
Deciphering Nicotine-Driven Oncogenesis in Head and Neck Cancer: Integrative Transcriptomics and Drug Repurposing Insights

 ,
,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Maintained Nicotine-Treated Sublines
2.2. Nicotine Tolerance
2.3. Colony Formation Assay
2.4. Matrigel Invasion Assay
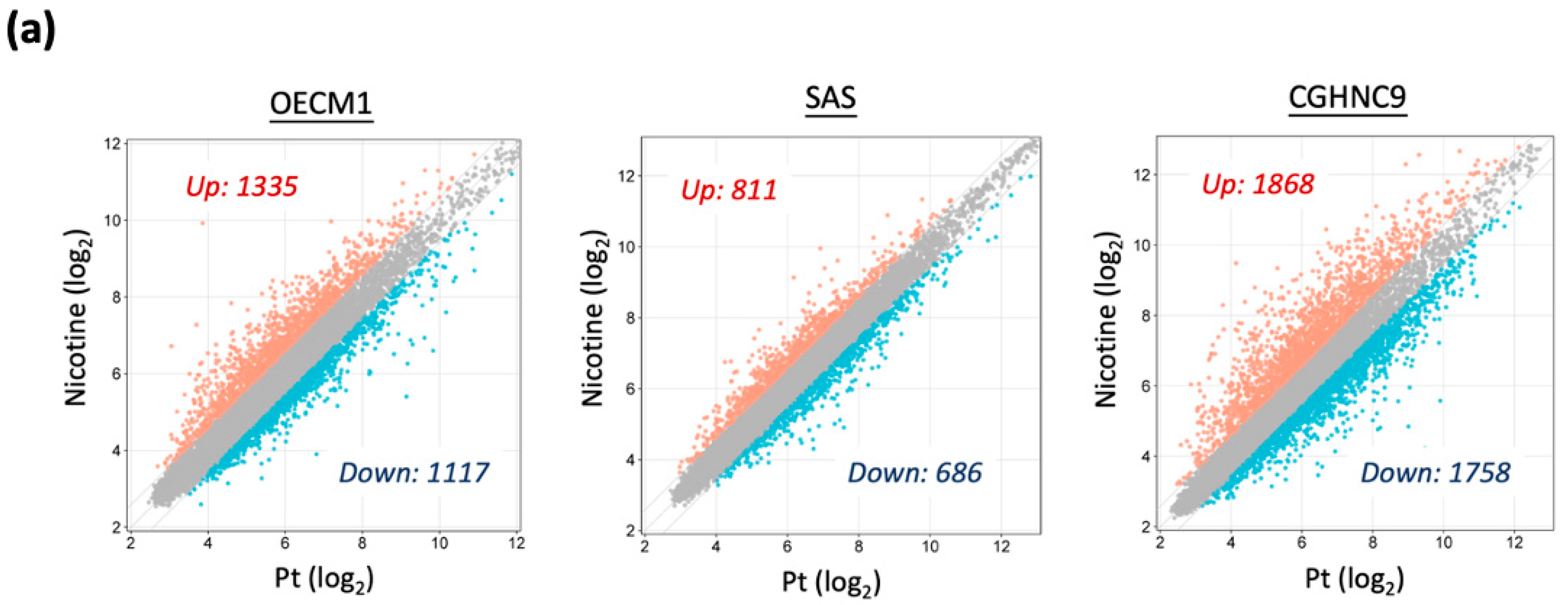
2.5. Transcriptomic and Pathway Enrichment Analysis
2.6. Drug Repurposing by In Silico Bioinformatic Analysis
3. Results
3.1. Establishment of Chronic Nicotine-Exposed Head and Neck Cancer Cell Sublines
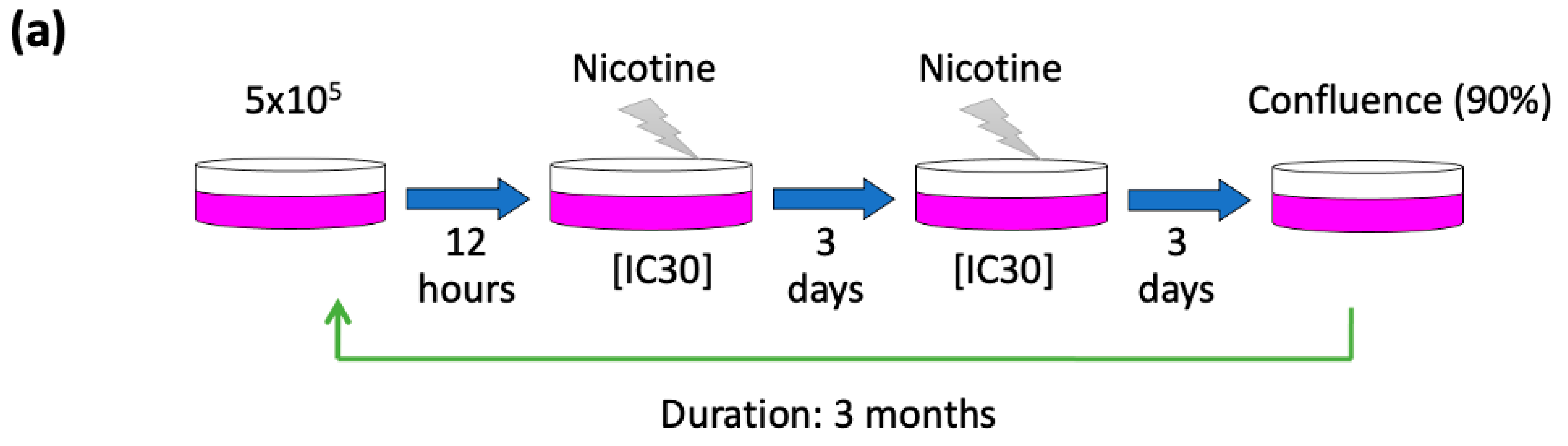
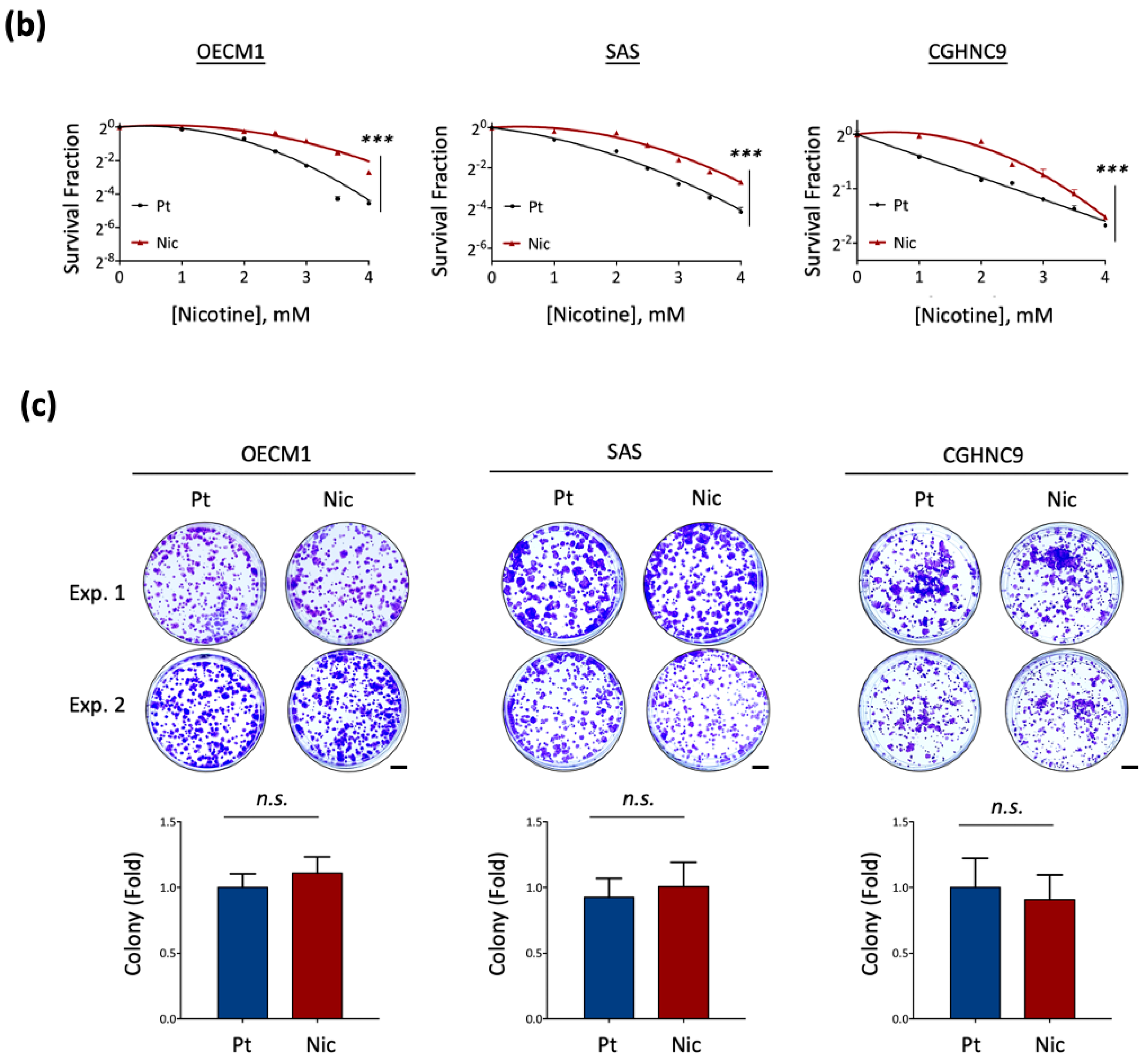
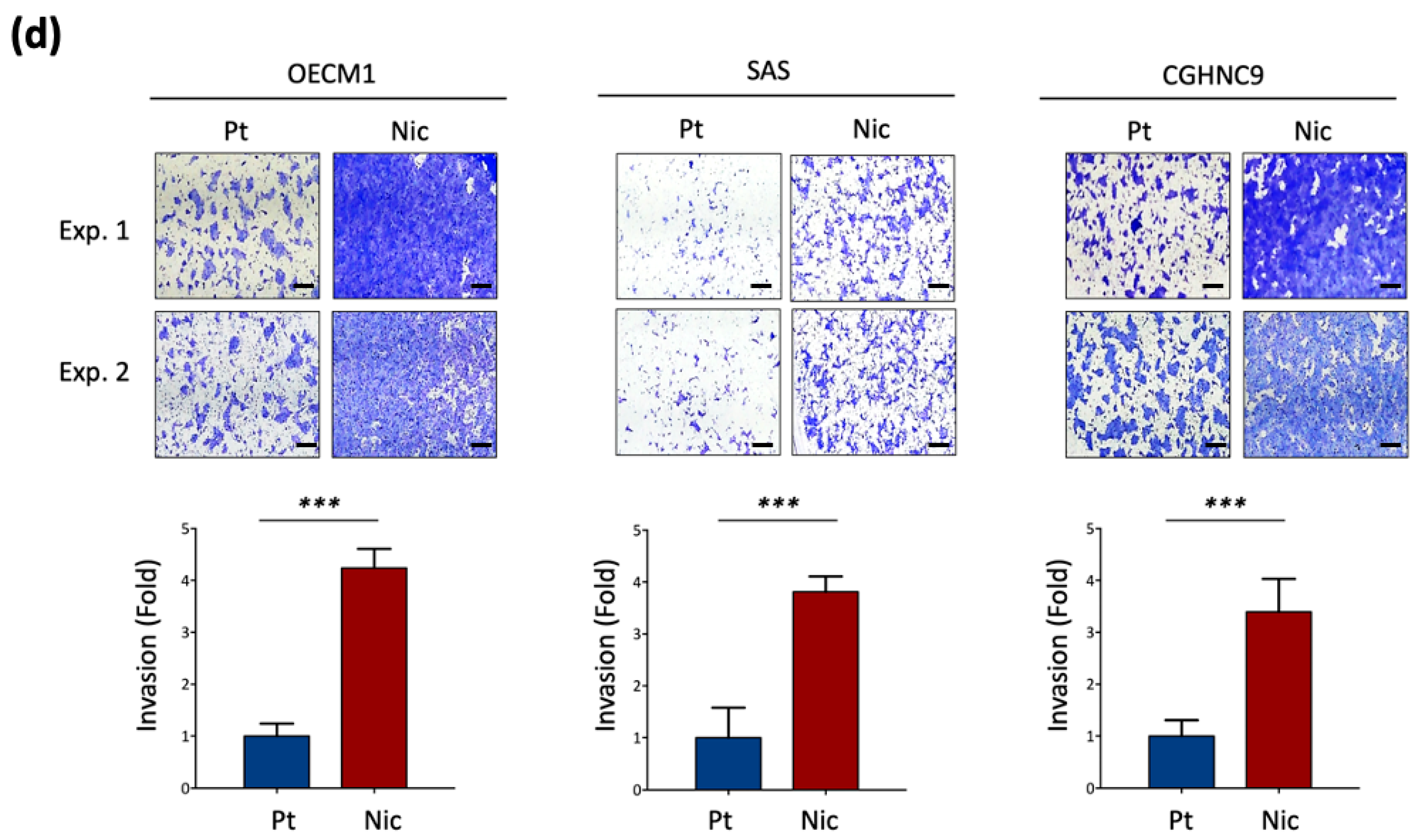
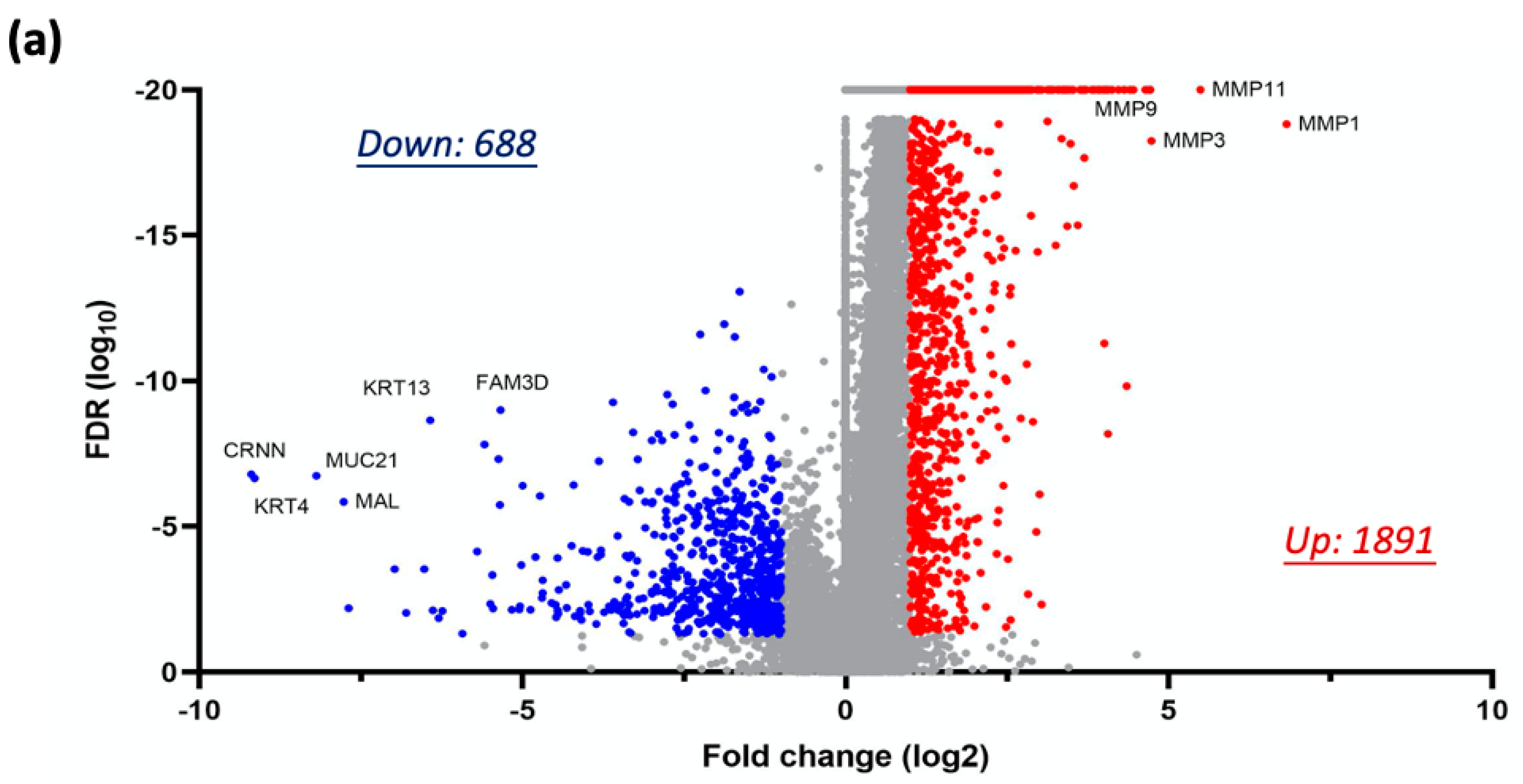
3.2. Global Transcriptomic Alterations Induced by Chronic Nicotine Exposure in HNC Cells
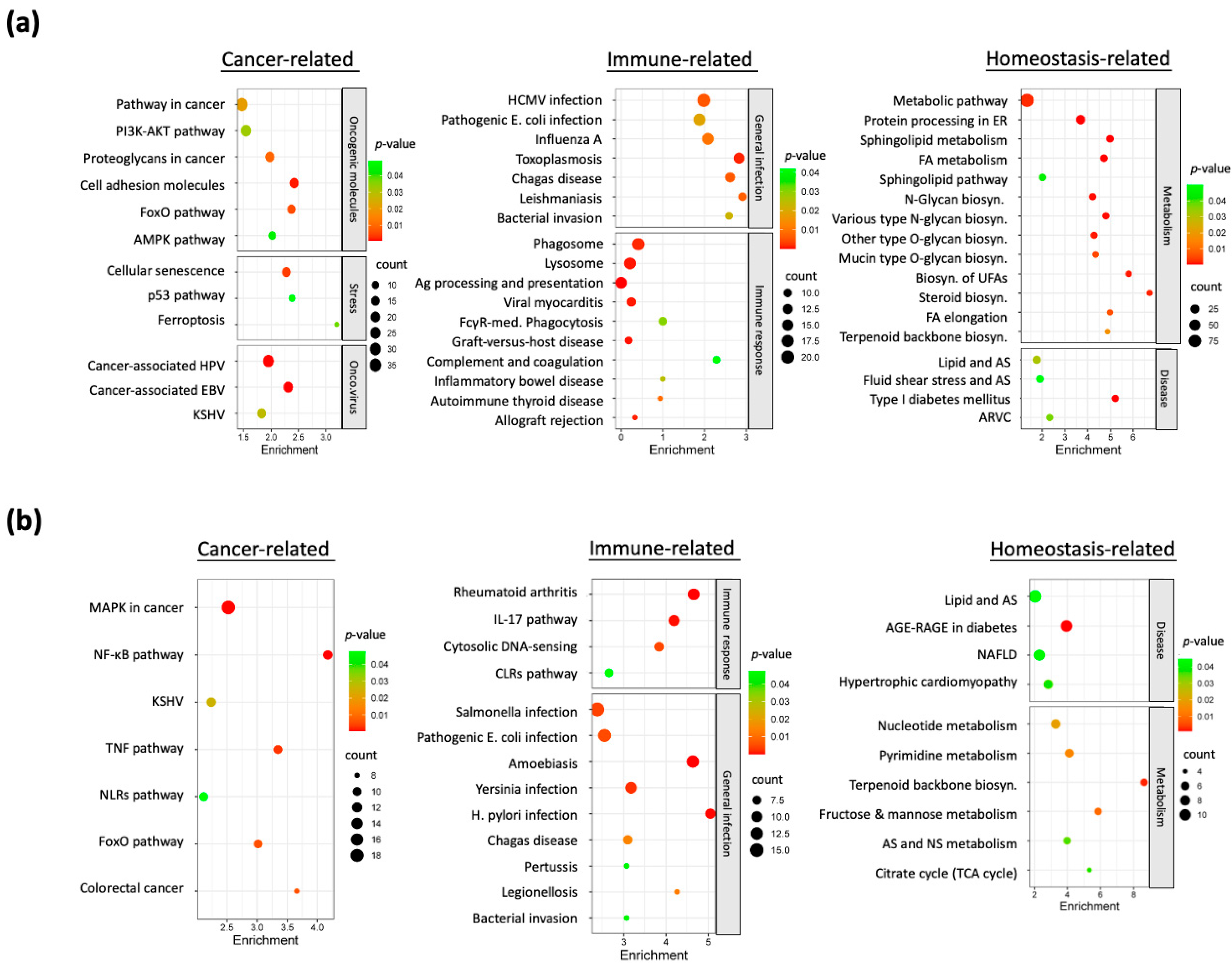
3.3. Dysregulated Molecular Pathways in Nicotine-Exposed HNC Cells
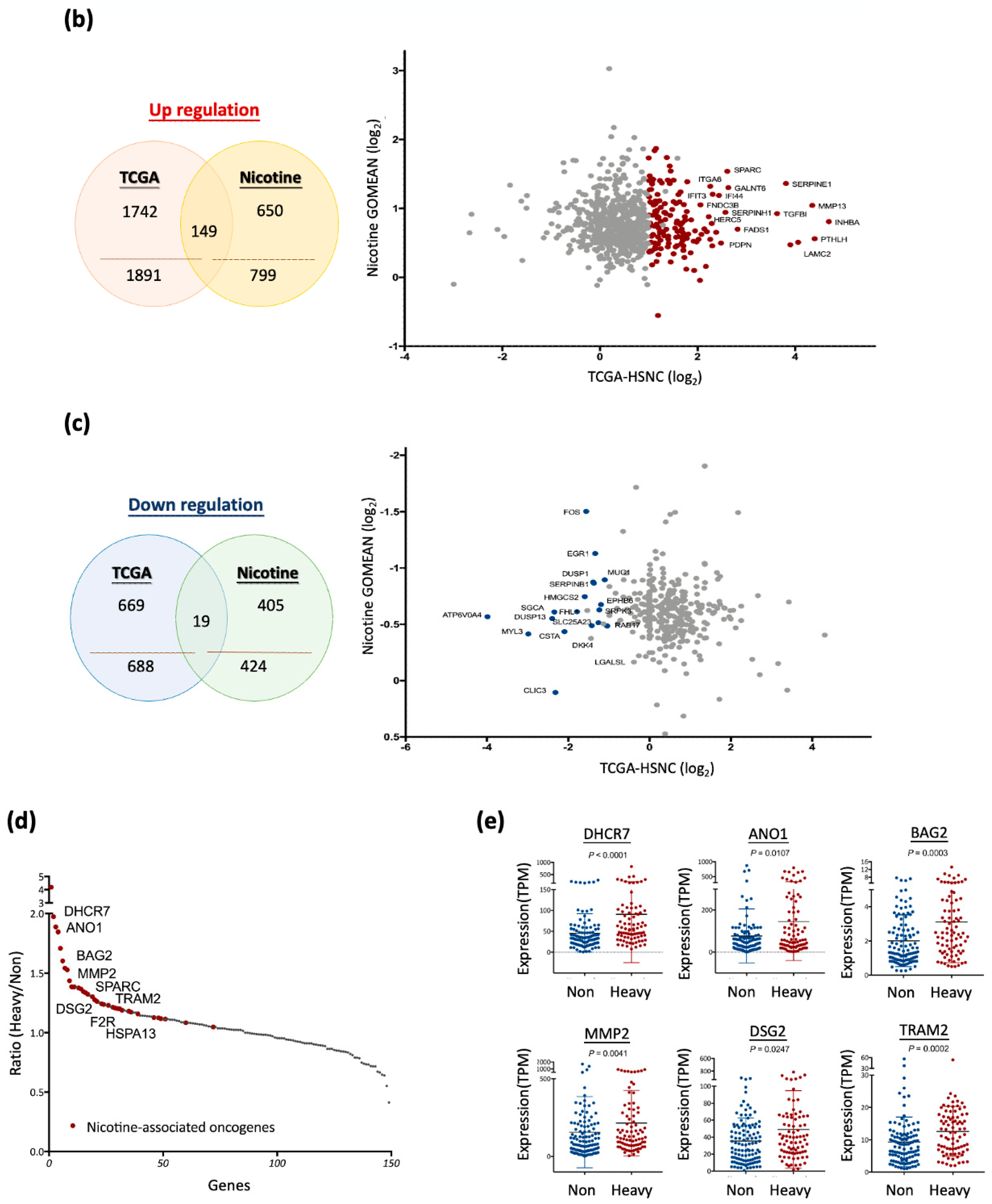
3.4. Nicotine-Driven Gene Signatures Associated with HNC Progression
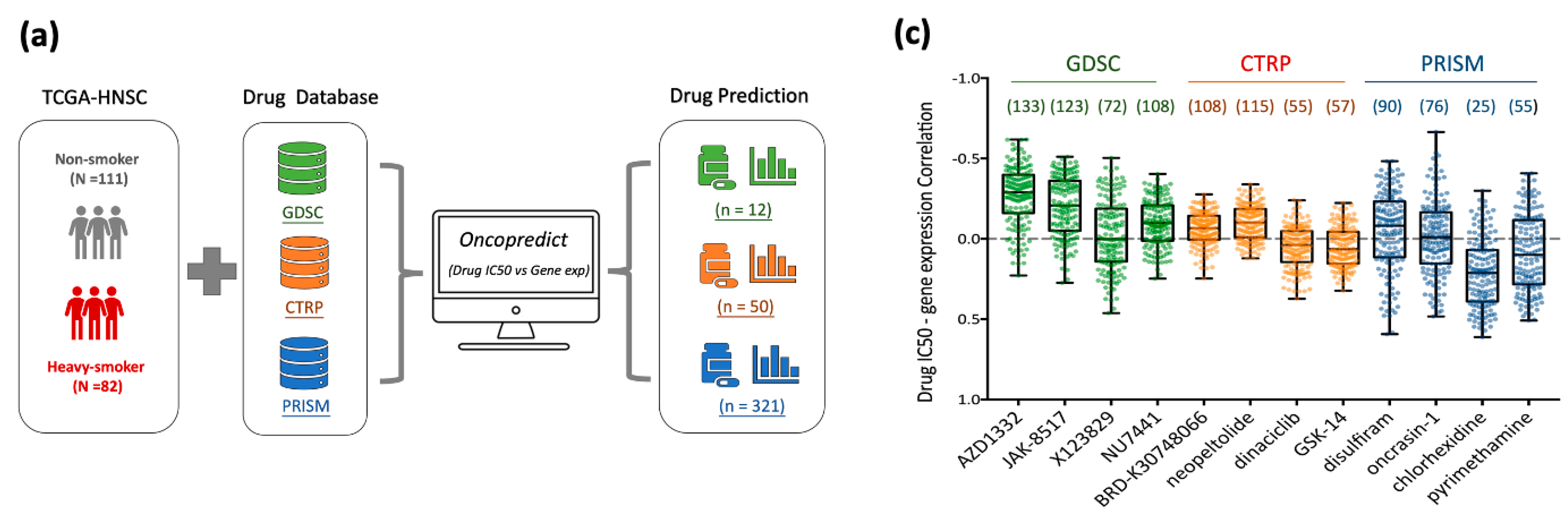
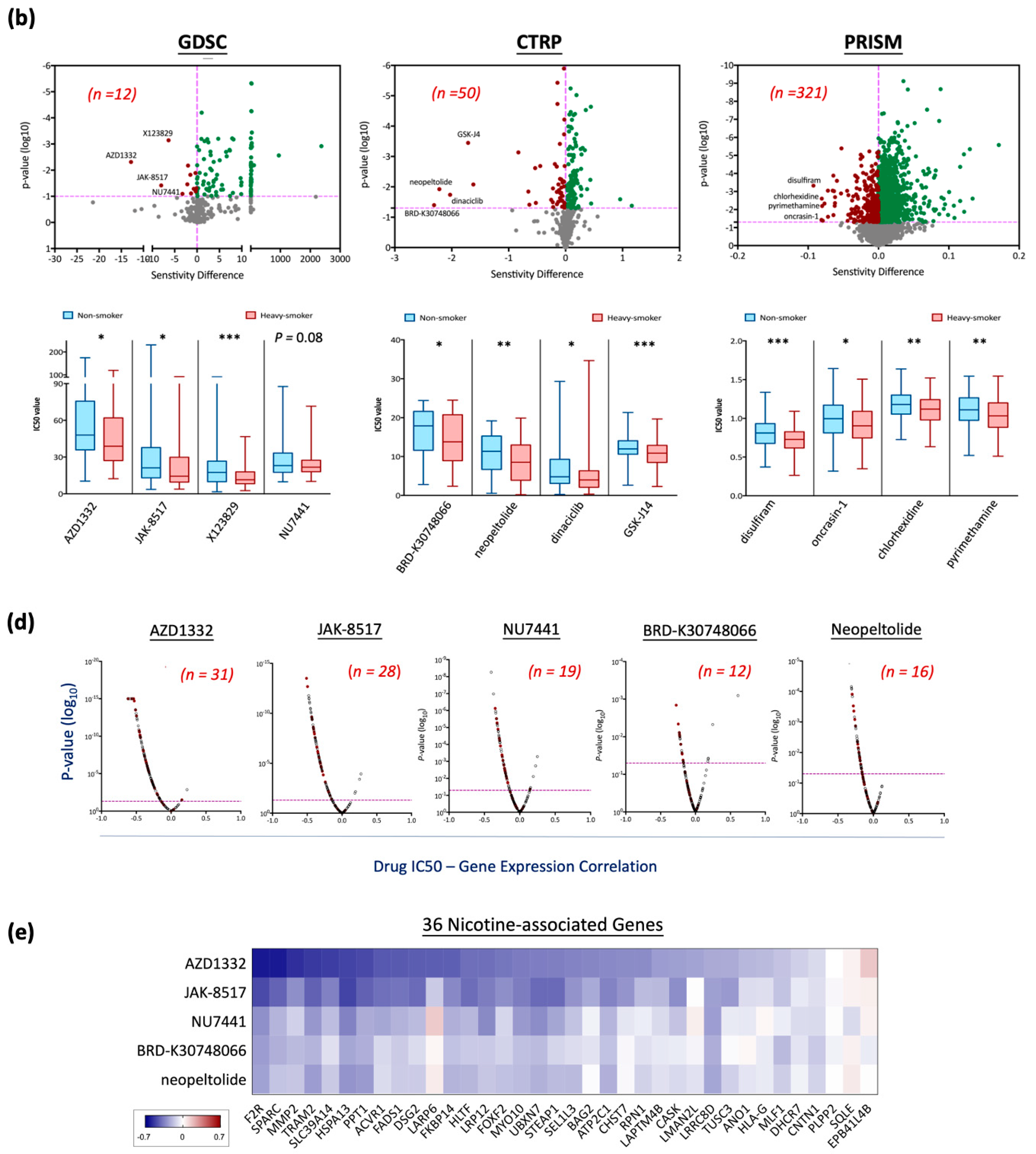
3.5. Drug Repurposing for Targeting Nicotine-Driven HNC: Identification of Potential Therapeutics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HNC | Head and neck cancer |
| DEGs | Differentially expressed genes |
| TCGA-HNSC | The Cancer Genome Atlas Head–Neck Squamous Cell Carcinoma |
| GDSC | Genomics of Drug Sensitivity in Cancer |
| CTRP | Cancer Therapeutics Response Portal |
| PRISM | Profiling Relative Inhibition Simultaneously in Mixtures |
| HPV | Human papillomavirus |
| nAChRs | Nicotinic acetylcholine receptors |
| RMA | Robust multiarray averaging |
| DAVID | Database for Annotation, Visualization, and Integrated Discovery |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GEOMEAN | Geometric mean |
| MMP | Matrix metalloproteinase |
