Simple Summary
Epstein–Barr virus (EBV) is implicated in the pathogenesis of a variety of malignancies, including Hodgkin’s lymphoma (HL). HL is associated with the gene expression program IIa of viral latency, and the expressed proteins and non-coding RNAs mediate both the malignant transformation of B-cells and evasion from immune surveillance. EBV targets pattern-recognition receptors and interferon-mediated signaling to evade innate immunity, hampers the processing and presentation of viral antigen to avoid recognition by CD4+ and CD8+ T cells, induces the expression of immune checkpoint molecules, and shapes the microenvironment to sustain the survival of the malignant cells. The aim of the current review is to shed light on the underlying molecular mechanisms, which is the first step in developing novel effective therapeutic interventions that would harness the immune system against HL.
Abstract
Epstein–Barr virus (EBV) constitutes a very common pathogen and a well-characterized carcinogen. EBV has the ability to establish a chronic latent infection, during which only a subset of the viral genes is expressed. EBV is implicated in multiple malignancies, including Hodgkin’s lymphoma (HL). HL mainly affects adolescents and young adults and has an overall favorable prognosis. However, relapsed or refractory disease still poses a therapeutic challenge. EBV does not only induce malignant transformation but also hinders the detection and clearance of the neoplastic cells by the immune system. The proteins and non-coding RNAs expressed in latency IIa, which is associated with HL, employ a variety of mechanisms to target different steps of innate and adaptive immunity, to take advantage of the immunosuppressant effect of immune checkpoints, and to shape the microenvironment to support the survival and proliferation of malignant cells. They suppress the expression or promote the degradation of pattern-recognition receptors, interfere with type I interferon and proinflammatory cytokine mediated signaling, and hinder the effector function of natural killer cells. The processing and presentation of peptides to CD4 and CD8 T cells are also hampered. EBV induces the expression of immune checkpoints, the secretion of immunosuppressive cytokines, and the efflux of regulatory T cells in the tumor microenvironment. The current review provides a comprehensive overview of the molecular mechanisms underlying this complex interplay between EBV and the immune system in HL with focus on clinical data from the pediatric population, which is the key for developing novel, effective therapeutic interventions.
1. Introduction
Approximately 13% of all cancer cases (excluding non-melanoma skin cancers) were attributed to infections in 2018 [1], and according to the International Agency for Research on Cancer, six viruses, namely hepatitis B virus, hepatitis C virus, human papillomavirus, Epstein–Barr virus (EBV), human herpesvirus type 8, also known as Kaposi sarcoma-associated herpesvirus, and human T cell lymphotropic virus type 1, are considered to be carcinogens [2]. Amongst them, EBV undoubtedly holds a special position, as it infects more than 90% of healthy individuals worldwide while being implicated in a wide range of malignancies, including nasopharyngeal carcinoma, Burkitt’s lymphoma and Hodgkin’s lymphoma (HL), diffuse large B cell lymphoma, plasmablastic lymphoma, primary effusion lymphoma, natural killer (NK)/T cell lymphoma, gastric carcinoma, leiomyosarcoma, and post-transplant lymphoproliferative disease [3,4]. EBV is a herpesvirus belonging to the γ-herpesvirus subfamily, also known as human herpesvirus 4, with a linear 172-kbp double-stranded DNA, which encodes approximately 85 proteins and 48 non-coding RNAs [5]. Another significant feature is the virus’s ability to establish a chronic latent infection, during which the viral genome persists in the form of episomes in the nucleus of host cells, and only a subset of the viral genes are expressed [6]. EBV latent infection includes distinct patterns (latency types 0, I, IIa, IIb, and III), each characterized by a specific gene expression signature under strict epigenetic regulation and associated with different EBV-associated malignancies [6]. During the course of EBV infection the pressure of EBV-specific cytotoxic T lymphocytes (CTLs) causes the virus to switch to latency type IIa, a more restricted state in terms of gene expression characterized by the expression of latent membrane proteins 1 and 2A (LMP1 and LMP2A), Epstein–Barr nuclear antigen 1 (EBNA1), and the expression of EBV encoded RNAs (EBER RNAs) and BamHI A rightward transcript (BART) miRNAs [7].
HL is a lymphoid malignancy originating from preapoptotic germinal center B cells [8] with an estimated incidence rate of 25 new cases/million/year in the United States per the Surveillance, Epidemiology, and End Results Program (2000–2022). It exhibits a bimodal distribution curve with the first, larger peak seen in adolescents and young adults (AYAs) [9]. Classical HL (cHL) represents the vast majority of cases. A strong association with EBV infection has been described in approximately 30% of cHL cases, with differences according to age and histological subtype, i.e., the mixed cellularity subtype is the most prevalent in children with a strong association with EBV infection (approximately 80%), while the nodular sclerosis subtype is the most common in older patients and exhibits a weaker association (<30%) [10,11,12]. EBV latency program IIa has been associated with HL, and the expressed genes contribute to HL pathogenesis [7]. LMP1 is a CD40 receptor homolog that stimulates nuclear factor κB (NF-κB) and Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) pathway activation [13,14,15] and shapes the tumor microenvironment (TME) to support the survival of the few Reed–Sternberg (RS) cells [16]. LMP2A substitutes the function of the B cell receptor, thus salvaging the RS cells from apoptosis [17]. EBNA1 is a viral transcription factor that supports the maintenance of EBV episomes and regulates viral and cellular gene expression [18]. HL has a generally favorable prognosis with a five-year relative survival of nearly 90% [9]. However, refractory disease occurs in 5–10% and relapse in 5–30% (depending on stage) of children and adolescents with HL [19].
Immune evasion constitutes one of the hallmarks of cancer [20], and immunotherapies have revolutionized the field of oncology, exhibiting extremely promising results even in heavily pretreated refractory or relapsed cases [21]. Therefore, understanding the complex interplay between known common carcinogens such as EBV, cancer cells, and the immune system constitutes the key to developing novel, effective therapeutic interventions. The current review summarizes the mechanisms underlying EBV-induced immune escape in HL (Figure 1).
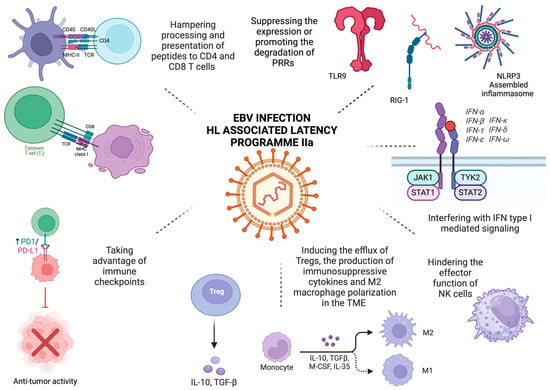
Figure 1.
EBV-mediated immune evasion in HL. The proteins and non-coding RNAs expressed in latency IIa, which is associated with HL, target different steps of innate and adaptive immunity, take advantage of the immunosuppressant effect of immune checkpoints, and shape the tumor microenvironment. They suppress the expression of TLR9 by hindering its promoter activity or by suppressing the N6-methyladenosine modification and that of NLRP3, thereby blocking the assembly and activation of the NLRP3 inflammasome. They also promote RIG-I degradation through the proteasome pathway, interfere with type I interferon and proinflammatory cytokine-mediated signaling, and hinder the effector function of NK cells. Processing, loading to MHC class I and II, and ultimately recognizing peptides by CD8 and CD4 T cells, respectively, are hampered. Increased expression of immune checkpoint molecules, namely PD-L1 and LAG-3, induces T cell exhaustion and impairs their effector function and thus the clearance of neoplastic cells. Finally, EBV shapes the TM by creating an immunosuppressive niche through attracting Tregs, inducing the expression of regulatory cytokines, such as IL-10 and TGF-β), affecting the polarization of macrophages, and upregulating immune checkpoints. Created in BioRender (accessed on 27 March 2025). Tsotridou, E. (2025).
