Propranolol and Capecitabine Synergy on Inducing Ferroptosis in Human Colorectal Cancer Cells: Potential Implications in Cancer Therapy

 , , , and
, , , and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability Analysis
2.3. Drug Combination Studies
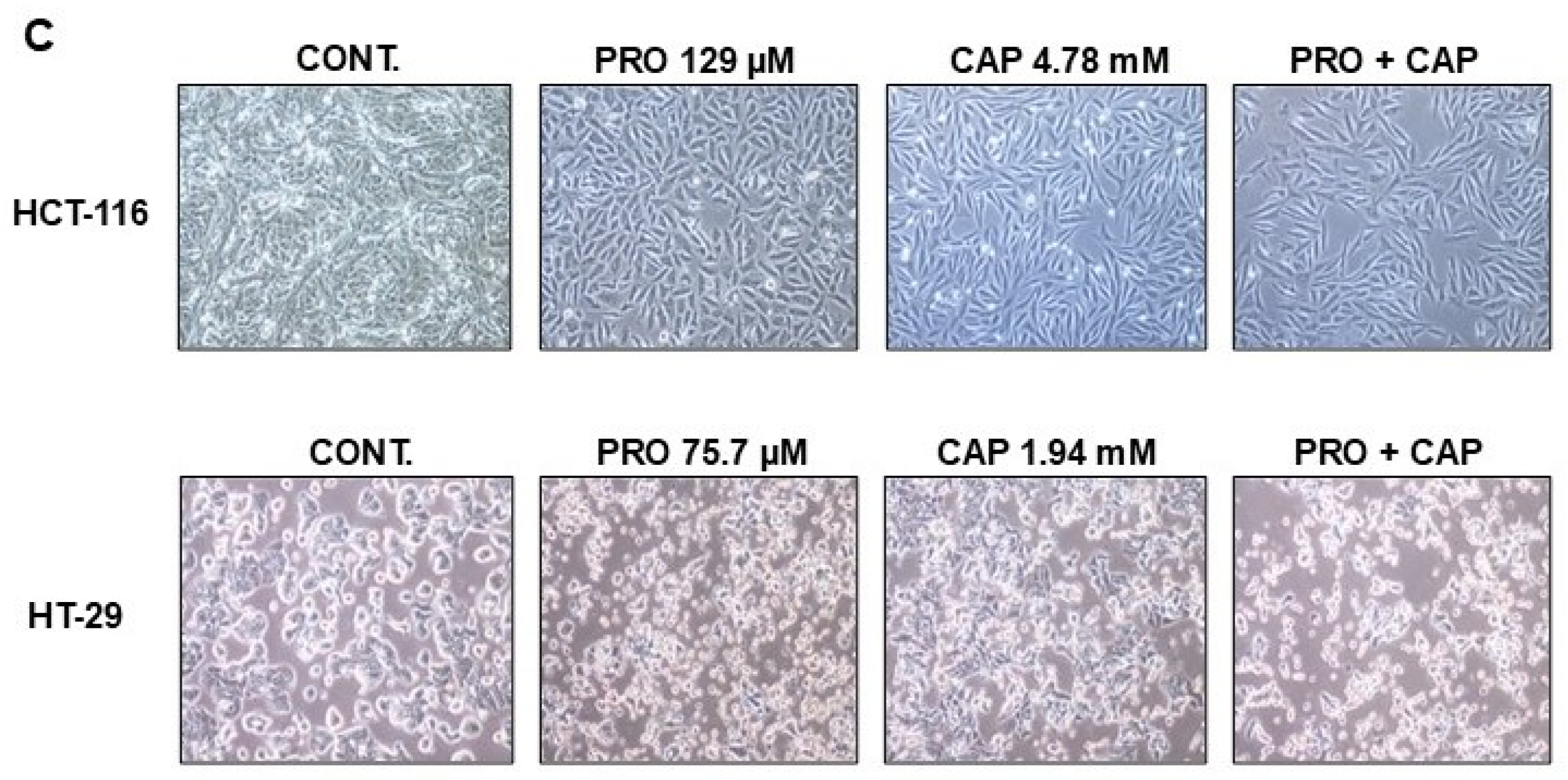
2.4. Morphological Images
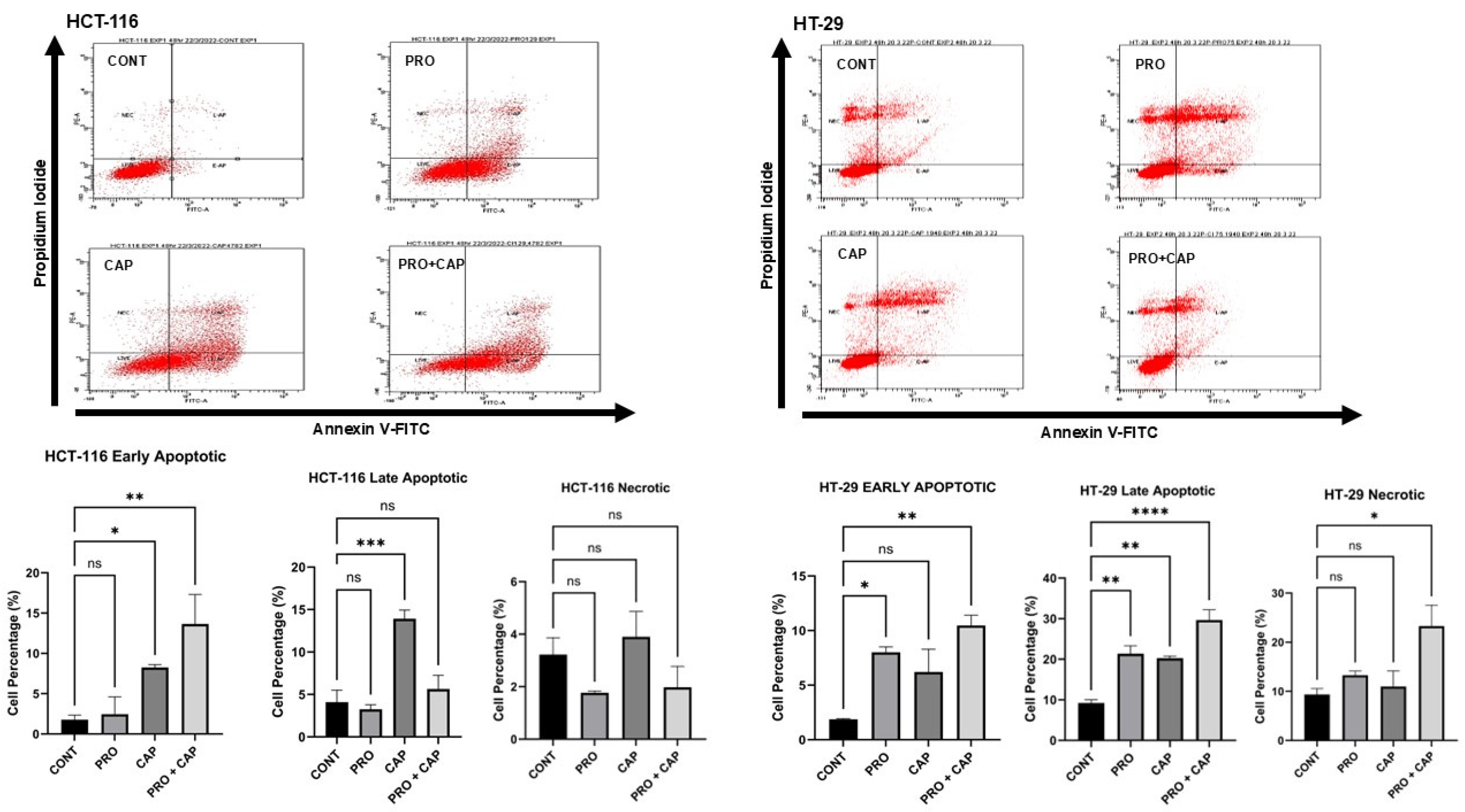
2.5. The Detection of Cell Death
2.6. Transcriptomic Study
2.6.1. RNA Extraction
2.6.2. RNA Library Construction
2.6.3. Identification of Differentially Expressed Genes (DEGs)
2.6.4. Enrichment Analysis
2.7. Global Untargeted Metabolomics Profiling Study Using LC-MS/MS
2.7.1. Metabolites Extraction
2.7.2. HPLC Workflow
2.7.3. HPLC-Mass Spectrometry
2.7.4. Data Processing and Analysis
2.8. Biochemical Validation Assays
2.8.1. Measurement of Intracellular Reactive Oxygen Species
2.8.2. Measurement of Mitochondrial Reactive Oxygen Species (Mito ROS)
2.8.3. Analysis of Mitochondrial Membrane Potential
2.8.4. TBARS Assay
2.8.5. Wound Healing Assay
2.9. Statistical Analysis
3. Results
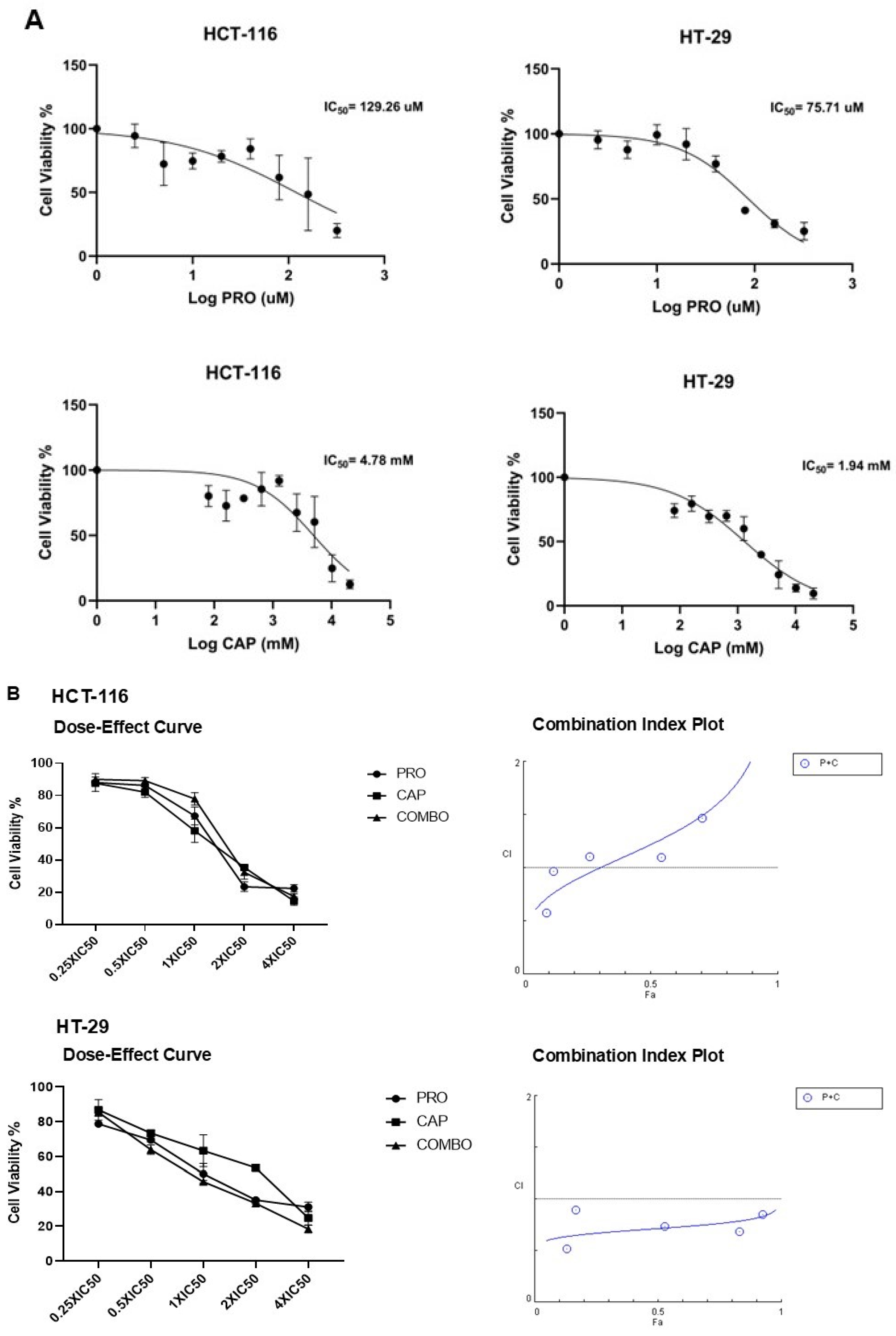
3.1. PRO and CAP Monotherapies Induced Cell Cytotoxicity in Cell Type-Specific and Dose-Dependent Manners
3.2. PRO Potentiates the Antiproliferative Effects of CAP in Cell Type-Specific Dependent Manner
3.3. Effects of PRO and/or CAP Treatments on Induction of the Cellular Death Mechanisms
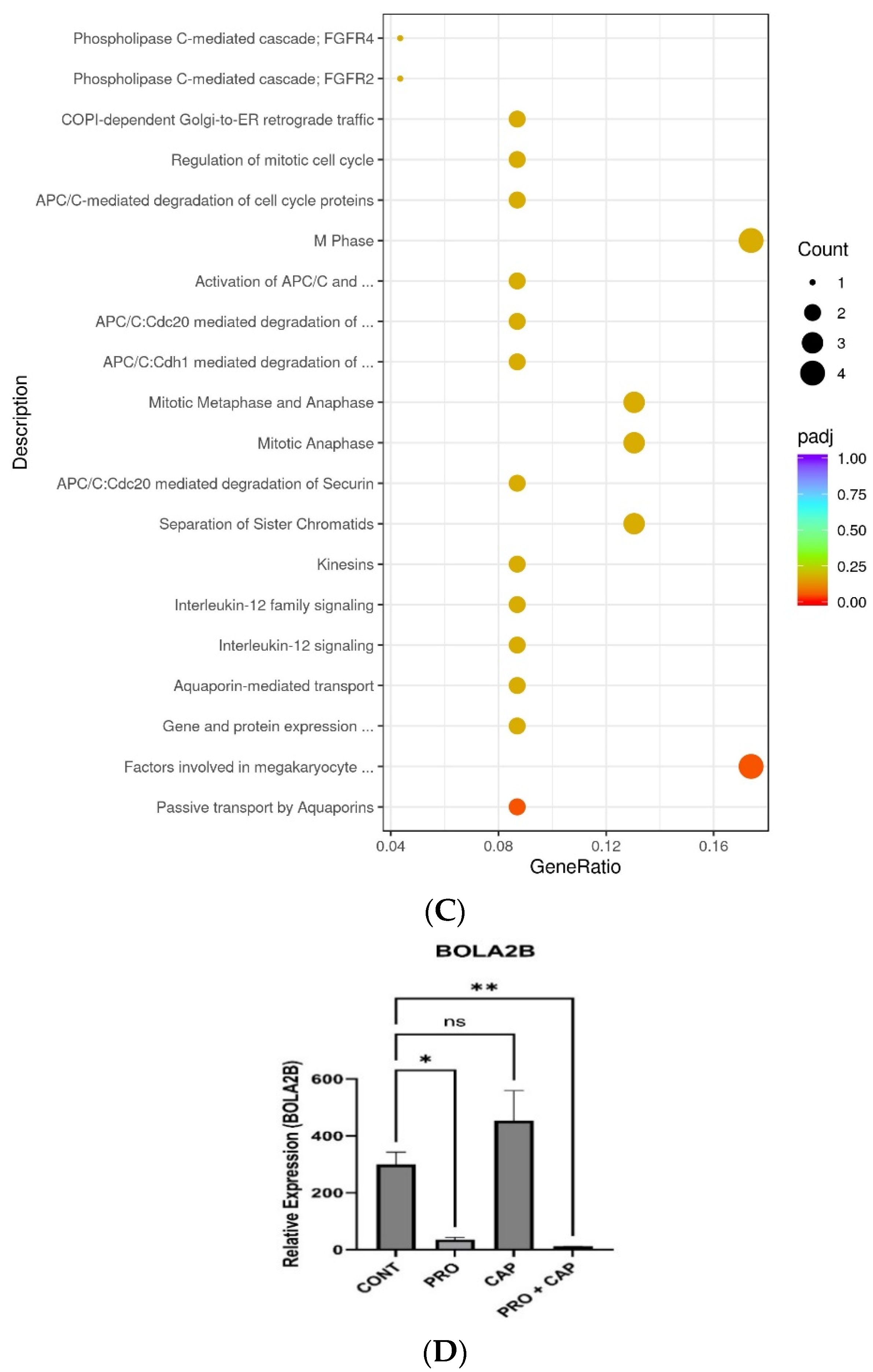
3.4. Effects of PRO and/or CAP Treatments on Transcriptomic Profile of HT-29 Cell Line
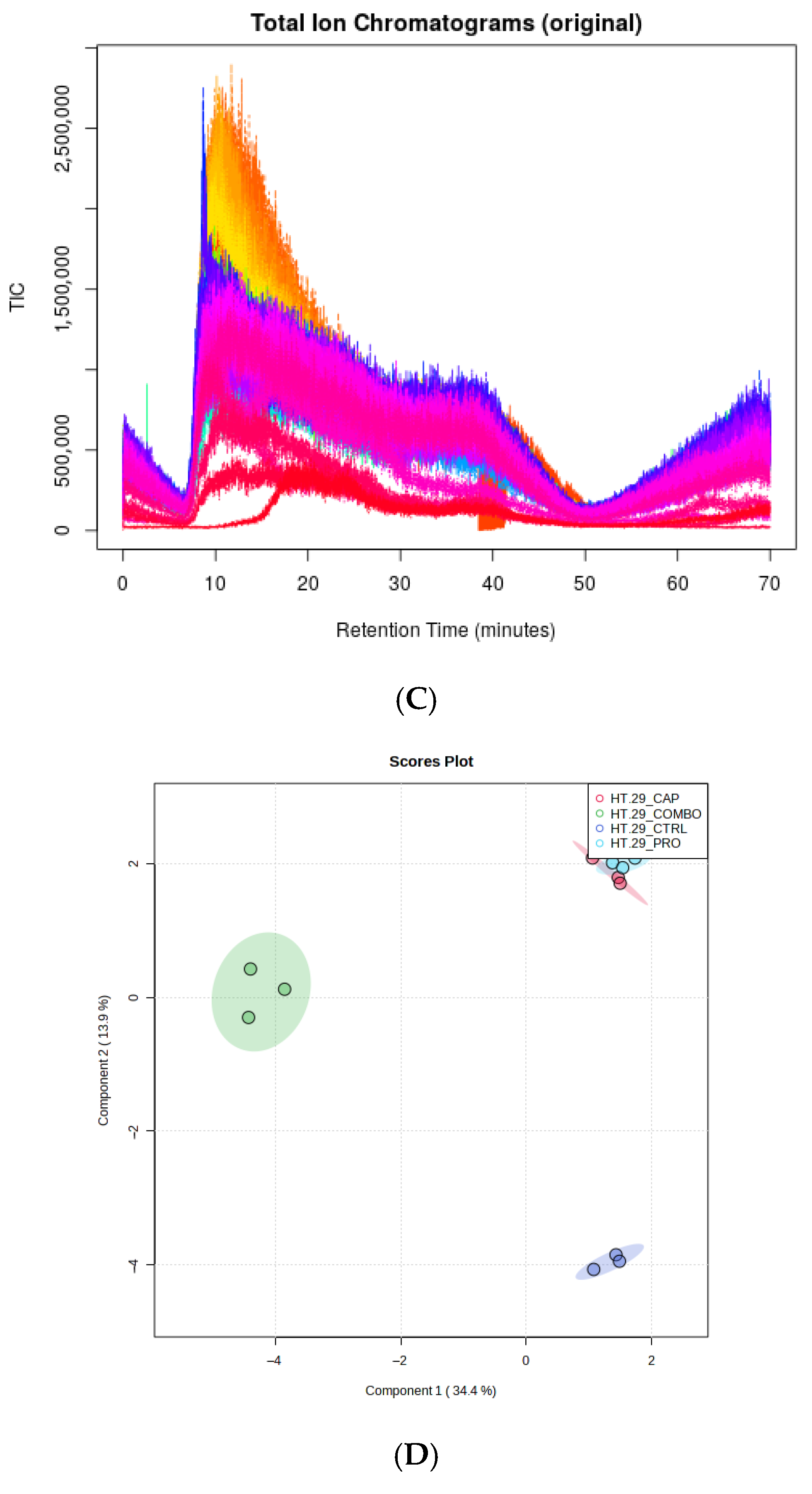
3.4.1. Overall View of the Transcriptome Profile
3.4.2. Identification of DEGs
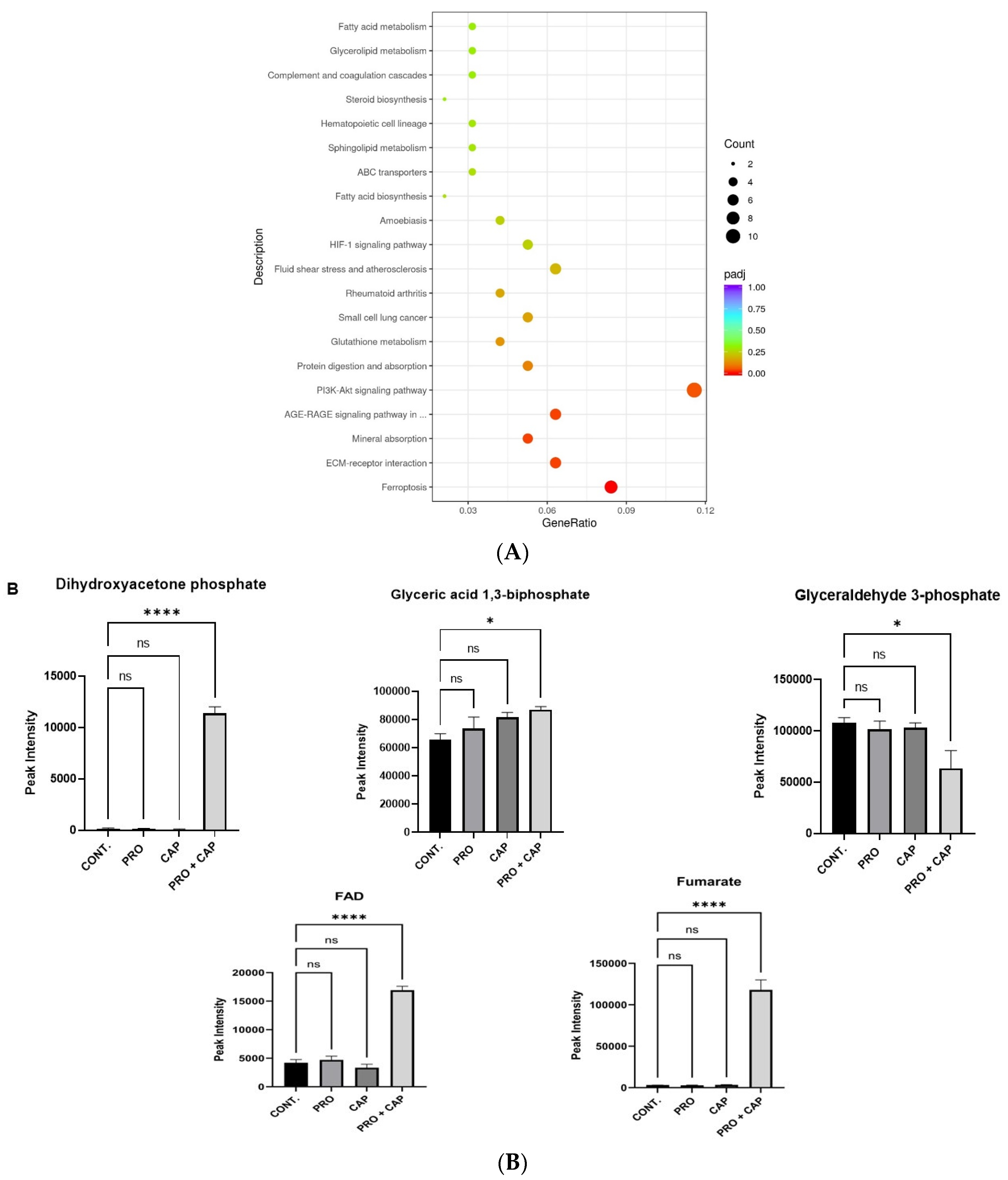
3.5. Effects of PRO and/or CAP Treatments on Cellular Metabolome Profile
Overall View of Metabolomic Profiles
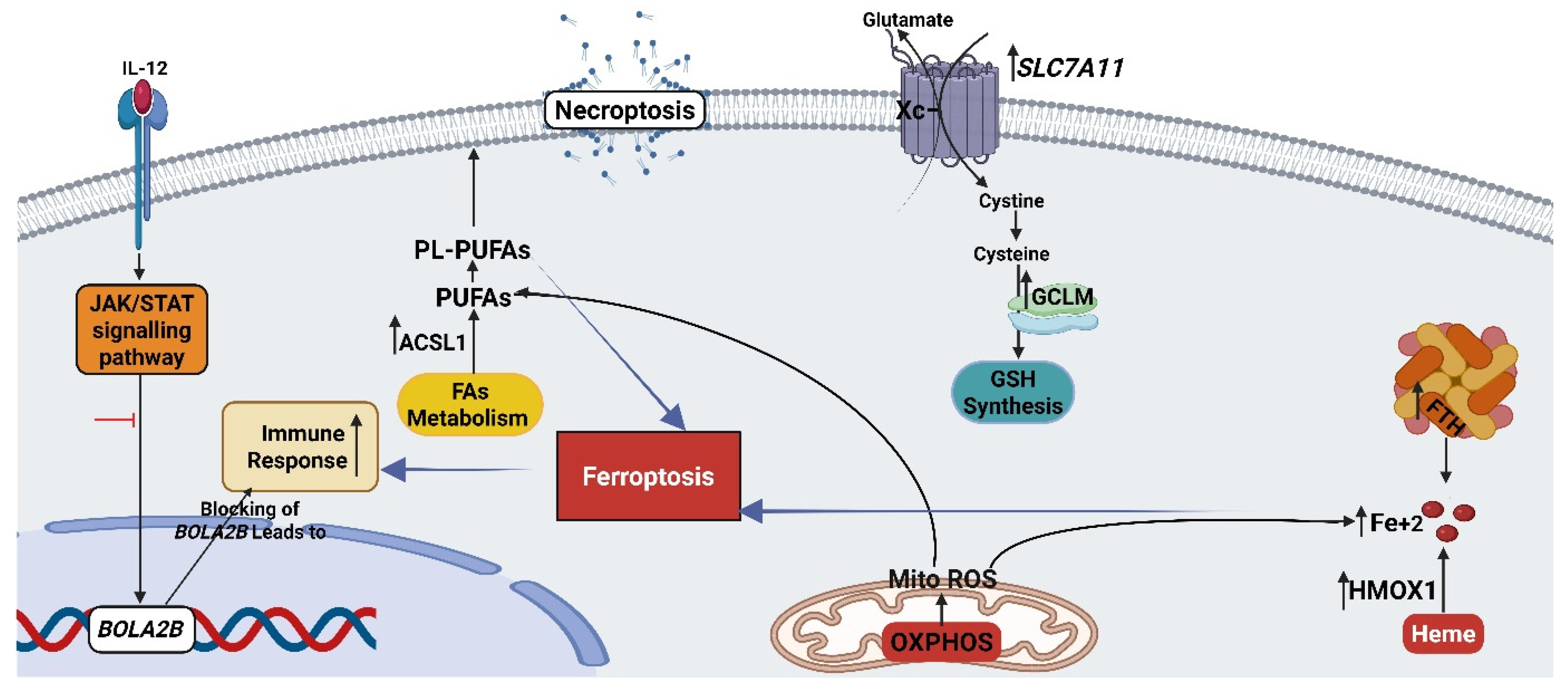
3.6. The Combination Treatment Induces the Ferroptosis in HT-29 Cells
3.6.1. Ferroptosis Pathway
3.6.2. Mitochondrial Oxidative Metabolism (OXPHOS)
3.6.3. Oxidative Stress-Related Genes
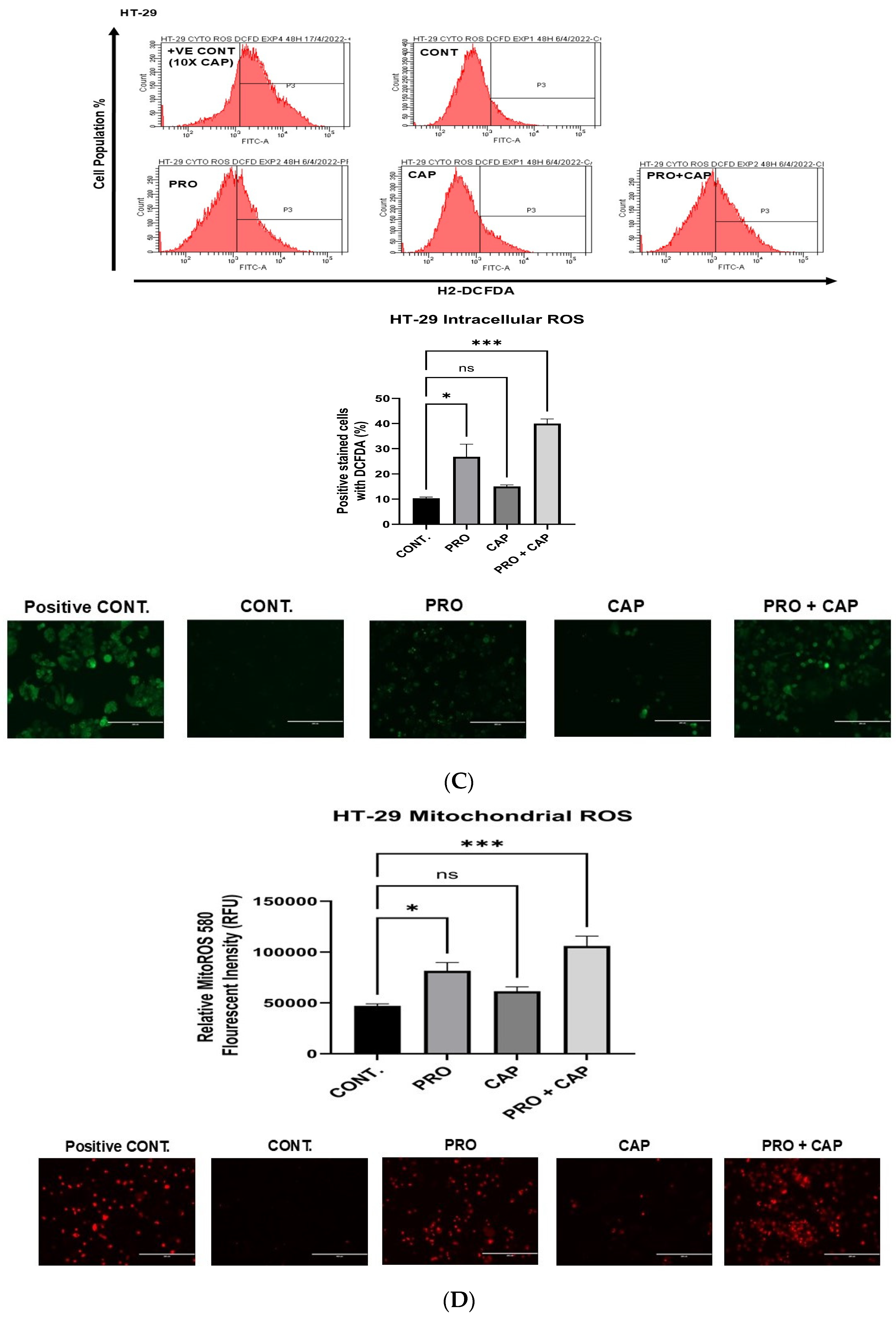
3.6.4. Generation of Intracellular Reactive Oxygen Species (ROS)
3.6.5. Generation of Mitochondrial Reactive Oxygen Species (Mito ROS)
3.6.6. The Analysis of Mitochondrial Membrane Potential
3.6.7. Lipid Peroxidation Level
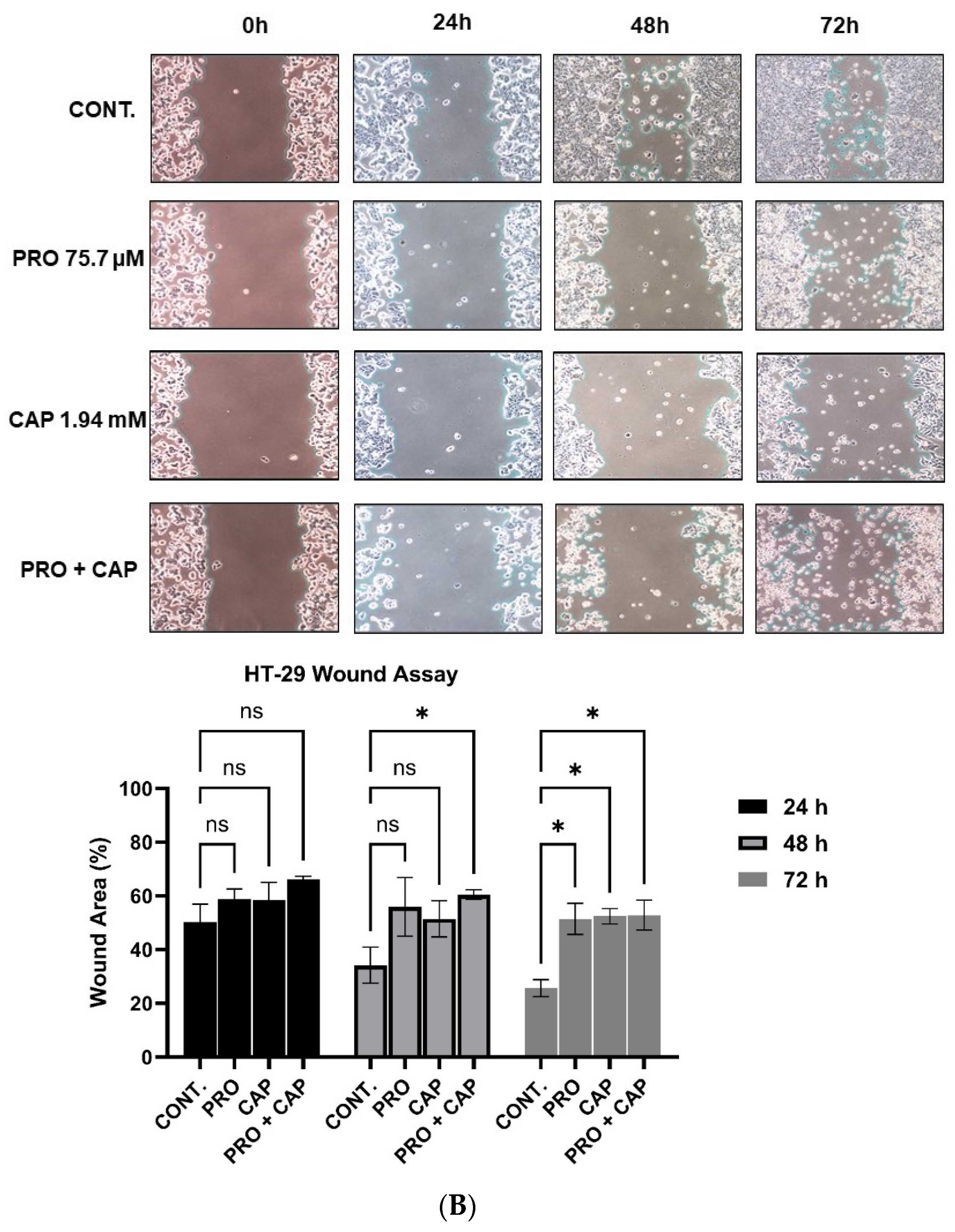
3.7. The Combination Treatment Inhibits the Cell Migration of HT-29 Cells
3.8. The Combination Treatment Triggers the Immune Response in HT-29 Cells via Blocking the JAK-STAT Signaling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, S.M.; Al Doghaither, H.A.; Al-Ghafari, A.B. General Insight into Cancer: An Overview of Colorectal Cancer (Review). Mol. Clin. Oncol. 2021, 15, 271. [Google Scholar] [CrossRef]
- Nenkov, M.; Ma, Y.; Gaßler, N.; Chen, Y. Metabolic Reprogramming of Colorectal Cancer Cells and the Microenvironment: Implication for Therapy. Int. J. Mol. Sci. 2021, 22, 6262. [Google Scholar] [CrossRef]
- Hutton, J.E.; Wang, X.; Zimmerman, L.J.; Slebos, R.J.; Trenary, I.A.; Young, J.D.; Li, M.; Liebler, D.C. Oncogenic KRAS and BRAF Drive Metabolic Reprogramming in Colorectal Cancer. Mol. Cell. Proteom. 2016, 15, 2924–2938. [Google Scholar] [CrossRef]
- Brown, R.E.; Short, S.P.; Williams, C.S. Colorectal Cancer and Metabolism. Curr. Color. Cancer Rep. 2018, 14, 226–241. [Google Scholar] [CrossRef]
- Zhang, Z.; Bao, C.; Jiang, L.; Wang, S.; Wang, K.; Lu, C.; Fang, H. When cancer drug resistance meets metabolomics (bulk, single-cell and/or spatial): Progress, potential, and perspective. Front. Oncol. 2023, 12, 1054233. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, G.V.; Zacharias, G.; Tsalafoutas, J.; Theodoridis, D.; Kouloulias, V. Capecitabine for Locally Advanced and Metastatic Colorectal Cancer: A Review. World J. Gastrointest. Oncol. 2010, 2, 311–321. [Google Scholar] [CrossRef]
- Aguado, C.; García-Paredes, B.; Sotelo, M.J.; Sastre, J.; Díaz-Rubio, E. Should Capecitabine Replace 5-Fluorouracil in the First-Line Treatment of Metastatic Colorectal Cancer? World J. Gastroenterol. 2014, 20, 6092–6101. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, S.M.; Al Doghaither, H.A.; Al-Ghafari, A.B.; Pushparaj, P.N. 5-Fluorouracil and Capecitabine Therapies for the Treatment of Colorectal Cancer (Review). Oncol. Rep. 2023, 50, 175. [Google Scholar] [CrossRef]
- Oneda, E.; Zaniboni, A. Adjuvant treatment of colon cancer with microsatellite instability—The state of the art. Crit. Rev. Oncol. Hematol. 2022, 169, 103537. [Google Scholar] [CrossRef]
- Anselmino, L.E.; Baglioni, M.V.; Malizia, F.; Laluce, N.C.; Etichetti, C.B.; Marignac, V.L.M.; Rozados, V.; Scharovsky, O.G.; Girardini, J.; Rico, M.J.; et al. Repositioning Metformin and Propranolol for Colorectal and Triple-Negative Breast Cancers Treatment. Sci. Rep. 2021, 11, 8091. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Wang, T.; Li, L.; Zhu, Z.; Zhang, W.; Cui, G.; Li, W. Co-delivery of Cisplatin (IV) and Capecitabine as an Effective and Non-toxic Cancer Treatment. Front. Pharmacol. 2019, 10, 110. [Google Scholar] [CrossRef] [PubMed]
- Verbaanderd, C.; Meheus, L.; Huys, I.; Pantziarka, P. Repurposing Drugs in Oncology: Next Steps. Trends Cancer 2017, 3, 543–546. [Google Scholar] [CrossRef]
- Nowak-Sliwinska, P.; Scapozza, L.; Ruiz i Altaba, A. Drug Repurposing in Oncology: Compounds, Pathways, Phenotypes and Computational Approaches for Colorectal Cancer. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 434–454. [Google Scholar] [CrossRef]
- Wolter, J.K.; Wolter, N.E.; Blanch, A.; Partridge, T.; Cheng, L.; Morgenstern, D.A.; Podkowa, M.; Kaplan, D.R.; Irwin, M.S. Anti-Tumor Activity of the Beta-Adrenergic Receptor Antagonist Propranolol in Neuroblastoma. Oncotarget 2014, 5, 161–172. [Google Scholar] [CrossRef]
- Chang, P.Y.; Huang, W.Y.; Lin, C.L.; Huang, T.C.; Wu, Y.Y.; Chen, J.H.; Kao, C.H. Propranolol Reduces Cancer Risk: A Population-Based Cohort Study. Medicine 2015, 94, e1097. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.; Moz, M.; Correia, G.; Teixeira, A.; Medeiros, R.; Ribeiro, L. Antiproliferative Effects of β-Blockers on Human Colorectal Cancer Cells. Oncol. Rep. 2015, 33, 2513–2520. [Google Scholar] [CrossRef]
- Wang, F.; Liu, H.; Wang, F.; Xu, R.; Wang, P.; Tang, F.; Zhang, X.; Zhu, Z.; Lv, H.; Han, T. Propranolol Suppresses the Proliferation and Induces the Apoptosis of Liver Cancer Cells. Mol. Med. Rep. 2018, 17, 5213–5221. [Google Scholar] [CrossRef]
- Liao, P.; Song, K.; Zhu, Z.; Liu, Z.; Zhang, W.; Li, W.; Hu, J.; Hu, Q.; Chen, C.; Chen, B.; et al. Propranolol Suppresses the Growth of Colorectal Cancer through Simultaneously Activating Autologous CD8+ T Cells and Inhibiting Tumor AKT/MAPK Pathway. Clin. Pharmacol. Ther. 2020, 108, 606–615. [Google Scholar] [CrossRef]
- Yarahmadi, A.; Zal, F.; Bolouki, A. Protective Effects of Quercetin on Nicotine-Induced Oxidative Stress in HepG2 Cells. Toxicol. Mech. Methods 2017, 27, 609–614. [Google Scholar] [CrossRef]
- Cole, S.W.; Sood, A.K. Molecular pathways: Beta-adrenergic signaling in cancer. Clin. Cancer. Res. 2012, 18, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Hoeger, P.H.; Harper, J.I.; Baselga, E.; Bonnet, D.; Boon, L.M.; Ciofi Degli Atti, M.; El Hachem, M.; Oranje, A.P.; Rubin, A.T.; Weibel, L.; et al. Treatment of Infantile Haemangiomas: Recommendations of a European Expert Group. Eur. J. Pediatr. 2015, 174, 855–865. [Google Scholar] [CrossRef]
- Pantziarka, P.; Bouche, G.; Sukhatme, V.; Meheus, L.; Rooman, I.; Sukhatme, V.P. Repurposing Drugs in Oncology (ReDO)—Propranolol as an Anti-Cancer Agent. Ecancermedicalscience 2016, 10, 680. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, A.M.; Gallardo-Vara, E.; Casado-Vela, J.; Recio-Poveda, L.; Botella, L.M.; Albiñana, V. The Role of Propranolol as a Repurposed Drug in Rare Vascular Diseases. Int. J. Mol. Sci. 2022, 23, 4217. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, R.; Pereira, M.L.; Oliveira, M. Beta-Blockers and Cancer: Where Are We? Pharmaceuticals 2020, 13, 105. [Google Scholar] [CrossRef]
- Puzderova, B.; Belvoncikova, P.; Grossmannova, K.; Csaderova, L.; Labudova, M.; Fecikova, S.; Pastorek, J.; Barathova, M. Propranolol, Promising Chemosensitizer and Candidate for the Combined Therapy through Disruption of Tumor Microenvironment Homeostasis by Decreasing the Level of Carbonic Anhydrase IX. Int. J. Mol. Sci. 2023, 24, 11094. [Google Scholar] [CrossRef]
- Lopez, J.S.; Banerji, U. Combine and conquer: Challenges for targeted therapy combinations in early phase trials. Nat. Rev. Clin. Oncol. 2017, 14, 57–66. [Google Scholar] [CrossRef]
- Zarewa, S.A.; Binobaid, L.; Sulaiman, A.A.A.; Sobeai, H.M.A.; Alotaibi, M.; Alhoshani, A.; Isab, A.A. Synthesis, Characterization, and Anticancer Activity of Phosphanegold(i) Complexes of 3-Thiosemicarbano-butan-2-one Oxime. Biomedicines 2023, 11, 2512. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative Analysis of Dose-Effect Relationships: The Combined Effects of Multiple Drugs or Enzyme Inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Kumar, R.; Saneja, A.; Panda, A.K. An Annexin V-FITC-Propidium Iodide-Based Method for Detecting Apoptosis in a Non-Small Cell Lung Cancer Cell Line. Methods Mol. Biol. 2021, 2279, 213–223. [Google Scholar] [CrossRef]
- Novogene. Next-Generation Sequencing Services for Genomics Research; Genomics Singapore Pte. Ltd.: Singapore; Available online: https://www.novogene.com (accessed on 1 May 2023).
- Alkhatabi, H.A.; Zohny, S.F.; Shait Mohammed, M.R.; Choudhry, H.; Rehan, M.; Ahmad, A.; Ahmed, F.; Khan, M.I. Venetoclax-Resistant MV4-11 Leukemic Cells Activate PI3K/AKT Pathway for Metabolic Reprogramming and Redox Adaptation for Survival. Antioxidants 2022, 11, 461. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.M.; Shait Mohammed, M.R.; Alghamdi, R.A.; Ahmad, A.; Zamzami, M.A.; Choudhry, H.; Khan, M.I. Urolithin A and B Alter Cellular Metabolism and Induce Metabolites Associated with Apoptosis in Leukemic Cells. Int. J. Mol. Sci. 2021, 22, 5465. [Google Scholar] [CrossRef] [PubMed]
- Shait Mohammed, M.R.; Alghamdi, R.A.; Alzahrani, A.M.; Zamzami, M.A.; Choudhry, H.; Khan, M.I. Compound C, a Broad Kinase Inhibitor Alters Metabolic Fingerprinting of Extracellular Matrix Detached Cancer Cells. Front. Oncol. 2021, 11, 612778. [Google Scholar] [CrossRef]
- Shait Mohammed, M.R.; Alzahrani, F.; Hosawi, S.; Choudhry, H.; Khan, M.I. Profiling the Effect of Targeting Wild Isocitrate Dehydrogenase 1 (IDH1) on the Cellular Metabolome of Leukemic Cells. Int. J. Mol. Sci. 2022, 23, 6653. [Google Scholar] [CrossRef]
- Bode, K.; Link, C.; Krammer, P.H.; Weyd, H. Flow-cytometric Detection of Low-level Reactive Oxygen Species in Cell Lines and Primary Immune Cells. Bio Protoc. 2020, 10, e3737. [Google Scholar] [CrossRef]
- Zhao, S.; Fan, S.; Shi, Y.; Ren, H.; Hong, H.; Gao, X.; Zhang, M.; Qin, Q.; Li, H. Propranolol Induced Apoptosis and Autophagy via the ROS/JNK Signaling Pathway in Human Ovarian Cancer. J. Cancer 2020, 11, 5900–5910. [Google Scholar] [CrossRef]
- Kimura, K.; Chun, J.H.; Lin, Y.L.; Liang, Y.C.; Jackson, T.L.; Huang, R.C.C. Tetra-O-Methyl-Nordihydroguaiaretic Acid Inhibits Energy Metabolism and Synergistically Induces Anticancer Effects with Temozolomide on LN229 Glioblastoma Tumors Implanted in Mice While Preventing Obesity in Normal Mice That Consume High-Fat Diets. PLoS ONE 2023, 18, e0285536. [Google Scholar] [CrossRef]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Analysis of the Mitochondrial Membrane Potential Using the Cationic JC-1 Dye as a Sensitive Fluorescent Probe. Bio-Protocol 2019, 9, e3128. [Google Scholar] [CrossRef] [PubMed]
- Kong, N.; Penaloza, N.; Agreda, G.; Nguyen, A.B.; Gutheinz, J.; Tran, A.; Nguyen, N.; Ho, T.V.; Marin, A.; Mellis, B.; et al. Oxidative impact on lipoprotein structure: Insights from dynamic light scattering. Biochem. Biophys. Rep. 2025, 41, 101945. [Google Scholar] [CrossRef]
- Abdulrahman, A.O.; Kuerban, A.; Alshehri, Z.A.; Abdulaal, W.H.; Khan, J.A.; Khan, M.I. Urolithins Attenuate Multiple Symptoms of Obesity in Rats Fed on a High-Fat Diet. Diabetes Metab. Syndr. Obes. 2020, 13, 3337–3348. [Google Scholar] [CrossRef]
- De Leon, J.A.D.; Borges, C.R. Evaluation of Oxidative Stress in Biological Samples Using the Thiobarbituric Acid Reactive Substances Assay. JoVE (J. Vis. Exp.) 2020, 159, e61122. [Google Scholar] [CrossRef]
- Miere, F.; Teușdea, A.C.; Laslo, V.; Cavalu, S.; Fritea, L.; Dobjanschi, L.; Zdrinca, M.; Zdrinca, M.; Ganea, M.; Pașc, P.; et al. Evaluation of In Vitro Wound-Healing Potential, Antioxidant Capacity, and Antimicrobial Activity of Stellaria media (L.) Vill. Appl. Sci. 2021, 11, 11526. [Google Scholar] [CrossRef]
- Liang, H.; He, X.; Tong, Y.; Bai, N.; Pu, Y.; Han, K.; Wang, Y. Ferroptosis Opens a New Door for Colorectal Cancer Treatment. Front. Oncol. 2023, 13, 1059520. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhou, X.; Xie, F.; Zhang, L.; Yan, H.; Huang, J.; Zhang, C.; Zhou, F.; Chen, J.; Zhang, L. Ferroptosis in Cancer and Cancer Immunotherapy. Cancer Commun. 2022, 42, 88–116. [Google Scholar] [CrossRef]
- Gotorbe, C.; Durivault, J.; Meira, W.; Cassim, S.; Ždralević, M.; Pouysségur, J.; Vučetić, M. Metabolic Rewiring toward Oxidative Phosphorylation Disrupts Intrinsic Resistance to Ferroptosis of the Colon Adenocarcinoma Cells. Antioxidants 2022, 11, 2412. [Google Scholar] [CrossRef] [PubMed]
- Roy Chowdhury, S.; Banerji, V. Targeting Mitochondrial Bioenergetics as a Therapeutic Strategy for Chronic Lymphocytic Leukemia. Oxid. Med. Cell. Longev. 2018, 2018, 2426712. [Google Scholar] [CrossRef]
- Cui, Z.; Houweling, M. Phosphatidylcholine and Cell Death. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2002, 1585, 87–96. [Google Scholar] [CrossRef]
- Zhu, J.; Meng, W.; Lam, S.M.; Shui, G.; Huang, X. Phosphatidylcholine Deficiency Increases Ferroptosis Susceptibility in the Caenorhabditis elegans Germline. J. Genet. Genom. 2023, 50, 318–329. [Google Scholar] [CrossRef]
- Abalsamo, L.; Spadaro, F.; Bozzuto, G.; Paris, L.; Cecchetti, S.; Lugini, L.; Iorio, E.; Molinari, A.; Ramoni, C.; Podo, F. Inhibition of phosphatidylcholine-specific phospholipase C results in loss of mesenchymal traits in metastatic breast cancer cells. Breast Cancer Res. 2012, 14, R50. [Google Scholar] [CrossRef]
- Kaya Çakir, H.; Eroglu, O. In Vitro Anti-Proliferative Effect of Capecitabine (Xeloda) Combined with Mocetinostat (MGCD0103) in 4T1 Breast Cancer Cell Line by Immunoblotting. Iran. J. Basic Med. Sci. 2021, 24, 1515–1522. [Google Scholar] [CrossRef]
- Ciccolini, J.; Fina, F.; Bezulier, K.; Giacometti, S.; Roussel, M.; Evrard, A.; Cuq, P.; Romain, S.; Martin, P.M.; Aubert, C. Transmission of Apoptosis in Human Colorectal Tumor Cells Exposed to Capecitabine, Xeloda, Is Mediated via Fas. Mol. Cancer Ther. 2002, 1, 923–927. [Google Scholar] [PubMed]
- Guichard, S.M.; Macpherson, J.S.; Mayer, I.; Reid, E.; Muir, M.; Dodds, M.; Alexander, S.; Jodrell, D.I. Gene Expression Predicts Differential Capecitabine Metabolism, Impacting on Both Pharmacokinetics and Antitumor Activity. Eur. J. Cancer 2008, 44, 310–317. [Google Scholar] [CrossRef]
- Chin, C.C.; Li, J.M.; Lee, K.F.; Huang, Y.C.; Wang, K.C.; Lai, H.C.; Cheng, C.C.; Kuo, Y.H.; Shi, C.S. Selective β2-AR Blockage Suppresses Colorectal Cancer Growth through Regulation of EGFR-Akt/ERK1/2 Signaling, G1-Phase Arrest, and Apoptosis. J. Cell. Physiol. 2016, 231, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, C.M.; Tjioe, K.C.; Oliveira, S.H.P.; Bernabé, D.G. Propranolol Inhibits Cell Viability and Expression of the Pro-Tumorigenic Proteins Akt, NF-ĸB, and VEGF in Oral Squamous Cell Carcinoma. Arch. Oral Biol. 2022, 136, 105383. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, E.; Ciccolini, J.; Carre, M.; Giacometti, S.; Fanciullino, R.; Pouchy, C.; Montero, M.P.; Serdjebi, C.; Kavallaris, M.; André, N. Propranolol Potentiates the Anti-Angiogenic Effects and Anti-Tumor Efficacy of Chemotherapy Agents: Implication in Breast Cancer Treatment. Oncotarget 2011, 2, 797–809. [Google Scholar] [CrossRef]
- Zeynali-Moghaddam, S.; Mohammadian, M.; Kheradmand, F.; Fathi-Azarbayjani, A.; Rasmi, Y.; Esna-Ashari, O.; Malekinejad, H. A Molecular Basis for the Synergy between 17-Allylamino-17-Demethoxy Geldanamycin with Capecitabine and Irinotecan in Human Colorectal Cancer Cells through VEGF and MMP-9 Gene Expression. Gene 2019, 684, 30–38. [Google Scholar] [CrossRef]
- Barathova, M.; Grossmannova, K.; Belvoncikova, P.; Kubasova, V.; Simko, V.; Skubla, R.; Csaderova, L.; Pastorek, J. Impairment of Hypoxia-Induced CA IX by Beta-Blocker Propranolol—Impact on Progression and Metastatic Potential of Colorectal Cancer Cells. Int. J. Mol. Sci. 2020, 21, 8760. [Google Scholar] [CrossRef]
- Övey, İ.S.; Güler, Y. Apoptotic Efficiency of Capecitabine and 5-Fluorouracil on Human Cancer Cells through TRPV1 Channels. Indian J. Biochem. Biophys. 2020, 57, 64–72. [Google Scholar] [CrossRef]
- Zhong, X.; He, X.; Wang, Y.; Hu, Z.; Huang, H.; Zhao, S.; Wei, P.; Li, D. Warburg Effect in Colorectal Cancer: The Emerging Roles in Tumor Microenvironment and Therapeutic Implications. J. Hematol. Oncol. 2022, 15, 160. [Google Scholar] [CrossRef]
- Du, P.; Fan, R.; Zhang, N.; Wu, C.; Zhang, Y. Advances in Integrated Multi-omics Analysis for Drug-Target Identification. Biomolecules 2024, 14, 692. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Chen, X.B.; Ying, M.D.; He, Q.J.; Cao, J.; Yang, B. The Role of Ferroptosis in Cancer Development and Treatment Response. Front. Pharmacol. 2018, 8, 992. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular Mechanisms and Health Implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Kong, Y.; Xian, Y.; Gao, P.; Xu, Y.; Wei, C.; Lin, P.; Ye, W.; Li, Z.; Zhu, X. The Mechanisms of Ferroptosis and the Applications in Tumor Treatment: Enemies or Friends? Front. Mol. Biosci. 2022, 9, 938677. [Google Scholar] [CrossRef]
- Jing, S.; Lu, Y.; Zhang, J.; Ren, Y.; Mo, Y.; Liu, D.; Duan, L.; Yuan, Z.; Wang, C.; Wang, Q. Levistilide A Induces Ferroptosis by Activating the Nrf2/HO-1 Signaling Pathway in Breast Cancer Cells. Drug Des. Dev. Ther. 2022, 16, 2981–2993. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, M. PM2.5 Induces Ferroptosis in Human Endothelial Cells through Iron Overload and Redox Imbalance. Environ. Pollut. 2019, 254 Pt A, 112937. [Google Scholar] [CrossRef]
- Beatty, A.; Singh, T.; Tyurina, Y.Y.; Tyurin, V.A.; Samovich, S.; Nicolas, E.; Maslar, K.; Zhou, Y.; Cai, K.Q.; Tan, Y.; et al. Ferroptotic Cell Death Triggered by Conjugated Linolenic Acids Is Mediated by ACSL1. Nat. Commun. 2021, 12, 2244. [Google Scholar] [CrossRef]
- Di Sanzo, M.; Quaresima, B.; Biamonte, F.; Palmieri, C.; Faniello, M.C. FTH1 Pseudogenes in Cancer and Cell Metabolism. Cells 2020, 9, 2554. [Google Scholar] [CrossRef]
- Stockwell, B.R. Ferroptosis Turns 10: Emerging Mechanisms, Physiological Functions, and Therapeutic Applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Hou, B.; Huang, H.; Li, Y.; Liang, J.; Xi, Z.; Jiang, X.; Liu, L.; Li, E. Grb2 Interacts with Necrosome Components and Is Involved in Rasfonin-Induced Necroptosis. Cell Death Dis. 2022, 8, 319. [Google Scholar] [CrossRef]
- Yuan, F.; Sun, Q.; Zhang, S.; Ye, L.; Xu, Y.; Deng, G.; Xu, Z.; Zhang, S.; Liu, B.; Chen, Q. The Dual Role of p62 in Ferroptosis of Glioblastoma According to p53 Status. Cell Biosci. 2022, 12, 20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xi, Q.; Wang, D.; Li, J.; Wang, M.; Li, D.; Zhu, L.; Jin, L. Mitochondrial Dysfunction and Endoplasmic Reticulum Stress Involved in Oocyte Aging: An Analysis Using Single-Cell RNA-Sequencing of Mouse Oocytes. J. Ovarian Res. 2019, 12, 53. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, H.; Liu, Y.; Duan, C.; Liu, X.; Xia, T.; Chen, D.; Piao, H.L.; Liu, H.X. The Double-Edged Roles of ROS in Cancer Prevention and Therapy. Theranostics 2021, 11, 4839–4857. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Ai, Y.; Sun, Q.; Ma, Y.; Cao, Y.; Wang, J.; Zhang, Z.; Wang, X. Membrane Damage During Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol. Cell 2021, 81, 355–369.e10. [Google Scholar] [CrossRef]
- Deng, M.; Tang, F.; Chang, X.; Zhang, Y.; Liu, P.; Ji, X.; Zhang, Y.; Yang, R.; Jiang, J.; He, J.; et al. A targetable OSGIN1 - AMPK - SLC2A3 axis controls the vulnerability of ovarian cancer to ferroptosis. npj Precis Oncol. 2025, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Seydi, E.; Tabbati, Y.; Pourahmad, J. Toxicity of Atenolol and Propranolol on Rat Heart Mitochondria. Drug Res. 2020, 70, 151–157. [Google Scholar] [CrossRef]
- Mishima, E.; Sato, E.; Ito, J.; Yamada, K.I.; Suzuki, C.; Oikawa, Y.; Matsuhashi, T.; Kikuchi, K.; Toyohara, T.; Suzuki, T.; et al. Drugs Repurposed as Antiferroptosis Agents Suppress Organ Damage, Including AKI, by Functioning as Lipid Peroxyl Radical Scavengers. J. Am. Soc. Nephrol. 2020, 31, 280–296. [Google Scholar] [CrossRef]
- Liang, D.; Minikes, A.M.; Jiang, X. Ferroptosis at the Intersection of Lipid Metabolism and Cellular Signaling. Mol. Cell 2022, 82, 2215–2227. [Google Scholar] [CrossRef]
- Bose, S.; Banerjee, S.; Mondal, A.; Chakraborty, U.; Pumarol, J.; Croley, C.R.; Bishayee, A. Targeting the JAK/STAT Signaling Pathway Using Phytocompounds for Cancer Prevention and Therapy. Cells 2020, 9, 1451. [Google Scholar] [CrossRef]
- Gotthardt, D.; Trifinopoulos, J.; Sexl, V.; Putz, E.M. JAK/STAT Cytokine Signaling at the Crossroad of NK Cell Development and Maturation. Front. Immunol. 2019, 10, 2590. [Google Scholar] [CrossRef]
- Limberg, J.; Egan, C.E.; Gray, K.D.; Singh, M.; Loewenstein, Z.; Yang, Y.; Riascos, M.C.; Al Asadi, H.; Safe, P.; El Eshaky, S.; et al. Activation of the JAK/STAT Pathway Leads to BRAF Inhibitor Resistance in BRAFV600E Positive Thyroid Carcinoma. Mol. Cancer Res. 2023, 21, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT Signaling Pathway: From Bench to Clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Chen, Y.; Fang, Z.M.; Yi, X.; Wei, X.; Jiang, D.S. The Interaction between Ferroptosis and Inflammatory Signaling Pathways. Cell Death Dis. 2023, 14, 205. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.; Wang, N.; Han, W.; Bao, W.; Lu, J. IFNγ-Mediated Repression of System xc- Drives Vulnerability to Induced Ferroptosis in Hepatocellular Carcinoma Cells. J. Leukoc. Biol. 2021, 110, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Liu, P.; Bao, R.; Chen, J.; Zhou, M.; Mo, Z.; Ma, Y.; Liu, H.; Zhou, Y.; Cai, X.; et al. A Dual PI3K/HDAC Inhibitor Induces Immunogenic Ferroptosis to Potentiate Cancer Immune Checkpoint Therapy. Cancer Res. 2021, 81, 6233–6245. [Google Scholar] [CrossRef]
- Liang, M.; Fei, Y.; Wang, Y.; Chen, W.; Liu, Z.; Xu, D.; Shen, H.; Zhou, H.; Tang, J. Integrative Analysis of the Role of BOLA2B in Human Pan-Cancer. Front. Genet. 2023, 14, 1077126. [Google Scholar] [CrossRef]
- Li, W.; Wan, J.; Chen, C.; Zhou, C.; Liao, P.; Hu, Q.; Hu, J.; Wang, Y.; Zhang, Y.; Peng, C.; et al. Dissecting the Role of Cell Signaling versus CD8+ T Cell Modulation in Propranolol Antitumor Activity. J. Mol. Med. 2022, 100, 1299–1306. [Google Scholar] [CrossRef]
- Tornesello, M.L. TP53 mutations in cancer: Molecular features and therapeutic opportunities (Review). Int. J. Mol. Med. 2025, 55, 7. [Google Scholar] [CrossRef]
- Colorectal Metastasis Prevention International Trial 2—Full Text View (No Date) Colorectal Metastasis Prevention International Trial 2—Full Text View—ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03919461 (accessed on 14 March 2023).
- Guerrero, R.M.; Labajos, V.A.; Ballena, S.L.; Macha, C.A.; Lezama, M.S.; Roman, C.P.; Beltran, P.M.; Torrejon, A.F. Targeting BRAF V600E in metastatic colorectal cancer: Where are we today? Ecancermedicalscience 2022, 16, 1489. [Google Scholar] [CrossRef]
- Martinelli, E.; Cremolini, C.; Mazard, T.; Vidal, J.; Virchow, I.; Tougeron, D.; Cuyle, P.J.; Chibaudel, B.; Kim, S.; Ghanem, I.; et al. Real-world first-line treatment of patients with BRAFV600E-mutant metastatic colorectal cancer: The CAPSTAN CRC study. ESMO Open 2022, 7, 100603. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | PRO (µM) | CAP (mM) |
|---|---|---|
| HCT-116 | 129.26 ± 5.45 | 4.78 ± 0.07 |
| HT-29 | 75.71 ± 12.58 | 1.94 ± 0.77 |
| Cell Line | CI (ED50) | CI (ED75) | CI (ED90) | CI (ED95) | Type of Interaction |
|---|---|---|---|---|---|
| HCT-116 | 1.06 ± 0.06 | 1.04 ± 0.04 | 1.01 ± 0.01 | 0.94 ± 0.04 | Additive |
| HT-29 | 0.85 ± 0.005 | 0.63 ± 0.04 | 0.47 ± 0.05 | 0.33 ± 0.002 | Highly synergism |
| Treatment | Gene Symbol | LFC | Expression Level Based on RNA-Seq | p-Value |
|---|---|---|---|---|
| PRO | ACSL1 | 0.88 | Up | 0.01 |
| FTH1P2 | 0.99 | Up | 0.04 | |
| CAP | FTH1P2 | 1.15 | Up | 0.03 |
| FTH1P23 | 1.12 | Up | 0.02 | |
| FTL | 1.03 | Up | 0.02 | |
| GCLM | 0.78 | Up | 0.03 | |
| HMOX1 | 1.93 | Up | 0.001 | |
| SLC3A2 | 0.70 | Up | 0.04 | |
| PRO + CAP | ACSL1 | 0.94 | Up | 0.008 |
| FTH1 | 0.86 | Up | 0.04 | |
| FTH1P2 | 1.18 | Up | 0.01 | |
| FTH1P8 | 1.10 | Up | 0.02 | |
| FTH1P23 | 1.00 | Up | 0.04 | |
| GCLM | 0.99 | Up | 0.007 | |
| HMOX1 | 1.39 | Up | 0.005 | |
| SLC7A11 | 0.80 | Up | 0.03 |
| Pathway Name | Total | Hits | p-Value | Relative Metabolites |
|---|---|---|---|---|
| Mitochondrial electron transport chain | 19 | 5 | 1.71 × 10−3 | DHAPH, GA3P, glyceric acid-1,3-bisphosphate, fumarate, and FAD. |
| Phosphatidylcholine biosynthesis | 14 | 5 | 6.65 × 10−3 | Cytosine triphosphate (CTP), cytosine monophosphate (CMP), SAM, and phosphocholines (PCs). |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alzahrani, S.M.; Al Doghaither, H.A.; Alkhatabi, H.A.; Basabrain, M.A.; Pushparaj, P.N. Propranolol and Capecitabine Synergy on Inducing Ferroptosis in Human Colorectal Cancer Cells: Potential Implications in Cancer Therapy. Cancers 2025, 17, 1470. https://doi.org/10.3390/cancers17091470
Alzahrani SM, Al Doghaither HA, Alkhatabi HA, Basabrain MA, Pushparaj PN. Propranolol and Capecitabine Synergy on Inducing Ferroptosis in Human Colorectal Cancer Cells: Potential Implications in Cancer Therapy. Cancers. 2025; 17(9):1470. https://doi.org/10.3390/cancers17091470
Chicago/Turabian StyleAlzahrani, Shiekhah Mohammad, Huda Abdulaziz Al Doghaither, Hind Ali Alkhatabi, Mohammad Abdullah Basabrain, and Peter Natesan Pushparaj. 2025. "Propranolol and Capecitabine Synergy on Inducing Ferroptosis in Human Colorectal Cancer Cells: Potential Implications in Cancer Therapy" Cancers 17, no. 9: 1470. https://doi.org/10.3390/cancers17091470
APA StyleAlzahrani, S. M., Al Doghaither, H. A., Alkhatabi, H. A., Basabrain, M. A., & Pushparaj, P. N. (2025). Propranolol and Capecitabine Synergy on Inducing Ferroptosis in Human Colorectal Cancer Cells: Potential Implications in Cancer Therapy. Cancers, 17(9), 1470. https://doi.org/10.3390/cancers17091470