| CCLE | Cancer Cell Line Encyclopedia |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Tsuge, H.; Kawakita, D.; Taniyama, Y.; Oze, I.; Koyanagi, Y.N.; Hori, M.; Nakata, K.; Sugiyama, H.; Miyashiro, I.; Oki, I.; et al. Subsite-specific trends in mid- and long-term survival for head and neck cancer patients in Japan: A population-based study. Cancer Sci. 2024, 115, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Hisamatsu, K.; Suzui, N.; Hara, A.; Tomita, H.; Miyazaki, T. A Review of HPV-Related Head and Neck Cancer. J. Clin. Med. 2018, 7, 241. [Google Scholar] [CrossRef]
- Turati, F.; Garavello, W.; Tramacere, I.; Pelucchi, C.; Galeone, C.; Bagnardi, V.; Corrao, G.; Islami, F.; Fedirko, V.; Boffetta, P.; et al. A meta-analysis of alcohol drinking and oral and pharyngeal cancers: Results from subgroup analyses. Alcohol. Alcohol. 2013, 48, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.; Mahmood, Q.; Mariottini, G.L.; Chiang, T.A.; Lee, K.W. Adverse Health Effects of Betel Quid and the Risk of Oral and Pharyngeal Cancers. BioMed Res. Int. 2017, 2017, 3904098. [Google Scholar] [CrossRef]
- Li, Y.C.; Cheng, A.J.; Lee, L.Y.; Huang, Y.C.; Chang, J.T. Multifaceted Mechanisms of Areca Nuts in Oral Carcinogenesis: The Molecular Pathology from Precancerous Condition to Malignant Transformation. J. Cancer 2019, 10, 4054–4062. [Google Scholar] [CrossRef]
- Khowal, S.; Wajid, S. Role of Smoking-Mediated molecular events in the genesis of oral cancers. Toxicol. Mech. Methods 2019, 29, 665–685. [Google Scholar] [CrossRef]
- Hecht, S.S.; Hatsukami, D.K. Smokeless tobacco and cigarette smoking: Chemical mechanisms and cancer prevention. Nat. Rev. Cancer 2022, 22, 143–155. [Google Scholar] [CrossRef]
- Inoue-Choi, M.; Shiels, M.S.; McNeel, T.S.; Graubard, B.I.; Hatsukami, D.; Freedman, N.D. Contemporary Associations of Exclusive Cigarette, Cigar, Pipe, and Smokeless Tobacco Use with Overall and Cause-Specific Mortality in the United States. JNCI Cancer Spectr. 2019, 3, pkz036. [Google Scholar] [CrossRef]
- Bhanvadia, R.; Bochner, E.; Popokh, B.; Taylor, J.; Franco, A.; Wu, Z.; Antonelli, A.; Ditonno, F.; Abdollah, F.; Simone, G.; et al. Cumulative smoking exposure impacts oncologic outcomes of upper tract urothelial carcinoma. Urol. Oncol. 2025, in press. [CrossRef]
- Awaisu, A.; Samsudin, S.; Amir, N.A.; Omar, C.G.; Hashim, M.I.; Mohamad, M.H.; Shafie, A.A.; Hassali, M.A. Measurement of nicotine withdrawal symptoms: Linguistic validation of the Wisconsin Smoking Withdrawal Scale (WSWS) in Malay. BMC Med. Res. Methodol. 2010, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Hosur, V.; Leppanen, S.; Abutaha, A.; Loring, R.H. Gene regulation of alpha4beta2 nicotinic receptors: Microarray analysis of nicotine-induced receptor up-regulation and anti-inflammatory effects. J. Neurochem. 2009, 111, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.R.; Chong, I.W.; Chen, C.C.; Lin, S.R.; Sheu, C.C.; Hwang, J.J. Mitogen-activated protein kinase pathway was significantly activated in human bronchial epithelial cells by nicotine. DNA Cell Biol. 2006, 25, 312–322. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, Y.; Ren, X. Nicotine promotes the development of non-small cell lung cancer through activating LINC00460 and PI3K/Akt signaling. Biosci. Rep. 2019, 39, BSR20182443. [Google Scholar] [CrossRef]
- Grando, S.A. Connections of nicotine to cancer. Nat. Rev. Cancer 2014, 14, 419–429. [Google Scholar] [CrossRef]
- Pfeifer, G.P.; Denissenko, M.F.; Olivier, M.; Tretyakova, N.; Hecht, S.S.; Hainaut, P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 2002, 21, 7435–7451. [Google Scholar] [CrossRef]
- Yu, M.A.; Kiang, A.; Wang-Rodriguez, J.; Rahimy, E.; Haas, M.; Yu, V.; Ellies, L.G.; Chen, J.; Fan, J.B.; Brumund, K.T.; et al. Nicotine promotes acquisition of stem cell and epithelial-to-mesenchymal properties in head and neck squamous cell carcinoma. PLoS ONE 2012, 7, e51967. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Shah, K.; Mirza, S.; Daga, A.; Rawal, R. Epigenetic regulators governing cancer stem cells and epithelial-mesenchymal transition in oral squamous cell carcinoma. Curr. Stem Cell Res. Ther. 2015, 10, 140–152. [Google Scholar] [CrossRef]
- Mayne, S.T.; Lippman, S.M. Cigarettes: A smoking gun in cancer chemoprevention. J. Natl. Cancer Inst. 2005, 97, 1319–1321. [Google Scholar] [CrossRef]
- Hamilton, M.; Wolf, J.L.; Rusk, J.; Beard, S.E.; Clark, G.M.; Witt, K.; Cagnoni, P.J. Effects of smoking on the pharmacokinetics of erlotinib. Clin. Cancer Res. 2006, 12, 2166–2171. [Google Scholar] [CrossRef]
- Fox, J.L.; Rosenzweig, K.E.; Ostroff, J.S. The effect of smoking status on survival following radiation therapy for non-small cell lung cancer. Lung Cancer 2004, 44, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Pantarotto, J.; Malone, S.; Dahrouge, S.; Gallant, V.; Eapen, L. Smoking is associated with worse outcomes in patients with prostate cancer treated by radical radiotherapy. BJU Int. 2007, 99, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Y.; Chen, J.; Yang, X.M.; Jiang, X.T.; Liu, P.; Li, M. Comparison of biological and transcriptomic effects of conventional cigarette and electronic cigarette smoke exposure at toxicological dose in BEAS-2B cells. Ecotoxicol. Environ. Saf. 2021, 222, 112472. [Google Scholar] [CrossRef]
- Boyle, J.O.; Gumus, Z.H.; Kacker, A.; Choksi, V.L.; Bocker, J.M.; Zhou, X.K.; Yantiss, R.K.; Hughes, D.B.; Du, B.; Judson, B.L.; et al. Effects of cigarette smoke on the human oral mucosal transcriptome. Cancer Prev. Res. 2010, 3, 266–278. [Google Scholar] [CrossRef]
- Huang, H.H.; You, G.R.; Tang, S.J.; Chang, J.T.; Cheng, A.J. Molecular Signature of Long Non-Coding RNA Associated with Areca Nut-Induced Head and Neck Cancer. Cells 2023, 12, 873. [Google Scholar] [CrossRef] [PubMed]
- You, G.R.; Chang, J.T.; Li, Y.L.; Huang, C.W.; Tsai, Y.L.; Fan, K.H.; Kang, C.J.; Huang, S.F.; Chang, P.H.; Cheng, A.J. MYH9 Facilitates Cell Invasion and Radioresistance in Head and Neck Cancer via Modulation of Cellular ROS Levels by Activating the MAPK-Nrf2-GCLC Pathway. Cells 2022, 11, 2855. [Google Scholar] [CrossRef]
- You, G.R.; Chang, J.T.; Li, Y.L.; Chen, Y.J.; Huang, Y.C.; Fan, K.H.; Chen, Y.C.; Kang, C.J.; Cheng, A.J. Molecular Interplays Between Cell Invasion and Radioresistance That Lead to Poor Prognosis in Head-Neck Cancer. Front. Oncol. 2021, 11, 681717. [Google Scholar] [CrossRef]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Goncalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef]
- Rees, M.G.; Seashore-Ludlow, B.; Cheah, J.H.; Adams, D.J.; Price, E.V.; Gill, S.; Javaid, S.; Coletti, M.E.; Jones, V.L.; Bodycombe, N.E.; et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 2016, 12, 109–116. [Google Scholar] [CrossRef]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef]
- Maeser, D.; Gruener, R.F.; Huang, R.S. oncoPredict: An R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief. Bioinform. 2021, 22, bbab260. [Google Scholar] [CrossRef]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.; Ramisetty, S.; Nair, M.; Kulkarni, P.; Horne, D.; Salgia, R.; Singhal, S.S. Strategic advancements in targeting the PI3K/AKT/mTOR pathway for Breast cancer therapy. Biochem. Pharmacol. 2025, 236, 116850. [Google Scholar] [CrossRef]
- Cheng, Y.; Chen, J.; Shi, Y.; Fang, X.; Tang, Z. MAPK Signaling Pathway in Oral Squamous Cell Carcinoma: Biological Function and Targeted Therapy. Cancers 2022, 14, 4625. [Google Scholar] [CrossRef]