2. Evading Innate Immunity
Innate immunity constitutes the first line of defense against pathogens activated by pathogen-associated molecular patterns or danger-associated molecular patterns. A plethora of pattern-recognition receptors (PRRs) take part in the initiation of the innate immunity response, including Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), Nod-like receptors, AIM2-like receptors, C-type lectin receptors, and other DNA sensors, such as cyclic GMP-AMP synthase, which activate the NF-κΒ signaling pathway and the production of type I interferons (IFNs) [22,23]. The EBV proteins expressed in latency type II (and subsequently in EBV-associated HL) interfere in different stages of this process in order to escape from immune control.
LMP1 suppresses TLR9 expression by hindering its promoter activity. LMP1 is known to induce NF-κB signaling, which is essential in TLR9 gene downregulation, as the TLR9 promoter contains four NF-κB binding sites [24]. Furthermore, LMP1 promotes RIG-I degradation through the proteasome pathway and specifically by recruiting E3 ligases to mark the protein for degradation by ubiquitination [25]. LMP1 also regulates IFN signaling by interacting with the non-receptor tyrosine-protein kinase Tyk2, a member of the JAK protein family [26]. Type I IFNs exert their function by binding to the heterodimeric Interferon Alpha/Beta Receptor 1/2 (IFNAR1/2). Tyk2 associates with IFNAR1 and, upon activation, induces the phosphorylation of IFNAR1 and recruits STAT1 and 2. STAT1/2 heterodimers, together with IFN-regulatory factor 9, form the IFN-stimulated gene factor 3 complex, which translocates to the nucleus and binds to IFN-stimulated response elements (ISRE) [27]. LMP1 inhibits Tyk2 phosphorylation and, subsequently, STAT2 phosphorylation and ISRE activation [26].
LMP2A/B have been shown to target the same pathway. Although they do not influence the levels of IFN receptors on the cell membrane, LMP2A/B attenuate interferon responses by inducing receptor degradation, either through ubiquitination or through trafficking from endosomes to lysosomes [28].
EBNA1 suppresses N6-methyladenosine modification of TLR9, thus affecting the encoded messenger RNA (mRNA) stability. Mechanistically, EBNA1 promotes the degradation of Methyltransferase 3, N6-Adenosine-Methyltransferase Complex Catalytic Subunit (METTL3), an enzyme catalyzing the formation of N6-methyladenosine, by increasing K48-linked ubiquitination mediated by parkin ubiquitin ligase [29]. EBNA1 targets another vital component of innate immunity, i.e., the function of NK cells [30]. EBNA1 downregulates the expression of UL16 Binding Proteins 1 and 5, both of which serve as Killer Cell Lectin-Like Receptor K1 (NKG2D) ligands. Apart from inhibiting the expression of ligands of NK cell receptors, EBNA1 binds to the c-Myc promoter, which hinders cellular responses to stress and/or DNA damage and apoptosis [30].
The role of EBV proteins expressed in latency IIa in evading innate immunity is summarized in Table 1.

Table 1.
The role of EBV proteins expressed in latency IIa in evading innate immunity.
Apart from EBV proteins, several viral miRNAs target PRRs and interfere with IFN- and other proinflammatory cytokine-mediated signaling [31,32,33,34,35,36,37]. miR-BART6-3p targets RIG-I [31,32], while miR-BART15 targets NLR Family Pyrin Domain Containing 3 (NLRP3), the most well-characterized of the Nod-like receptors, thereby inhibiting the production of interleukin 1β (IL-1β) by the inflammasome, as NLRP3 activation results in the assembly and activation of the NLRP3 inflammasome and, in turn, in the release of proinflammatory cytokines [33,38]. Interestingly, this effect was observed both in infected and neighboring non-infected cells, suggesting miRNA trafficking via exosomes [33]. MiR-BART16 targets the histone acetyltransferase cAMP Responsive Element Binding Protein (CREB), which interacts with positive regulatory domains of the IFNβ promoter [34,39]. Skinner et al. demonstrated that the BamHI fragment H rightward open reading frame BHRF1-2-5p blocks IL-1 signaling by targeting receptor 1 of IL-1 (IL1R1) in latently infected B cells [35]. Taking into consideration that this cell surface cytokine receptor initiates a signaling cascade upon activation, which leads to the NF-κB-mediated induction of IL-1α and IL-1β production, BHRF1-2-5p also impacts the expression levels of cytokines [35]. Indeed, upon BHRF1-2-5p inhibition, IL-1β treatment resulted in a two-fold increase in the levels of IL-1α, IL-1β, and IL-6, thus proving the disruption of a positive feedback loop of autocrine/paracrine signaling [35]. Using RNA-induced silencing complex immunoprecipitation followed by polymerase chain reaction target validation, Dölken et al. proved that EBV miR-BART3 targets importin 7, resulting in the reduced production of IL-6 upon lipopolysaccharide challenge, thus also confirming the previous findings of other groups [36,37]. Finally, miR-BART-2p targets MHC Class I Polypeptide-Related Sequence B (MICB), a function preserved amongst herpesviruses [40]. MICB constitutes an NKG2D ligand, which is expressed upon the activation of cellular stress pathways following malignant transformation, thus marking them for recognition by NK cells [41].
3. Evading Adaptive Immunity
Although the main EBV proteins that facilitate evasion of adaptive immunity mechanisms are early lytic cycle proteins, the proteins expressed in latency IIa have also been reported to contribute [42].
LMP1 limits its self-presentation to CD8+ T cells [43] and hijacks the cellular transcription program by activating multiple signal transduction pathways, including NF-κB, all three Mitogen-Activated Protein Kinase (MAPK) pathways, namely c-Jun N-terminal kinase, extracellular signal-regulated kinases 1/2 (ERK1/2) and p38, JAK/STAT, and Phosphoinositide 3-kinase/Protein Kinase B (PI3K/Akt) [44]. LMP1 induces the production of IL-10 in B cells [45], which acts not only as an autocrine growth factor for B cells [46] but also as an immunosuppressant [47,48]. Increased IL-10 production results in the downregulation of Antigen Peptide Transporter 1 (TAP1), a part of the heterodimeric complex responsible for peptide transport to the lumen of the endoplasmic reticulum and, subsequently, loading to Major Histocompatibility Complex (MHC) class I molecules [47]. Taking into consideration that molecules lacking loaded peptides also lack stability, it is evident that the reduction in TAP1 expression levels directly influences the steady-state levels of MHC class I molecules [47]. At the same time, increased IL-10 production modifies cell surface glycosylation, thereby increasing the antigenic threshold required for T cell activation [48]. This is achieved through the upregulation of the glycosyltransferase Mgat5, which enhances N-glycan branching on surface glycoproteins, which in turn leads to the formation of a galectin 3-mediated membrane lattice [48]. Mechanistically, LMP1 induces the activation of both PI3K/Akt and p38 pathways through distinct intracellular signaling regions to enhance IL-10 production [45]. P38 mediates the phosphorylation and activation of CREB. The PI3K pathway activation has secondary effects on CREB, as it induces the phosphorylation and inactivation of the Serine/Threonine-Protein Kinase GSK3B, thus hindering its inhibitory effect on CREB-dependent IL-10 production [45]. LMP1 also induces the ectopic expression of CD137 on RS cells and the subsequent secretion of IL-13, which, like IL-10, is a potent growth factor for the neoplastic cells and mediates immune escape by reducing IFNγ production [49,50]. The ectopic expression of CD137 on RS cells, which is a result of the LMP1 mediated activation of the PI3K/Akt/Mechanistic Target of Rapamycin Kinase (mTOR) pathway, eliminates its ligand CD137 from adjacent antigen presenting cells either by internalization and degradation or by trogocytosis, thereby also eliminating the costimulatory signal normally provided by CD137/CD137L signaling for T cell activation [49,50].
LMP2A hinders antigen presentation to CD4+ T cells by reducing the expression of MHC class II molecules [51]. Immunoreceptor tyrosine-based activation motifs within LMP2A interact with the kinases Spleen-Associated Tyrosine Kinase (Syk) and SRC Proto-Oncogene, Non-Receptor Tyrosine Kinase (Src), which mediate the activation of the PI3K/Akt pathway, leading to the reduced promoter activity of E47 and PU.1 [51]. These transcription factors bind to promoter III of the Class II MHC Transactivator, thus regulating its expression levels [51]. There are also reports that LMP2A interferes with the effector function of CD8+ T cells through multiple mechanisms: it downregulates (to a lesser extent) the expression of MHC class I molecules, reduces the recognition of EBV+ neoplastic B cells by reducing the expression of NKG2D ligands, and finally, again through the initial activation of Syk, leads to the activation of PI3K and Bruton kinase-mediated signaling to induce the phosphorylation of STAT3 and ultimately the production of IL-10 [52,53].
EBNA1 acts as an inhibitor of both ribosomal and proteosomal activity, thereby preventing the presentation of its epitopes by MHC class I molecules and their recognition by CD8+ T cells. The glycine-alanine repeat domain (GAr) within EBNA1 acts in cis and inhibits the translation of its own mRNA [54,55,56]. Interestingly, this effect is location-dependent since it is required for the GAr domain to be located within the coding sequence of the gene to have an effect on the process of translation [55]. EBNA1 has also been reported to inhibit its own degradation by interfering with the initial steps of substrate unfolding [57]. The extent to which each process contributes to immune evasion remains unclear, although there are reports that the inhibition of translation is sufficient to prevent the presentation of viral peptides to cytotoxic cells [56]. However, it should be noted that a low turnover rate is also required for a low rate of protein synthesis.
The role of EBV proteins expressed in latency IIa in evading adaptive immunity is summarized in Table 2.
Table 2.
The role of EBV proteins expressed in latency IIa in evading adaptive immunity.
The role of EBV miRNAs in evading adaptive immunity was initially demonstrated by experiments with miRNA-deficient EBV strains. Upon the in vivo infection of mice with reconstituted human immune system components with EBV strains expressing or lacking miRNAs, the absence of miRNAs resulted in reduced viral loads, lymphomagenesis, and T cell proliferation [58]. However, antibody-mediated CD8+ T cell depletion caused an increase in viral loads, which was more evident in miRNA-deficient (200-fold increase) compared to wild-type EBV-infected cells (40-fold increase) [58]. Moreover, more than 50% of animals developed tumors, while no tumors were reported prior to CD8+ T cell depletion in the miRNA-deficient group [58]. These results suggest that EBV-encoded miRNAs mediate immune evasion by hindering the clearance of EBV-infected B cells by CD8+ T cells, thus also contributing to lymphomagenesis [58]. EBV miRNAs target LMP1 and interfere with the release of proinflammatory cytokines, antigen processing, and presentation either by MHC class I or class II molecules [59,60]. MiR-BART3 and miR-BART16 directly target LMP1, thus limiting its expression and, consequently, the EBV-induced antigenic stimuli [59]. The production of proinflammatory cytokines is also hindered [59]. Particularly, IL-12, which is targeted by miR-BART1, miR-BART2, miR-BART10, miR-BART22 and BHRF1-2, plays a critical role in the differentiation of CD4+ to Th1 T cells [59,60]. MiR-BART17 and BHRF1-3 target Antigen Peptide Transporter 2 (TAP2), a part of the heterodimeric complex responsible for peptide transport and loading to MHC class I molecules [60]. Several EBV miRNAs also target cathepsin B (CTSB), Lysosomal Thiol Reductase (IFI30), and Asparaginyl Endopeptidase (AEP), lysosomal enzymes involved in antigen processing and MHC class II-mediated presentation [59,60].
The miRNAs, along with their targets implicated in evasion of both innate and adaptive immunity, are presented in Table 3.
Table 3.
MiRNAs and their targets implicated in evasion of innate and adaptive immunity.
4. Taking Advantage of Immune Checkpoints
Programmed death receptor 1 (PD-1) is a transmembrane glycoprotein that is expressed on the surface of activated CD4+ and CD8+ T lymphocytes, B lymphocytes, dendritic cells (DCs), and NK cells [61,62]. PD-1 activation via binding of its PD-L1 and PD-L2 ligands inhibits T cell growth, proliferation, and effector functions via the dephosphorylation of essential components of the T cell receptor (TCR) signaling cascade, the inhibition of the activation of the PI3K/Akt/mTOR and Ras/MEK/Erk pathways, and the hampering of T cell motility and interaction with antigen presenting cells [61,62]. PD-1 signaling eventually inhibits the entry of T cells into the S phase of the cell cycle, causes a metabolic shift towards increased fatty acid oxidation, and increases the differentiation of T cells into induced regulatory T cells (Tregs) [61,62]. According to a recent meta-analysis, the expression levels of PD-L1 were higher not only on tumor cells but also on immune cells in the TME in EBV-positive compared to EBV-negative cases of cHL, with risk ratios of 1.66 and 1.43, respectively [63]. Alterations of 9p24.1/CD274(PD-L1)/PDCD1LG2(PD-L2), which constitute a defining feature of cHL and increase PD-L1 or PD-L2 expression, have been shown to be similarly distributed in patients with EBV-negative and EBV-positive cHL, but EBV-positive cHLs displayed higher PD-L1 H-scores (percentage of RS cells with positive staining multiplied by the average intensity of positive staining), suggesting further induction of PD-L1 expression by EBV infection [64]. Mechanistically, EBV LMP1 induces PD-L1 expression in two ways: JAK/STAT signaling mediated enhanced promoter activity and activator protein 1-mediated enhancer activity [65,66]. Of note, the LMP1-associated induction of PD-L1 is fine-tuned by viral miRNA BHRF1-2-5p, which binds to the 3’untranslated region, thus inhibiting gene expression [67]. Carey et al. demonstrated that PD-L1 is mostly expressed by tumor-associated macrophages (TAMs) and that these PD-L1-positive TAMs tend to colocalize with PD-L1-positive RS cells. The vicinity of this cellular niche is enriched with PD-1-positive T cells, thus facilitating T cell exhaustion and immunosuppression [68].
Cytotoxic T lymphocyte-associated protein 4 (CTLA-4) is a CD28 homolog. Binding to its ligands, CD80 and CD86, inhibits their interaction with CD28 and thus the necessary co-stimulatory signal in the immune synapse for T cell activation [69]. Evidence regarding the role of CTLA-4 in EBV-related malignancies and particularly Hodgkin’s lymphoma remains scarce. However, data from a cord blood-humanized mouse model suggest that both PD-1 and CTLA-4 are expressed on T cells and that PD-1/CTLA-4 blockade increases T cell infiltration of tumors, promotes T cell activation, and eventually leads to a drastic reduction of the size of EBV-induced lymphomas [70].
Other immune checkpoints, including the protein encoded by lymphocyte activation gene-3 (LAG-3), T cell immunoglobulin-3 (TIM-3), T cell immunoglobulin and ITIM domain (TIGIT), V-Domain Ig suppressor of T cell activation (VISTA), the B7 homolog 3 protein (B7-H3), the B and T cell lymphocyte attenuator (BTLA), and the sialic acid-binding immunoglobulin-like lectin 15 (Siglec-15), are constantly gaining attention. However, questions about their ligands and mechanisms of action still remain, and there are limited preclinical and clinical data [62].
LAG-3 is expressed on CD4+ and CD8+ T cells, regulatory T cells (T-regs), a subpopulation of NKs, B cells, and plasmacytoid DCs and binds to MHC class II with a 100-fold higher affinity than CD4, thus hampering TCR-mediated signaling [71]. Gandhi et al. demonstrated that LAG-3 is expressed on tumor-infiltrating lymphocytes adjacent to the malignant RS cells and that higher expression levels correlate with EBV positivity. Furthermore, they reported that the immunological responses against LMP1 epitopes are impaired in newly diagnosed or relapsed HL patients and that the observed T cell functional impairment is proportional to the degree of LAG-3 and forkhead box protein P3 (FOXP3), a marker of Tregs. These results suggest a significant role of LAG-3-expressing Tregs in the microenvironment of EBV-positive RS cells in dampening T cell responses and leading to immune evasion [72].
Specifically in the pediatric population, Dilly-Feldis et al. assessed ligand PD-L1 in 42 children with cHL and reported higher expression levels in EBV-positive cases [73]. Based on these findings, Uccini et al. used immunostaining to measure the expression of PD-1 and PD-L1 in 53 cases of cHL in children under 14 years of age and, apart from confirming that PD-L1 levels are higher in EBV cases, proved that increased PD-L1 expression is independent of 9p24.1 amplification, as all of the EBV positive cases were negative for 9p24.1 amplification by fluorescent in situ hybridization [74]. Recently, Oscar et al. demonstrated that LAG-3 positive cells have a positive correlation with PD-1 positive cells in cases of cHL, which remains significant when analyzing exclusively EBV positive cases, but is lost in EBV negative ones [75]. EBV-positive cases with LAG-3 and PD-1 co-expression had strikingly lower 5-year survival rates compared to either LAG-3 and PD-1 negative cases or cases with expression of one of the exhaustion markers (54% versus 100%) [75].
5. Shaping the Microenvironment
Despite the fact that RS cells constitute the defining feature of HL, they comprise only 1% of the tumor, while the rest represents a plethora of immune cells including T and B lymphocytes, neutrophils, eosinophils, macrophages, plasma cells, NK cells, dendritic cells and mast cells, as well as stromal cells, fibroblasts, and endothelial cells. This diverse cellular infiltrate is in a continuous crosstalk with the few neoplastic cells and plays a pivotal role in sustaining tumor growth and facilitating immune escape [76,77,78]. The significance of the composition of the TME is evident by its great prognostic and predictive value in terms of response to treatment and survival [79,80]. EBV seems to play a unique role in shaping the TME and creating an immunosuppressive niche by attracting Tregs, inducing the expression of regulatory cytokines, affecting the polarization of macrophages, and upregulating immune checkpoints [65,66,81,82,83,84,85,86,87,88,89]. Morales et al. compared gene expression in the lymph nodes and the peripheral blood between EBV-positive and -negative HL patients aged between 8 and 71 years. In this mixed-age patient population, EBV-positive cases were characterized by an upregulation of CD4, FOXP3, which is a marker of Tregs, adhesive molecules, such as integrin B2 and p-selectine, and immune checkpoints, such as CTLA-4 and LAG-3 [82]. Furthermore, the increased expression of immunosuppressive cytokines (IL-10) and transforming growth factor beta (TGF-β) was detected both in the lymph nodes and the sera of EBV-positive patients [82]. The efflux of Tregs was attributed to an increased production of cc motif chemokine (CC) ligands 4, 5, 17, 19, and 20 and their receptors CCR5 and CCR7 [82]. A distinct subpopulation of Tregs, namely T regulatory 1 cells, secreted IL-10 both in the lymph nodes and the periphery [82], which has also been confirmed by other groups [83,84]. EBV-encoded proteins LMP1 and EBNA1, by upregulating CCL20, have been reported to attract Tregs in the TME [83,85,90]. Furthermore, stromal cells in the TME express indoleamine 2,3-dioxygenase (IDO), an enzyme that suppresses T cell effector functions while at the same time enhancing the immunosuppressive effect of Tregs [91,92]. Higher IDO expression has been reported in EBV-positive cases and has been associated with inferior survival [91]. The composition of the TME also differs between adults and children and even varies among different age groups in the pediatric population [93]. Interestingly, Barros et al. demonstrated that in the pediatric population, EBV positivity was not associated with increased infiltration of FOXP3 cells, but rather CD8, Cytotoxic Granule-Associated RNA Binding Protein TIA1, Granzyme B, and T-Box Transcription Factor 21-positive cells, suggesting a cytotoxic Th1 profile [93]. This is further reinforced by the distinct macrophage composition and their M1 polarization in young children with EBV-associated HL, which greatly influences survival rates [88,89,94]. However, Jimenez et al. reported the recruitment of PD-L1-positive cells in the microenvironment of pediatric HL cases [81]. Despite the presence of cytotoxic T lymphocytes [86,87,93] both in pediatric and adult patients, the concomitant upregulation of immune checkpoint molecules [65,66,81] probably induces exhaustion and hinders their effector functions.
6. Conclusions
EBV latency program II is associated with Hodgkin’s lymphoma, and the virus plays a critical role not only in malignant transformation but also in the maintenance of the neoplastic cells by facilitating escape from anti-tumor immune control. The proteins and non-coding RNAs expressed target all the frontiers of the immune response to support the survival of the malignant cells [95]. The current review provides a comprehensive overview of the molecular mechanisms underlying this complex interplay. Understanding these mechanisms would be the first step in the process to develop novel therapeutic interventions, which would harness the immune system to clear the neoplastic cells. Immune checkpoint inhibitors have already gained approval and proven their efficacy both in the adult [96,97] and in the pediatric population [98,99]. Strategies, such as the use of EBV-specific CTLs in the autologous [100,101,102] or the allogeneic setting [103], EBNA-1 targeted inhibitors [104], and Chimeric Antigen Receptor T cells are currently being investigated [105,106,107]. Creating an EBV-targeted vaccine has been a long-lasting effort, with research focusing on the gp340 glycoprotein [108]. Combining agents that target different evasion mechanisms aimed at different steps of the immune response could provide a promising alternative. However, this course of action also requires attention to the possibility of off-target effects and increased associated toxicities. Children and adolescents undoubtedly constitute a diverse and unique patient population, especially in the context of immunotherapy. There is a pressing need for new clinical trials in HL with different age groups of the so-called pediatric population to explore the potential of novel immunotherapeutics.
Author Contributions
Conceptualization, E.T. and E.H.; methodology, E.T.; software, E.T.; validation, E.T. and E.H.; formal analysis, E.T.; investigation, E.T.; resources, E.T.; data curation, E.T. and E.H.; writing—original draft preparation, E.T.; writing—review and editing, E.H.; visualization, E.T.; supervision, E.H.; project administration, E.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| EBV | Epstein–Barr virus |
| HL | Hodgkin’s lymphoma |
| NK | Natural killer |
| CTLs | Cytotoxic T lymphocytes |
| LMP1 | Latent membrane protein 1 |
| LMP2A | Latent membrane protein 2A |
| EBNA1 | Epstein–Barr nuclear antigen 1 |
| EBERs | EBV-encoded RNAs |
| BART | BamHI A rightward transcript |
| AYAs | Adolescents and young adults |
| cHL | classical HL |
| NF-κB | Nuclear factor κB |
| JAK | Janus kinase |
| STAT | Signal Transducer and Activator of Transcription |
| TME | Tumor microenvironment |
| RS | Reed–Sternberg |
| PRRs | Pattern-recognition receptors |
| TLRs | Toll-like receptors |
| RLRs | RIG-I-like receptors |
| IFNs | Interferons |
| IFNAR1/2 | Interferon Alpha/Beta Receptor 1/2 |
| ISRE | IFN-stimulated response elements |
| mRNA | messenger RNA |
| METTL3 | Methyltransferase 3, N6-Adenosine-Methyltransferase Complex Catalytic Subunit |
| NKG2D | Killer Cell Lectin-Like Receptor K1 |
| NLRP3 | NLR Family Pyrin Domain Containing 3 |
| IL-1β | Interleukin 1β |
| CREB | cAMP Responsive Element Binding Protein |
| BHRF | BamHI fragment H rightward open reading frame |
| IL1R1 | Receptor 1 of IL-1 |
| MICB | MHC Class I Polypeptide-Related Sequence B |
| MAPK | Mitogen-Activated Protein Kinase |
| ERK | Extracellular signal-regulated kinases |
| PI3K | Phosphoinositide 3-kinase |
| Akt | Protein Kinase B |
| TAP1/2 | Antigen Peptide Transporter 1/2 |
| MHC | Major Histocompatibility Complex |
| mTOR | Mechanistic Target of Rapamycin Kinase |
| Syk | Spleen-Associated Tyrosine Kinase |
| Src | SRC Proto-Oncogene, Non-Receptor Tyrosine Kinase |
| GAr | Glycine-alanine repeat |
| CTSB | Cathepsin B |
| IFI30 | Lysosomal Thiol Reductase |
| AEP | Asparaginyl Endopeptidase |
| PD-1 | Programmed death receptor 1 |
| PD-L1/2 | Programmed death receptor ligand 1/2 |
| DCs | Dendritic cells |
| TCR | T cell receptor |
| Tregs | Regulatory T cells |
| TAMs | Tumor-associated macrophages |
| CTLA-4 | Cytotoxic T lymphocyte-associated protein 4 |
| LAG-3 | Lymphocyte activation gene-3 |
| TIM-3 | T cell immunoglobulin the (BTLA) and the (Siglec-15) |
| TIGIT | T cell immunoglobulin and ITIM domain |
| VISTA | V-Domain Ig suppressor of T cell activation |
| B7-H3 | B7 homolog 3 protein |
| BTLA | B and T cell lymphocyte attenuator |
| Siglec-15 | sialic acid-binding immunoglobulin-like lectin 15 |
| FOXP3 | Forkhead box protein P3 |
| TGF-β | Transforming growth factor beta |
| CC | cc motif chemokine |
| CCR | cc motif chemokine receptor |
| IDO | Indoleamine 2,3-dioxygenase |
References
- de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global Burden of Cancer Attributable to Infections in 2018: A Worldwide Incidence Analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A Review of Human Carcinogens—Part B: Biological Agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Smatti, M.K.; Al-Sadeq, D.W.; Ali, N.H.; Pintus, G.; Abou-Saleh, H.; Nasrallah, G.K. Epstein-Barr Virus Epidemiology, Serology, and Genetic Variability of LMP-1 Oncogene Among Healthy Population: An Update. Front. Oncol. 2018, 8, 211. [Google Scholar] [CrossRef]
- Shannon-Lowe, C.; Rickinson, A. The Global Landscape of EBV-Associated Tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef]
- Tarbouriech, N.; Buisson, M.; Géoui, T.; Daenke, S.; Cusack, S.; Burmeister, W.P. Structural Genomics of the Epstein-Barr Virus. Biol. Crystallogr. 2006, 62, 1276–1285. [Google Scholar] [CrossRef]
- Kong, I.Y.; Giulino-Roth, L. Targeting Latent Viral Infection in EBV-Associated Lymphomas. Front. Immunol. 2024, 15, 1342455. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A.; Gross, A. Persistence of the Epstein-Barr Virus and the Origins of Associated Lymphomas. N. Engl. J. Med. 2004, 350, 1328–1337. [Google Scholar] [CrossRef]
- Kanzler, H.; Küppers, R.; Hansmann, M.L.; Rajewsky, K. Hodgkin and Reed-Sternberg Cells in Hodgkin’s Disease Represent the Outgrowth of a Dominant Tumor Clone Derived from (Crippled) Germinal Center B Cells. J. Exp. Med. 1996, 184, 1495–1505. [Google Scholar] [CrossRef]
- Surveillance, Epidemiology, and End Results (SEER) Program. SEER*Stat Database: Incidence—SEER Research Data, 8 Registries, Nov 2023 Sub (1975–2021)—Linked To County Attributes—Time Dependent (1990–2022) Income/Rurality, 1969–2022 Counties, National Cancer Institute, DCCPS, Surveillance Research Program, Released April 2024, Based on the November 2023 Submission. Available online: https://seer.cancer.gov/statfacts/html/hodg.html (accessed on 27 February 2025).
- Donzel, M.; Bonjour, M.; Combes, J.-D.; Broussais, F.; Sesques, P.; Traverse-Glehen, A.; de Martel, C. Lymphomas Associated with Epstein-Barr Virus Infection in 2020: Results from a Large, Unselected Case Series in France. eClinicalMedicine 2022, 54, 101674. [Google Scholar] [CrossRef] [PubMed]
- Cickusić, E.; Mustedanagić-Mujanović, J.; Iljazović, E.; Karasalihović, Z.; Skaljić, I. Association of Hodgkin’s Lymphoma with Epstein Barr Virus Infection. Bosn. J. Basic Med. Sci. 2007, 7, 58–65. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Armstrong, A.A.; Alexander, F.E.; Cartwright, R.; Angus, B.; Krajewski, A.S.; Wright, D.H.; Brown, I.; Lee, F.; Kane, E.; Jarrett, R.F. Epstein-Barr Virus and Hodgkin’s Disease: Further Evidence for the Three Disease Hypothesis. Leukemia 1998, 12, 1272–1276. [Google Scholar] [CrossRef]
- Gires, O.; Kohlhuber, F.; Kilger, E.; Baumann, M.; Kieser, A.; Kaiser, C.; Zeidler, R.; Scheffer, B.; Ueffing, M.; Hammerschmidt, W. Latent Membrane Protein 1 of Epstein-Barr Virus Interacts with JAK3 and Activates STAT Proteins. EMBO J. 1999, 18, 3064–3073. [Google Scholar] [CrossRef]
- Izumi, K.M.; Kaye, K.M.; Kieff, E.D. The Epstein-Barr Virus LMP1 Amino Acid Sequence That Engages Tumor Necrosis Factor Receptor Associated Factors Is Critical for Primary B Lymphocyte Growth Transformation. Proc. Natl. Acad. Sci. USA 1997, 94, 1447–1452. [Google Scholar] [CrossRef] [PubMed]
- Cheerathodi, M.R.; Meckes, D.G.J. The Epstein-Barr Virus LMP1 Interactome: Biological Implications and Therapeutic Targets. Future Virol. 2018, 13, 863–887. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.G.; Young, L.S. An Etiological Role for the Epstein-Barr Virus in the Pathogenesis of Classical Hodgkin Lymphoma. Blood 2019, 134, 591–596. [Google Scholar] [CrossRef]
- Merchant, M.; Swart, R.; Katzman, R.B.; Ikeda, M.; Ikeda, A.; Longnecker, R.; Dykstra, M.L.; Pierce, S.K. The Effects of the Epstein-Barr Virus Latent Membrane Protein 2A on B Cell Function. Int. Rev. Immunol. 2001, 20, 805–835. [Google Scholar] [CrossRef]
- Westhoff Smith, D.; Sugden, B. Potential Cellular Functions of Epstein-Barr Nuclear Antigen 1 (EBNA1) of Epstein-Barr Virus. Viruses 2013, 5, 226–240. [Google Scholar]
- Shankar, A.; Hayward, J.; Kirkwood, A.; McCarthy, K.; Hewitt, M.; Morland, B.; Daw, S. Treatment Outcome in Children and Adolescents with Relapsed Hodgkin Lymphoma--Results of the UK HD3 Relapse Treatment Strategy. Br. J. Haematol. 2014, 165, 534–544. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Shahid, K.; Khalife, M.; Dabney, R.; Phan, A.T. Immunotherapy and Targeted Therapy—The New Roadmap in Cancer Treatment. Ann. Transl. Med. 2019, 7, 595. [Google Scholar] [CrossRef]
- Cui, J.; Chen, Y.; Wang, H.Y.; Wang, R.-F. Mechanisms and Pathways of Innate Immune Activation and Regulation in Health and Cancer. Hum. Vaccines Immunother. 2014, 10, 3270–3285. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Fathallah, I.; Parroche, P.; Gruffat, H.; Zannetti, C.; Johansson, H.; Yue, J.; Manet, E.; Tommasino, M.; Sylla, B.S.; Hasan, U.A. EBV Latent Membrane Protein 1 Is a Negative Regulator of TLR9. J. Immunol. 2010, 185, 6439–6447. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Sun, L.; Liu, W.; Duan, Z. Latent Membrane Protein 1 of Epstein-Barr Virus Promotes RIG-I Degradation Mediated by Proteasome Pathway. Front. Immunol. 2018, 9, 1446. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.R.; Martin, J.M. The Epstein-Barr Virus-Encoded LMP-1 Oncoprotein Negatively Affects Tyk2 Phosphorylation and Interferon Signaling in Human B Cells. J. Virol. 2006, 80, 11638–11650. [Google Scholar] [CrossRef]
- Sadler, A.J.; Williams, B.R.G. Interferon-Inducible Antiviral Effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef]
- Shah, K.M.; Stewart, S.E.; Wei, W.; Woodman, C.B.J.; O’Neil, J.D.; Dawson, C.W.; Young, L.S. The EBV-Encoded Latent Membrane Proteins, LMP2A and LMP2B, Limit the Actions of Interferon by Targeting Interferon Receptors for Degradation. Oncogene 2009, 28, 3903–3914. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Z.; Peng, Q.; Liu, C.; Wu, Y.; Wen, Y.; Zheng, R.; Xu, C.; Tian, J.; Zheng, X.; et al. Epstein-Barr Virus Suppresses N(6)-Methyladenosine Modification of TLR9 to Promote Immune Evasion. J. Biol. Chem. 2024, 300, 107226. [Google Scholar] [CrossRef]
- Westhoff Smith, D.; Chakravorty, A.; Hayes, M.; Hammerschmidt, W.; Sugden, B. The Epstein-Barr Virus Oncogene EBNA1 Suppresses Natural Killer Cell Responses and Apoptosis Early after Infection of Peripheral B Cells. MBio 2021, 12, e0224321. [Google Scholar] [CrossRef]
- Bouvet, M.; Voigt, S.; Tagawa, T.; Albanese, M.; Chen, Y.-F.A.; Chen, Y.; Fachko, D.N.; Pich, D.; Göbel, C.; Skalsky, R.L.; et al. Multiple Viral MicroRNAs Regulate Interferon Release and Signaling Early during Infection with Epstein-Barr Virus. MBio 2021, 12, e03440-20. [Google Scholar] [CrossRef]
- Lu, Y.; Qin, Z.; Wang, J.; Zheng, X.; Lu, J.; Zhang, X.; Wei, L.; Peng, Q.; Zheng, Y.; Ou, C.; et al. Epstein-Barr Virus MiR-BART6-3p Inhibits the RIG-I Pathway. J. Innate Immun. 2017, 9, 574–586. [Google Scholar] [CrossRef]
- Haneklaus, M.; Gerlic, M.; Kurowska-Stolarska, M.; Rainey, A.-A.; Pich, D.; McInnes, I.B.; Hammerschmidt, W.; O’Neill, L.A.J.; Masters, S.L. Cutting Edge: MiR-223 and EBV MiR-BART15 Regulate the NLRP3 Inflammasome and IL-1β Production. J. Immunol. 2012, 189, 3795–3799. [Google Scholar] [CrossRef] [PubMed]
- Hooykaas, M.J.G.; van Gent, M.; Soppe, J.A.; Kruse, E.; Boer, I.G.J.; van Leenen, D.; Groot Koerkamp, M.J.A.; Holstege, F.C.P.; Ressing, M.E.; Wiertz, E.J.H.J.; et al. EBV MicroRNA BART16 Suppresses Type I IFN Signaling. J. Immunol. 2017, 198, 4062–4073. [Google Scholar] [CrossRef] [PubMed]
- Skinner, C.M.; Ivanov, N.S.; Barr, S.A.; Chen, Y.; Skalsky, R.L. An Epstein-Barr Virus MicroRNA Blocks Interleukin-1 (IL-1) Signaling by Targeting IL-1 Receptor 1. J. Virol. 2017, 91, e00530-17. [Google Scholar] [CrossRef] [PubMed]
- Dölken, L.; Malterer, G.; Erhard, F.; Kothe, S.; Friedel, C.C.; Suffert, G.; Marcinowski, L.; Motsch, N.; Barth, S.; Beitzinger, M.; et al. Systematic Analysis of Viral and Cellular MicroRNA Targets in Cells Latently Infected with Human Gamma-Herpesviruses by RISC Immunoprecipitation Assay. Cell Host Microbe 2010, 7, 324–334. [Google Scholar] [CrossRef]
- Yang, I.V.; Wade, C.M.; Kang, H.M.; Alper, S.; Rutledge, H.; Lackford, B.; Eskin, E.; Daly, M.J.; Schwartz, D.A. Identification of Novel Genes That Mediate Innate Immunity Using Inbred Mice. Genetics 2009, 183, 1535–1544. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Fan, J.; Li, Q.; Liang, J.; Chen, Z.; Chen, L.; Lai, J.; Chen, Q. Regulation of IFNβ Expression: Focusing on the Role of Its Promoter and Transcription Regulators. Front. Microbiol. 2023, 14, 1158777. [Google Scholar] [CrossRef]
- Nachmani, D.; Stern-Ginossar, N.; Sarid, R.; Mandelboim, O. Diverse Herpesvirus MicroRNAs Target the Stress-Induced Immune Ligand MICB to Escape Recognition by Natural Killer Cells. Cell Host Microbe 2009, 5, 376–385. [Google Scholar] [CrossRef]
- Xing, S.; Ferrari de Andrade, L. NKG2D and MICA/B Shedding: A “tag Game” between NK Cells and Malignant Cells. Clin. Transl. Immunol. 2020, 9, e1230. [Google Scholar] [CrossRef]
- Ressing, M.E.; van Gent, M.; Gram, A.M.; Hooykaas, M.J.G.; Piersma, S.J.; Wiertz, E.J.H.J. Immune Evasion by Epstein-Barr Virus. Curr. Top. Microbiol. Immunol. 2015, 391, 355–381. [Google Scholar] [CrossRef]
- Smith, C.; Wakisaka, N.; Crough, T.; Peet, J.; Yoshizaki, T.; Beagley, L.; Khanna, R. Discerning Regulation of Cis- and Trans-Presentation of CD8+ T-Cell Epitopes by EBV-Encoded Oncogene LMP-1 through Self-Aggregation. Blood 2009, 113, 6148–6152. [Google Scholar] [CrossRef] [PubMed]
- Šimičić, P.; Batović, M.; Stojanović Marković, A.; Židovec-Lepej, S. Deciphering the Role of Epstein-Barr Virus Latent Membrane Protein 1 in Immune Modulation: A Multifaced Signalling Perspective. Viruses 2024, 16, 564. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.L.; Martinez, O.M. Latent Membrane Protein 1 of EBV Activates Phosphatidylinositol 3-Kinase to Induce Production of IL-10. J. Immunol. 2007, 179, 8225–8234. [Google Scholar] [CrossRef] [PubMed]
- Beatty, P.R.; Krams, S.M.; Martinez, O.M. Involvement of IL-10 in the Autonomous Growth of EBV-Transformed B Cell Lines. J. Immunol. 1997, 158, 4045–4051. [Google Scholar] [CrossRef]
- Zeidler, R.; Eissner, G.; Meissner, P.; Uebel, S.; Tampé, R.; Lazis, S.; Hammerschmidt, W. Downregulation of TAP1 in B Lymphocytes by Cellular and Epstein-Barr Virus-Encoded Interleukin-10. Blood 1997, 90, 2390–2397. [Google Scholar] [CrossRef]
- Smith, L.K.; Boukhaled, G.M.; Condotta, S.A.; Mazouz, S.; Guthmiller, J.J.; Vijay, R.; Butler, N.S.; Bruneau, J.; Shoukry, N.H.; Krawczyk, C.M.; et al. Interleukin-10 Directly Inhibits CD8(+) T Cell Function by Enhancing N-Glycan Branching to Decrease Antigen Sensitivity. Immunity 2018, 48, 299–312.e5. [Google Scholar] [CrossRef]
- Aravinth, S.P.; Rajendran, S.; Li, Y.; Wu, M.; Yi Wong, A.H.; Schwarz, H. Epstein-Barr Virus-Encoded LMP1 Induces Ectopic CD137 Expression on Hodgkin and Reed-Sternberg Cells via the PI3K-AKT-MTOR Pathway. Leuk. Lymphoma 2019, 60, 2697–2704. [Google Scholar] [CrossRef]
- Rajendran, S.; Ho, W.T.; Schwarz, H. CD137 Signaling in Hodgkin and Reed-Sternberg Cell Lines Induces IL-13 Secretion, Immune Deviation and Enhanced Growth. Oncoimmunology 2016, 5, e1160188. [Google Scholar] [CrossRef]
- Lin, J.-H.; Lin, J.-Y.; Chou, Y.-C.; Chen, M.-R.; Yeh, T.-H.; Lin, C.-W.; Lin, S.-J.; Tsai, C.-H. Epstein-Barr Virus LMP2A Suppresses MHC Class II Expression by Regulating the B-Cell Transcription Factors E47 and PU.1. Blood 2015, 125, 2228–2238. [Google Scholar] [CrossRef]
- Incrocci, R.; Barse, L.; Stone, A.; Vagvala, S.; Montesano, M.; Subramaniam, V.; Swanson-Mungerson, M. Epstein-Barr Virus Latent Membrane Protein 2A (LMP2A) Enhances IL-10 Production through the Activation of Bruton’s Tyrosine Kinase and STAT3. Virology 2017, 500, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Rancan, C.; Schirrmann, L.; Hüls, C.; Zeidler, R.; Moosmann, A. Latent Membrane Protein LMP2A Impairs Recognition of EBV-Infected Cells by CD8+ T Cells. PLoS Pathog. 2015, 11, e1004906. [Google Scholar] [CrossRef] [PubMed]
- Levitskaya, J.; Coram, M.; Levitsky, V.; Imreh, S.; Steigerwald-Mullen, P.M.; Klein, G.; Kurilla, M.G.; Masucci, M.G. Inhibition of Antigen Processing by the Internal Repeat Region of the Epstein-Barr Virus Nuclear Antigen-1. Nature 1995, 375, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Manoury, B.; Fåhraeus, R. Self-Inhibition of Synthesis and Antigen Presentation by Epstein-Barr Virus-Encoded EBNA1. Science 2003, 301, 1371–1374. [Google Scholar] [CrossRef]
- Apcher, S.; Daskalogianni, C.; Manoury, B.; Fåhraeus, R. Epstein Barr Virus-Encoded EBNA1 Interference with MHC Class I Antigen Presentation Reveals a Close Correlation between MRNA Translation Initiation and Antigen Presentation. PLoS Pathog. 2010, 6, e1001151. [Google Scholar] [CrossRef]
- Daskalogianni, C.; Apcher, S.; Candeias, M.M.; Naski, N.; Calvo, F.; Fåhraeus, R. Gly-Ala Repeats Induce Position- and Substrate-Specific Regulation of 26 S Proteasome-Dependent Partial Processing. J. Biol. Chem. 2008, 283, 30090–30100. [Google Scholar] [CrossRef]
- Murer, A.; Rühl, J.; Zbinden, A.; Capaul, R.; Hammerschmidt, W.; Chijioke, O.; Münz, C. MicroRNAs of Epstein-Barr Virus Attenuate T-Cell-Mediated Immune Control In Vivo. MBio 2019, 10, e01941-18. [Google Scholar] [CrossRef]
- Tagawa, T.; Albanese, M.; Bouvet, M.; Moosmann, A.; Mautner, J.; Heissmeyer, V.; Zielinski, C.; Lutter, D.; Hoser, J.; Hastreiter, M.; et al. Epstein-Barr Viral MiRNAs Inhibit Antiviral CD4+ T Cell Responses Targeting IL-12 and Peptide Processing. J. Exp. Med. 2016, 213, 2065–2080. [Google Scholar] [CrossRef]
- Albanese, M.; Tagawa, T.; Bouvet, M.; Maliqi, L.; Lutter, D.; Hoser, J.; Hastreiter, M.; Hayes, M.; Sugden, B.; Martin, L.; et al. Epstein-Barr Virus MicroRNAs Reduce Immune Surveillance by Virus-Specific CD8+ T Cells. Proc. Natl. Acad. Sci. USA 2016, 113, E6467–E6475. [Google Scholar] [CrossRef]
- Boussiotis, V.A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N. Engl. J. Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef]
- Tsotridou, E.; Vasileiou, E.; Mantadakis, E.; Tragiannidis, A. Safety and Efficacy of Immune Checkpoint Inhibitors in Children and Young Adults with Haematological Malignancies: Review and Future Perspectives. Cardiovasc. Hematol. Agents Med. Chem. 2022, 20, 20–33. [Google Scholar] [CrossRef]
- Yu, J.; Jin, S.; Yin, X.; Du, H. Expression of the Immune Checkpoint Molecules PD-L1 and PD-1 in EBV-associated Lymphoproliferative Disorders: A Meta-analysis. Exp. Ther. Med. 2024, 27, 7. [Google Scholar] [CrossRef]
- Roemer, M.G.M.; Advani, R.H.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 2690–2697. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative Analysis Reveals Selective 9p24.1 Amplification, Increased PD-1 Ligand Expression, and Further Induction via JAK2 in Nodular Sclerosing Hodgkin Lymphoma and Primary Mediastinal Large B-Cell Lymphoma. Blood 2010, 116, 3268–3277. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 Activity and EBV Infection Induce PD-L1 in Hodgkin Lymphomas and Posttransplant Lymphoproliferative Disorders: Implications for Targeted Therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef]
- Cristino, A.S.; Nourse, J.; West, R.A.; Sabdia, M.B.; Law, S.C.; Gunawardana, J.; Vari, F.; Mujaj, S.; Thillaiyampalam, G.; Snell, C.; et al. EBV MicroRNA-BHRF1-2-5p Targets the 3′UTR of Immune Checkpoint Ligands PD-L1 and PD-L2. Blood 2019, 134, 2261–2270. [Google Scholar] [CrossRef] [PubMed]
- Carey, C.D.; Gusenleitner, D.; Lipschitz, M.; Roemer, M.G.M.; Stack, E.C.; Gjini, E.; Hu, X.; Redd, R.; Freeman, G.J.; Neuberg, D.; et al. Topological Analysis Reveals a PD-L1-Associated Microenvironmental Niche for Reed-Sternberg Cells in Hodgkin Lymphoma. Blood 2017, 130, 2420–2430. [Google Scholar] [CrossRef]
- Chikuma, S. CTLA-4, an Essential Immune-Checkpoint for T-Cell Activation. Curr. Top. Microbiol. Immunol. 2017, 410, 99–126. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.-D.; Xu, X.; Jones, R.; Delecluse, H.-J.; Zumwalde, N.A.; Sharma, A.; Gumperz, J.E.; Kenney, S.C. PD-1/CTLA-4 Blockade Inhibits Epstein-Barr Virus-Induced Lymphoma Growth in a Cord Blood Humanized-Mouse Model. PLoS Pathog. 2016, 12, e1005642. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel Immune Checkpoint Targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef]
- Gandhi, M.K.; Lambley, E.; Duraiswamy, J.; Dua, U.; Smith, C.; Elliott, S.; Gill, D.; Marlton, P.; Seymour, J.; Khanna, R. Expression of LAG-3 by Tumor-Infiltrating Lymphocytes Is Coincident with the Suppression of Latent Membrane Antigen-Specific CD8+ T-Cell Function in Hodgkin Lymphoma Patients. Blood 2006, 108, 2280–2289. [Google Scholar] [CrossRef] [PubMed]
- Dilly-Feldis, M.; Aladjidi, N.; Refait, J.K.; Parrens, M.; Ducassou, S.; Rullier, A. Expression of PD-1/PD-L1 in Children’s Classical Hodgkin Lymphomas. Pediatr. Blood Cancer 2019, 66, e27571. [Google Scholar] [CrossRef]
- Uccini, S.; Al-Jadiry, M.F.; Pepe, G.; Scarpino, S.; Al-Hadad, S.A.; Ruco, L. PD-L1 Expression in Pediatric Epstein-Barr Virus Positive Classic Hodgkin Lymphoma Is Not Associated with 9p24.1 Amplification. Pediatr. Blood Cancer 2019, 66, e27757. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, O.; Mangiaterra, T.; Colli, S.; García Lombardi, M.; Preciado, M.V.; De Matteo, E.; Chabay, P. PD-1 and LAG-3 Expression in EBV-Associated Pediatric Hodgkin Lymphoma Has Influence on Survival. Front. Oncol. 2022, 12, 957208. [Google Scholar] [CrossRef]
- Weniger, M.A.; Küppers, R. Molecular Biology of Hodgkin Lymphoma. Leukemia 2021, 35, 968–981. [Google Scholar] [CrossRef] [PubMed]
- Georgoulis, V.; Papoudou-Bai, A.; Makis, A.; Kanavaros, P.; Hatzimichael, E. Unraveling the Immune Microenvironment in Classic Hodgkin Lymphoma: Prognostic and Therapeutic Implications. Biology 2023, 12, 862. [Google Scholar] [CrossRef]
- Henry, M.; Buck, S.; Savaşan, S. Flow Cytometry for Assessment of the Tumor Microenvironment in Pediatric Hodgkin Lymphoma. Pediatr. Blood Cancer 2018, 65, e27307. [Google Scholar] [CrossRef]
- Menéndez, V.; Solórzano, J.L.; García-Cosío, M.; Alonso-Alonso, R.; Rodríguez, M.; Cereceda, L.; Fernández, S.; Díaz, E.; Montalbán, C.; Estévez, M.; et al. Immune and Stromal Transcriptional Patterns That Influence the Outcome of Classic Hodgkin Lymphoma. Sci. Rep. 2024, 14, 710. [Google Scholar] [CrossRef]
- Aoki, T.; Jiang, A.; Xu, A.; Yin, Y.; Gamboa, A.; Milne, K.; Takata, K.; Miyata-Takata, T.; Chung, S.; Rai, S.; et al. Spatially Resolved Tumor Microenvironment Predicts Treatment Outcomes in Relapsed/Refractory Hodgkin Lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2024, 42, 1077–1087. [Google Scholar] [CrossRef]
- Jimenez, O.; Colli, S.; Garcia Lombardi, M.; Preciado, M.V.; De Matteo, E.; Chabay, P. Epstein-Barr Virus Recruits PDL1-Positive Cells at the Microenvironment in Pediatric Hodgkin Lymphoma. Cancer Immunol. Immunother. 2021, 70, 1519–1526. [Google Scholar] [CrossRef]
- Morales, O.; Mrizak, D.; François, V.; Mustapha, R.; Miroux, C.; Depil, S.; Decouvelaere, A.-V.; Lionne-Huyghe, P.; Auriault, C.; de Launoit, Y.; et al. Epstein-Barr Virus Infection Induces an Increase of T Regulatory Type 1 Cells in Hodgkin Lymphoma Patients. Br. J. Haematol. 2014, 166, 875–890. [Google Scholar] [CrossRef]
- Baumforth, K.R.N.; Birgersdotter, A.; Reynolds, G.M.; Wei, W.; Kapatai, G.; Flavell, J.R.; Kalk, E.; Piper, K.; Lee, S.; Machado, L.; et al. Expression of the Epstein-Barr Virus-Encoded Epstein-Barr Virus Nuclear Antigen 1 in Hodgkin’s Lymphoma Cells Mediates Up-Regulation of CCL20 and the Migration of Regulatory T Cells. Am. J. Pathol. 2008, 173, 195–204. [Google Scholar] [CrossRef]
- Marshall, N.A.; Christie, L.E.; Munro, L.R.; Culligan, D.J.; Johnston, P.W.; Barker, R.N.; Vickers, M.A. Immunosuppressive Regulatory T Cells Are Abundant in the Reactive Lymphocytes of Hodgkin Lymphoma. Blood 2004, 103, 1755–1762. [Google Scholar] [CrossRef]
- Marshall, N.A.; Culligan, D.J.; Tighe, J.; Johnston, P.W.; Barker, R.N.; Vickers, M.A. The Relationships between Epstein-Barr Virus Latent Membrane Protein 1 and Regulatory T Cells in Hodgkin’s Lymphoma. Exp. Hematol. 2007, 35, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Adam, M.; Bekuretsion, Y.; Gebremedhin, A.; Kwiecinska, A.; Howe, R.; Petros, B.; Jerkeman, M. Evidence for Distinct Mechanisms of Immune Suppression in EBV-Positive and EBV-Negative Hodgkin Lymphoma. J. Clin. Exp. Hematop. 2023, 63, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Pavlović, A.; Miljak, A.; Brzica, K.; Glavina Durdov, M. The Abundance of FOXP3, FOXP3/CD4 and CD8 Cells in the Microenvironment of Nodular Sclerosis and Mixed Cellularity Subtypes Is Associated with the Epstein-Barr Virus Status of Classic Hodgkin Lymphoma. Biomedicines 2024, 12, 1680. [Google Scholar] [CrossRef]
- Jimenez, O.; Barros, M.H.; De Matteo, E.; Garcia Lombardi, M.; Preciado, M.V.; Niedobitek, G.; Chabay, P. M1-like Macrophage Polarization Prevails in Young Children with Classic Hodgkin Lymphoma from Argentina. Sci. Rep. 2019, 9, 12687. [Google Scholar] [CrossRef] [PubMed]
- Barros, M.H.M.; Segges, P.; Vera-Lozada, G.; Hassan, R.; Niedobitek, G. Macrophage Polarization Reflects T Cell Composition of Tumor Microenvironment in Pediatric Classical Hodgkin Lymphoma and Has Impact on Survival. PLoS ONE 2015, 10, e0124531. [Google Scholar] [CrossRef]
- Vistarop, A.; Jimenez, O.; Cohen, M.; De Matteo, E.; Preciado, M.V.; Chabay, P. Differences in Epstein-Barr Virus Characteristics and Viral-Related Microenvironment Could Be Responsible for Lymphomagenesis in Children. Pathogens 2020, 9, 68. [Google Scholar] [CrossRef]
- Choe, J.-Y.; Yun, J.Y.; Jeon, Y.K.; Kim, S.H.; Park, G.; Huh, J.R.; Oh, S.; Kim, J.E. Indoleamine 2,3-Dioxygenase (IDO) Is Frequently Expressed in Stromal Cells of Hodgkin Lymphoma and Is Associated with Adverse Clinical Features: A Retrospective Cohort Study. BMC Cancer 2014, 14, 335. [Google Scholar] [CrossRef]
- Andersen, M.H. The Specific Targeting of Immune Regulation: T-Cell Responses against Indoleamine 2,3-Dioxygenase. Cancer Immunol. Immunother. 2012, 61, 1289–1297. [Google Scholar] [CrossRef]
- Barros, M.H.M.; Vera-Lozada, G.; Soares, F.A.; Niedobitek, G.; Hassan, R. Tumor Microenvironment Composition in Pediatric Classical Hodgkin Lymphoma Is Modulated by Age and Epstein-Barr Virus Infection. Int. J. Cancer 2012, 131, 1142–1152. [Google Scholar] [CrossRef]
- Barros, M.H.M.; Hassan, R.; Niedobitek, G. Tumor-Associated Macrophages in Pediatric Classical Hodgkin Lymphoma: Association with Epstein-Barr Virus, Lymphocyte Subsets, and Prognostic Impact. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 3762–3771. [Google Scholar] [CrossRef]
- Massini, G.; Siemer, D.; Hohaus, S. EBV in Hodgkin Lymphoma. Mediterr. J. Hematol. Infect. Dis. 2009, 1, e2009013. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Bröckelmann, P.J.; von Keudell, G.; Lee, H.J.; Santoro, A.; Zinzani, P.L.; Collins, G.P.; Cohen, J.B.; de Boer, J.P.; Kuruvilla, J.; et al. Nivolumab for Relapsed/Refractory Classical Hodgkin Lymphoma: 5-Year Survival from the Pivotal Phase 2 CheckMate 205 Study. Blood Adv. 2023, 7, 6266–6274. [Google Scholar] [CrossRef]
- Ramchandren, R.; Domingo-Domènech, E.; Rueda, A.; Trněný, M.; Feldman, T.A.; Lee, H.J.; Provencio, M.; Sillaber, C.; Cohen, J.B.; Savage, K.J.; et al. Nivolumab for Newly Diagnosed Advanced-Stage Classic Hodgkin Lymphoma: Safety and Efficacy in the Phase II CheckMate 205 Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Harker-Murray, P.; Leblanc, T.; Mascarin, M.; Mauz-Körholz, C.; Michel, G.; Cooper, S.; Beishuizen, A.; Leger, K.; Garaventa, A.; Buffardi, S.; et al. Response-Adapted Therapy with Nivolumab and Brentuximab Vedotin (BV), Followed By BV and Bendamustine for Suboptimal Response, in Children, Adolescents, and Young Adults with Standard-Risk Relapsed/Refractory Classical Hodgkin Lymphoma. Blood 2018, 132, 927. [Google Scholar] [CrossRef]
- Davis, K.L.; Fox, E.; