panels in hereditary cancer predisposition testing: Analysis of more than 2,000 patients. Genet. Med. 2014, 16, 830–837. [Google Scholar] [CrossRef]
- Neben, C.L.; Zimmer, A.D.; Stedden, W.; van den Akker, J.; O’Connor, R.; Chan, R.C.; Chen, E.; Tan, Z.; Leon, A.; Ji, J.; et al. Multi-Gene Panel Testing of 23,179 Individuals for Hereditary Cancer Risk Identifies Pathogenic Variant Carriers Missed by Current Genetic Testing Guidelines. J. Mol. Diagn. 2019, 21, 646–657. [Google Scholar] [CrossRef]
- Carlsson, L.; Bedard, P.L.; Kim, R.H.; Metcalfe, K. Psychological distress following multi-gene panel testing for hereditary breast and ovarian cancer risk. J. Genet. Couns. 2024, 34, e1940. [Google Scholar] [CrossRef]
- Slavin, T.P.; Niell-Swiller, M.; Solomon, I.; Nehoray, B.; Rybak, C.; Blazer, K.R.; Weitzel, J.N. Clinical Application of Multigene Panels: Challenges of Next-Generation Counseling and Cancer Risk Management. Front. Oncol. 2015, 5, 208. [Google Scholar] [CrossRef]
- Katz, A.E.; Nussbaum, R.L.; Solomon, B.D.; Rehm, H.L.; Williams, M.S.; Biesecker, L.G. Management of Secondary Genomic Findings. Am. J. Hum. Genet. 2020, 107, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.E.; Jackson, S.A.; Susswein, L.R.; Zeinomar, N.; Ma, X.; Marshall, M.L.; Stettner, A.R.; Milewski, B.; Xu, Z.; Solomon, B.D.; et al. MSH6 and PMS2 germ-line pathogenic variants implicated in Lynch syndrome are associated with breast cancer. Genet. Med. 2018, 20, 1167–1174. [Google Scholar] [CrossRef]
- Chan, S.H.; Chiang, J.; Ngeow, J. CDKN2A germline alterations and the relevance of genotype-phenotype associations in cancer predisposition. Hered. Cancer. Clin. Pract. 2021, 19, 21. [Google Scholar] [CrossRef]
- Miller, D.T.; Lee, K.; Abul-Husn, N.S.; Amendola, L.M.; Brothers, K.; Chung, W.K.; Gollob, M.H.; Gordon, A.S.; Harrison, S.M.; Hershberger, R.E.; et al. ACMG SF v3.2 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2023, 25, 100866. [Google Scholar] [CrossRef]
- Schmidt, R.J.; Steeves, M.; Bayrak-Toydemir, P.; Benson, K.A.; Coe, B.P.; Conlin, L.K.; Ganapathi, M.; Garcia, J.; Gollob, M.H.; Jobanputra, V.; et al. ClinGen Low Penetrance/Risk Allele Working Group. Recommendations for risk allele evidence curation, classification, and reporting from the ClinGen Low Penetrance/Risk Allele Working Group. Genet. Med. 2024, 26, 101036. [Google Scholar] [CrossRef] [PubMed]
- Fortier, N.; Rudy, G.; Scherer, A. Detection of CNVs in NGS Data Using VS-CNV. Methods Mol. Biol. 2018, 1833, 115–127. [Google Scholar] [CrossRef]
- Incorvaia, L.; Fanale, D.; Bono, M.; Calò, V.; Fiorino, A.; Brando, C.; Corsini, L.R.; Cutaia, S.; Cancelliere, D.; Pivetti, A.; et al. BRCA1/2 pathogenic variants in triple-negative versus luminal-like breast cancers: Genotype-phenotype correlation in a cohort of 531 patients. Ther. Adv. Med. Oncol. 2020, 12, 1758835920975326. [Google Scholar] [CrossRef] [PubMed]