- Dajee, M.; Lazarov, M.; Zhang, J.Y.; Cai, T.; Green, C.L.; Russell, A.J.; Marinkovich, M.P.; Tao, S.; Lin, Q.; Kubo, Y.; et al. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature 2003, 421, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. NF-kappaB: Tumor promoter or suppressor? Trends Cell Biol. 2004, 14, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Netskar, H.; Pfefferle, A.; Goodridge, J.P.; Sohlberg, E.; Dufva, O.; Teichmann, S.A.; Brownlie, D.; Michaelsson, J.; Marquardt, N.; Clancy, T.; et al. Pan-cancer profiling of tumor-infiltrating natural killer cells through transcriptional reference mapping. Nat. Immunol. 2024, 25, 1445–1459. [Google Scholar] [CrossRef]
- Pavon, M.A.; Arroyo-Solera, I.; Cespedes, M.V.; Casanova, I.; Leon, X.; Mangues, R. uPA/uPAR and SERPINE1 in head and neck cancer: Role in tumor resistance, metastasis, prognosis and therapy. Oncotarget 2016, 7, 57351–57366. [Google Scholar] [CrossRef]
- Li, S.; Pritchard, D.M.; Yu, L.G. Regulation and Function of Matrix Metalloproteinase-13 in Cancer Progression and Metastasis. Cancers 2022, 14, 3263. [Google Scholar] [CrossRef]
- Wu, Z.; Chen, J.; Yang, L.; Sun, K.; Jiang, Q.; Dong, F.; Lu, W.; Chen, R.; Chen, Y. Elevated INHBA Promotes Tumor Progression of Cervical Cancer. Technol. Cancer Res. Treat. 2024, 23, 15330338241234798. [Google Scholar] [CrossRef]
- Rossi, M.K.; Gnanamony, M.; Gondi, C.S. The ‘SPARC’ of life: Analysis of the role of osteonectin/SPARC in pancreatic cancer (Review). Int. J. Oncol. 2016, 48, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Zhang, L.; Li, N. ANO1: More Than Just Calcium-Activated Chloride Channel in Cancer. Front. Oncol. 2022, 12, 922838. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Chen, G.; Sun, A.; Wang, Z.; Huang, H.; Gao, Z.; Liang, W.; Liu, C.; Li, K. BAG2 Promotes Proliferation and Metastasis of Gastric Cancer via ERK1/2 Signaling and Partially Regulated by miR186. Front. Oncol. 2020, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, Y.; Huang, M.; Wang, Z.; Liu, D.; Liu, J.; Fu, X.; Yang, S.; Shan, S.; Yang, L.; et al. DHCR7 promotes tumorigenesis via activating PI3K/AKT/mTOR signalling pathway in bladder cancer. Cell Signal 2023, 102, 110553. [Google Scholar] [CrossRef]
- Deng, W.; Peng, W.; Wang, T.; Chen, J.; Zhu, S. Overexpression of MMPs Functions as a Prognostic Biomarker for Oral Cancer Patients: A Systematic Review and Meta-analysis. Oral Health Prev. Dent. 2019, 17, 505–514. [Google Scholar] [CrossRef]
- Gao, X.; Jiang, W.; Ke, Z.; Huang, Q.; Chen, L.; Zhang, G.; Li, C.; Yu, X. TRAM2 promotes the malignant progression of glioma through PI3K/AKT/mTOR pathway. Biochem. Biophys. Res. Commun. 2022, 586, 34–41. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Non-Smoker Average (TPM) | Heavy Smoker Average (TPM) | Heavy/Non-Smoker (Ratio) | Wilcoxon Test (p) |
|---|---|---|---|---|
| ACVR1 | 19.8436 | 24.6753 | 1.2435 | 0.0042 |
| ANO1 | 76.7027 | 144.7553 | 1.8872 | 0.0107 |
| ATP2C1 | 66.6058 | 77.0756 | 1.1572 | 0.0237 |
| BAG2 | 2.0117 | 3.1045 | 1.5432 | 0.0003 |
| CASK | 20.5336 | 28.0910 | 1.3681 | 0.0067 |
| CHST7 | 3.2940 | 6.0847 | 1.8472 | 0.0001 |
| CNTN1 | 7.9286 | 13.5521 | 1.7093 | 0.0008 |
| DHCR7 | 45.8835 | 90.5598 | 1.9737 | <0.0001 |
| DSG2 | 35.3876 | 49.0542 | 1.3862 | 0.0247 |
| EPB41L4B | 6.8675 | 7.7425 | 1.1274 | 0.0474 |
| F2R | 16.2279 | 19.5841 | 1.2068 | 0.0138 |
| FADS1 | 11.6135 | 16.6838 | 1.4366 | 0.0003 |
| FKBP14 | 7.2604 | 8.5677 | 1.1801 | 0.0222 |
| FOXF2 | 4.3628 | 5.5117 | 1.2633 | 0.0096 |
| HLA-G | 2.4634 | 10.3195 | 4.1891 | 0.0035 |
| HLTF | 11.4617 | 12.0126 | 1.0481 | 0.0039 |
| HSPA13 | 8.3831 | 10.0846 | 1.2030 | 0.0091 |
| LAPTM4B | 79.5637 | 106.3585 | 1.3368 | <0.0001 |
| LARP6 | 11.9819 | 14.3860 | 1.2006 | 0.0276 |
| LMAN2L | 13.0224 | 15.2664 | 1.1723 | 0.0011 |
| LRP12 | 9.2280 | 14.1074 | 1.5288 | 0.0006 |
| LRRC8D | 21.0277 | 26.0205 | 1.2374 | 0.0194 |
| MLF1 | 20.5659 | 32.9833 | 1.6038 | <0.0001 |
| MMP2 | 156.0294 | 214.7444 | 1.3763 | 0.0041 |
| MYO10 | 23.2765 | 25.2124 | 1.0832 | 0.0480 |
| PLPP2 | 59.7515 | 77.8394 | 1.3027 | 0.0003 |
| PPT1 | 63.3588 | 87.7912 | 1.3856 | 0.0164 |
| RPN1 | 202.3942 | 226.2346 | 1.1178 | 0.0052 |
| SEL1L3 | 20.1116 | 23.9188 | 1.1893 | 0.0196 |
| SLC39A14 | 22.6472 | 27.4927 | 1.2140 | 0.0210 |
| SPARC | 1358.7691 | 1798.5699 | 1.3237 | 0.0111 |
| SQLE | 68.2900 | 76.8158 | 1.1248 | 0.0299 |
| STEAP1 | 19.8731 | 22.1430 | 1.1142 | 0.0003 |
| TRAM2 | 9.3543 | 12.5970 | 1.3466 | 0.0002 |
| TUSC3 | 32.7518 | 41.8073 | 1.2765 | 0.0089 |
| UBXN7 | 8.9994 | 11.0759 | 1.2307 | 0.0092 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, G.-R.; Chang, D.Y.; Huang, H.-H.; Chen, Y.-J.; Chang, J.T.; Cheng, A.-J. Deciphering Nicotine-Driven Oncogenesis in Head and Neck Cancer: Integrative Transcriptomics and Drug Repurposing Insights. Cancers 2025, 17, 1430. https://doi.org/10.3390/cancers17091430
You G-R, Chang DY, Huang H-H, Chen Y-J, Chang JT, Cheng A-J. Deciphering Nicotine-Driven Oncogenesis in Head and Neck Cancer: Integrative Transcriptomics and Drug Repurposing Insights. Cancers. 2025; 17(9):1430. https://doi.org/10.3390/cancers17091430
Chicago/Turabian StyleYou, Guo-Rung, Daniel Yu Chang, Hung-Han Huang, Yin-Ju Chen, Joseph T. Chang, and Ann-Joy Cheng. 2025. "Deciphering Nicotine-Driven Oncogenesis in Head and Neck Cancer: Integrative Transcriptomics and Drug Repurposing Insights" Cancers 17, no. 9: 1430. https://doi.org/10.3390/cancers17091430
APA StyleYou, G.-R., Chang, D. Y., Huang, H.-H., Chen, Y.-J., Chang, J. T., & Cheng, A.-J. (2025). Deciphering Nicotine-Driven Oncogenesis in Head and Neck Cancer: Integrative Transcriptomics and Drug Repurposing Insights. Cancers, 17(9), 1430. https://doi.org/10.3390/cancers17091430