- Plon, S.E.; Eccles, D.M.; Easton, D.; Foulkes, W.D.; Genuardi, M.; Greenblatt, M.S.; Hogervorst, F.B.; Hoogerbrugge, N.; Spurdle, A.B.; Tavtigian, S.V.; et al. Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008, 29, 1282–1291. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Kurian, A.W.; Hare, E.E.; Mills, M.A.; Kingham, K.E.; McPherson, L.; Whittemore, A.S.; McGuire, V.; Ladabaum, U.; Kobayashi, Y.; Lincoln, S.E.; et al. Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J. Clin. Oncol. 2014, 32, 2001–2009. [Google Scholar] [CrossRef]
- Samadder, N.J.; Riegert-Johnson, D.; Boardman, L.; Rhodes, D.; Wick, M.; Okuno, S.; Kunze, K.L.; Golafshar, M.; Uson, P.L.S., Jr.; Mountjoy, L.; et al. Comparison of Universal Genetic Testing vs. Guideline-Directed Targeted Testing for Patients with Hereditary Cancer Syndrome. JAMA Oncol. 2021, 7, 230–237. [Google Scholar] [CrossRef]
- Desmond, A.; Kurian, A.W.; Gabree, M.; Mills, M.A.; Anderson, M.J.; Kobayashi, Y.; Horick, N.; Yang, S.; Shannon, K.M.; Tung, N.; et al. Clinical Actionability of Multigene Panel Testing for Hereditary Breast and Ovarian Cancer Risk Assessment. JAMA Oncol. 2015, 1, 943–951. [Google Scholar] [CrossRef]
- Innella, G.; Ferrari, S.; Miccoli, S.; Turchetti, D. Light and shade of multigene panel testing for hereditary cancer: Examples from the real world. Tumori J. 2024, epub ahead of print. [Google Scholar] [CrossRef]
- Li, J.; Wen, W.X.; Eklund, M.; Kvist, A.; Eriksson, M.; Christensen, H.N.; Torstensson, A.; Bajalica-Lagercrantz, S.; Dunning, A.M.; Decker, B.; et al. Prevalence of BRCA1 and BRCA2 pathogenic variants in a large, unselected breast cancer cohort. Int. J. Cancer 2019, 144, 1195–1204. [Google Scholar] [CrossRef]
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J.; et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N. Engl. J. Med. 2021, 384, 440–451. [Google Scholar] [CrossRef]
- Tischkowitz, M.; Capanu, M.; Sabbaghian, N.; Li, L.; Liang, X.; Vallée, M.P.; Tavtigian, S.V.; Concannon, P.; Foulkes, W.D.; Bernstein, L.; et al. Rare germline mutations in PALB2 and breast cancer risk: A population-based study. Hum. Mutat. 2012, 33, 674–680. [Google Scholar] [CrossRef]
- Damiola, F.; Schultz, I.; Barjhoux, L.; Sornin, V.; Dondon, M.G.; Eon-Marchais, S.; Marcou, M.; GENESIS Study Investigators; Caron, O.; Gauthier-Villars, M.; et al. Mutation analysis of PALB2 gene in French breast cancer families. Breast Cancer Res. Treat. 2015, 154, 463–471. [Google Scholar] [CrossRef]
- Hu, C.; Polley, E.C.; Yadav, S.; Lilyquist, J.; Shimelis, H.; Na, J.; Hart, S.N.; Goldgar, D.E.; Shah, S.; Pesaran, T.; et al. The Contribution of Germline Predisposition Gene Mutations to Clinical Subtypes of Invasive Breast Cancer from a Clinical Genetic Testing Cohort. J. Natl. Cancer Inst. 2020, 112, 1231–1241. [Google Scholar] [CrossRef]
- Lu, H.M.; Li, S.; Black, M.H.; Lee, S.; Hoiness, R.; Wu, S.; Mu, W.; Huether, R.; Chen, J.; Sridhar, S.; et al. Association of Breast and Ovarian Cancers with Predisposition Genes Identified by Large-Scale Sequencing. JAMA Oncol. 2019, 5, 51–57. [Google Scholar] [CrossRef]
- Win, A.K.; Lindor, N.M.; Jenkins, M.A. Risk of breast cancer in Lynch syndrome: A systematic review. Breast Cancer Res. 2013, 15, R27. [Google Scholar] [CrossRef]
- Weber-Lassalle, N.; Hauke, J.; Ramser, J.; Richters, L.; Groß, E.; Blümcke, B.; Gehrig, A.; Kahlert, A.K.; Müller, C.R.; Hackmann, K.; et al. BRIP1 loss-of-function mutations confer high risk for familial ovarian cancer, but not familial breast cancer. Breast Cancer Res. 2018, 20, 7. [Google Scholar] [CrossRef]
- Lowstuter, K.; Espenschied, C.R.; Sturgeon, D.; Ricker, C.; Karam, R.; LaDuca, H.; Culver, J.O.; Dolinsky, J.S.; Chao, E.; Sturgeon, J.; et al. Unexpected CDH1 Mutations Identified on Multigene Panels Pose Clinical Management Challenges. JCO Precis. Oncol. 2017, 1, 1–12. [Google Scholar] [CrossRef]
- Pal, T.; Brzosowicz, J.; Valladares, A.; Wiesner, G.L.; Laronga, C. Identification and Management of TP53 Gene Carriers Detected Through Multigene Panel Testing. South Med. J. 2017, 110, 643–648. [Google Scholar] [CrossRef]
- Thompson, A.B.; Sutcliffe, E.G.; Arvai, K.; Roberts, M.E.; Susswein, L.R.; Marshall, M.L.; Torene, R.; Postula, K.J.V.; Hruska, K.S.; Bai, S. Monoallelic MUTYH pathogenic variants ascertained via multi-gene hereditary cancer panels are not associated with colorectal, endometrial, or breast cancer. Fam. Cancer 2022, 21, 415–422. [Google Scholar] [CrossRef]
- Cleary, S.P.; Cotterchio, M.; Jenkins, M.A.; Kim, H.; Bristow, R.; Green, R.; Haile, R.; Hopper, J.L.; LeMarchand, L.; Lindor, N.; et al. Germline MutY human homologue mutations and colorectal cancer: A multisite case-control study. Gastroenterology 2009, 136, 1251–1260. [Google Scholar] [CrossRef]
- Liang, J.; Lin, C.; Hu, F.; Wang, F.; Zhu, L.; Yao, X.; Wang, Y.; Zhao, Y. APC Polymorphisms and the Risk of Colorectal Neoplasia: A HuGE Review and Meta-Analysis. Am. J. Epidemiol. 2013, 177, 1169–1179. [Google Scholar] [CrossRef]
- Forkosh, E.; Bergel, M.; Hatchell, K.E.; Nielsen, S.M.; Heald, B.; Benson, A.A.; Friedman, E.; Esplin, E.D.; Katz, L.H. Ashkenazi Jewish and Other White APC I1307K Carriers Are at Higher Risk for Multiple Cancers. Cancers 2022, 14, 5875. [Google Scholar] [CrossRef]
- Weischer, M.; Bojesen, S.E.; Ellervik, C.; Tybjaerg-Hansen, A.; Nordestgaard, B.G. CHEK2*1100delC Genotyping for Clinical Assessment of Breast Cancer Risk: Meta-Analyses of 26,000 Patient Cases and 27,000 Controls. J. Clin. Oncol. 2008, 26, 542–548. [Google Scholar] [CrossRef]
- Dorling, L.; Carvalho, S.; Allen, J.; Parsons, M.T.; Fortuno, C.; González-Neira, A.; Heijl, S.M.; Adank, M.A.; Ahearn, T.U.; Andrulis, I.L.; et al. Breast cancer risks associated with missense variants in breast cancer susceptibility genes. Genome Med. 2022, 14, 51. [Google Scholar] [CrossRef]
- Bychkovsky, B.L.; Agaoglu, N.B.; Horton, C.; Zhou, J.; Yussuf, A.; Hemyari, P.; Richardson, M.E.; Young, C.; LaDuca, H.; McGuinness, D.L.; et al. Differences in Cancer Phenotypes Among Frequent CHEK2 Variants and Implications for Clinical Care-Checking CHEK2. JAMA Oncol. 2022, 8, 1598–1606. [Google Scholar] [CrossRef]
- Lebo, M.S.; Zakoor, K.R.; Chun, K.; Speevak, M.D.; Waye, J.S.; McCready, E.; Parboosingh, J.S.; Lamont, R.E.; Feilotter, H.; Bosdet, I.; et al. Data sharing as a national quality improvement program: Reporting on BRCA1 and BRCA2 variant-interpretation comparisons through the Canadian Open Genetics Repository (COGR). Genet. Med. 2018, 20, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Tsai, G.J.; Rañola, J.M.O.; Smith, C.; Garrett, L.T.; Bergquist, T.; Casadei, S.; Bowen, D.J.; Shirts, B.H. Outcomes of 92 patient-driven family studies for reclassification of variants of uncertain significance. Genet. Med. 2019, 21, 1435–1442. [Google Scholar] [CrossRef]
- Chen, E.; Facio, F.M.; Aradhya, K.W.; Rojahn, S.; Hatchell, K.E.; Aguilar, S.; Ouyang, K.; Saitta, S.; Hanson-Kwan, A.K.; Capurro, N.N.; et al. Rates and Classification of Variants of Uncertain Significance in Hereditary Disease Genetic Testing. JAMA Netw. Open 2023, 6, e2339571. [Google Scholar] [CrossRef]
- Mavaddat, N.; Barrowdale, D.; Andrulis, I.L.; Domchek, S.M.; Eccles, D.; Nevanlinna, H.; Ramus, S.J.; Spurdle, A.; Robson, M.; Sherman, M.; et al. Pathology of breast and ovarian cancers among BRCA1 and BRCA2 mutation carriers: Results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol. Biomark. Prev. 2012, 21, 134–147. [Google Scholar] [CrossRef]
- Larsen, M.J.; Kruse, T.A.; Tan, Q.; Lænkholm, A.V.; Bak, M.; Lykkesfeldt, A.E.; Sørensen, K.P.; Hansen, T.V.; Ejlertsen, B.; Gerdes, A.M.; et al. Classifications within molecular subtypes enables identification of BRCA1/BRCA2 mutation carriers by RNA tumor profiling. PLoS ONE 2013, 8, e64268. [Google Scholar] [CrossRef]
- Kwong, A.; Chen, J.; Shin, V.Y.; Ho, J.C.; Law, F.B.; Au, C.H.; Chan, T.L.; Ma, E.S.; Ford, J.M. The importance of analysis of long-range rearrangement of BRCA1 and BRCA2 in genetic diagnosis of familial breast cancer. Cancer Genet. 2015, 208, 448–454. [Google Scholar] [CrossRef]
- Wang, Y.; Bernhardy, A.J.; Nacson, J.; Krais, J.J.; Tan, Y.F.; Nicolas, E.; Radke, M.R.; Handorf, E.; Llop-Guevara, A.; Balmaña, J.; et al. BRCA1 intronic Alu elements drive gene rearrangements and PARP inhibitor resistance. Nat. Commun. 2019, 10, 5661. [Google Scholar] [CrossRef]
- Janatova, M.; Kleibl, Z.; Stribrna, J.; Panczak, A.; Vesela, K.; Zimovjanova, M.; Kleiblova, P.; Dundr, P.; Soukupova, J.; Pohlreich, P. The PALB2 gene is a strong candidate for clinical testing in BRCA1- and BRCA2-negative hereditary breast cancer. Cancer Epidemiol. Biomark. Prev. 2013, 22, 2323–2332. [Google Scholar] [CrossRef]
- Richardson, M.E.; Chong, H.; Mu, W.; Conner, B.R.; Hsuan, V.; Willett, S.; Lam, S.; Tsai, P.; Pesaran, T.; Chamberlin, A.C.; et al. DNA breakpoint assay reveals a majority of gross duplications occur in tandem reducing VUS classifications in breast cancer predisposition genes. Genet. Med. 2019, 21, 683–693. [Google Scholar] [CrossRef]
- Yang, X.; Leslie, G.; Doroszuk, A.; Schneider, S.; Allen, J.; Decker, B.; Dunning, A.M.; Redman, J.; Scarth, J.; Plaskocinska, I.; et al. Cancer Risks Associated with Germline PALB2 Pathogenic Variants: An International Study of 524 Families. J. Clin. Oncol. 2020, 38, 674–685. [Google Scholar] [CrossRef]
- Sidoti, D.; Margotta, V.; Calosci, D.; Fiorentini, E.; Bacci, C.; Gensini, F.; Papi, L.; Montini, M. Alu-Mediated Duplication and Deletion of Exon 11 Are Frequent Mechanisms of PALB2 Inactivation, Predisposing Individuals to Hereditary Breast-Ovarian Cancer Syndrome. Cancers 2024, 16, 4022. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Arnold, A.G.; Trottier, M.; Sonoda, Y.; Abu-Rustum, N.R.; Zivanovic, O.; Robson, M.E.; Stadler, Z.K.; Walsh, M.F.; Hyman, D.M.; et al. Characterization of a novel germline PALB2 duplication in a hereditary breast and ovarian cancer family. Breast Cancer Res. Treat. 2016, 160, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zethoven, M.; McInerny, S.; Healey, E.; DeSilva, D.; Devereux, L.; Scott, R.J.; James, P.A.; Campbell, I.G. Contribution of large genomic rearrangements in PALB2 to familial breast cancer: Implications for genetic testing. J. Med. Genet. 2023, 60, 112–118. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| SGT Results | Patients (N = 308) | % | |
|---|---|---|---|
| Positive | 42 | 13.6% | |
| PFs | 42 | 13.6% | |
| BRCA1 | 32 | 10.4% | |
| BRCA2 | 10 | 3.2% | |
| Uncertain | 21 | 6.8% | |
| Uninformative | 245 | 79.5% | |
| MGPT Results | Patients (N = 776) | % | |
| Positive | 123 | 15.9% | |
| PFs | 89 | 11.5% | |
| BRCA1 | 13 | 1.7% | |
| BRCA2 | 31 | 4.0% | |
| BRCA1 + BRCA2 | 1 | 0.1% | |
| BRCA2 + BRCA2 | 1 | 0.1% | |
| BRCA2 + APC | 1 | 0.1% | |
| BRCA2 + CHEK2 | 1 | 0.1% | |
| BRCA2 + RAD51C | 1 | 0.1% | |
| ATM | 9 | 1.2% | |
| ATM + RAD51D | 1 | 0.1% | |
| BARD1 | 1 | 0.1% | |
| CDH1 | 1 | 0.1% | |
| CDKN2A + MUTYH | 1 | 0.1% | |
| CHEK2 | 10 | 1.3% | |
| MLH1 | 2 | 0.3% | |
| MUTYH (biallelic) | 1 | 0.1% | |
| PALB2 | 10 | 1.3% | |
| PMS2 | 1 | 0.1% | |
| RAD51C | 3 | 0.4% | |
| SFs | 5 | 0.6% | |
| CDH1 | 2 | 0.3% | |
| CDKN2A | 1 | 0.1% | |
| BRIP1 | 2 | 0.3% | |
| lowPVs | 10 | 1.3% | |
| APC | 6 | 0.8% | |
| CHEK2 | 4 | 0.5% | |
| hetPVs | 18 | 2.3% | |
| MUTYH (monoallelic) | 13 | 1.7% | |
| NBN + MRE11A | 1 | 0.1% | |
| RAD50 | 4 | 0.5% | |
| lowPV + hetPV | 1 | 0.1% | |
| APC + EPCAM | 1 | 0.1% | |
| Uncertain | 266 | 34.3% | |
| Uninformative | 387 | 49.9% | |
| Results | Overall (%) | SGT | MGPT | p-Value a |
|---|---|---|---|---|
| Positive | 165 (15.2%) | 42 (13.6%) | 123 (15.9%) | <0.001 |
| Uncertain | 287 (26.5%) | 21 (6.8%) | 266 (34.3%) | |
| Uninformative | 632 (58.3%) | 245 (79.5%) | 387 (49.9%) | |
| PFs | 131 (12.1%) | 42 (13.6%) | 89 (11.5%) | <0.001 |
| Complex | 321 (29.6%) | 21 (6.8%) | 300 (38.7%) | |
| Uninformative | 632 (58.3%) | 245 (79.5%) | 387 (49.9%) | |
| 1084 (100%) | 308 (100%) | 776 (100%) |
| Variable | Contrast | Testing Approach | End-Point | OR | Low 95% CI | Up 95% CI | p-Value e |
|---|---|---|---|---|---|---|---|
| Genetic testing results | Actionable vs. Inconclusive | SGT a | TNBC b | 5.97 | 2.458 | 15.818 | <0.001 |
| ER positive c | 0.29 | 0.115 | 0.677 | 0.006 | |||
| MGPT a | TNBC b | 2.46 | 1.055 | 5.277 | 0.026 | ||
| ER positive d | 0.42 | 0.211 | 0.882 | 0.016 |
| Overall Patients (N = 467) | BRCA1/BRCA2 a Carriers (N = 47) | ATM/CHEK2 b,c Carriers (N = 19) | BARD1/ PALB2/RAD51C c Carriers (N = 14) | WT Patients (N = 387) | p-Value d | |
|---|---|---|---|---|---|---|
| ER | 0.004 | |||||
| Positive | 370 (79.2%) | 35 (74.5%) | 16 (84.2%) | 6 (42.9%) | 313 (80.9%) | |
| Negative | 74 (15.8%) | 10 (21.3%) | 3 (15.8%) | 7 (50.0%) | 54 (14.0%) | |
| Unknown | 23 (4.9%) | 2 (4.3%) | 0 (0.0%) | 1 (7.1%) | 20 (5.2%) | |
| HER2 | 0.599 | |||||
| Positive | 52 (11.1%) | 3 (6.4%) | 3 (15.8%) | 1 (7.1%) | 45 (11.6%) | |
| Negative | 374 (80.1%) | 41 (87.2%) | 14 (73.7%) | 12 (85.7%) | 307 (79.3%) | |
| Unknown | 41 (8.8%) | 3 (6.4%) | 2 (10.5%) | 1 (7.1%) | 35 (9.0%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marabelli, M.; Calvello, M.; Marino, E.; Morocutti, C.; Gandini, S.; Dal Molin, M.; Zanzottera, C.; Mannucci, S.; Fava, F.; Feroce, I.; et al. Germline Testing in Breast Cancer: A Single-Center Analysis Comparing Strengths and Challenges of Different Approaches. Cancers 2025, 17, 1419. https://doi.org/10.3390/cancers17091419
Marabelli M, Calvello M, Marino E, Morocutti C, Gandini S, Dal Molin M, Zanzottera C, Mannucci S, Fava F, Feroce I, et al. Germline Testing in Breast Cancer: A Single-Center Analysis Comparing Strengths and Challenges of Different Approaches. Cancers. 2025; 17(9):1419. https://doi.org/10.3390/cancers17091419
Chicago/Turabian StyleMarabelli, Monica, Mariarosaria Calvello, Elena Marino, Chiara Morocutti, Sara Gandini, Matteo Dal Molin, Cristina Zanzottera, Sara Mannucci, Francesca Fava, Irene Feroce, and et al. 2025. "Germline Testing in Breast Cancer: A Single-Center Analysis Comparing Strengths and Challenges of Different Approaches" Cancers 17, no. 9: 1419. https://doi.org/10.3390/cancers17091419
APA StyleMarabelli, M., Calvello, M., Marino, E., Morocutti, C., Gandini, S., Dal Molin, M., Zanzottera, C., Mannucci, S., Fava, F., Feroce, I., Lazzeroni, M., Guerrieri-Gonzaga, A., Bertolini, F., & Bonanni, B. (2025). Germline Testing in Breast Cancer: A Single-Center Analysis Comparing Strengths and Challenges of Different Approaches. Cancers, 17(9), 1419. https://doi.org/10.3390/cancers17091419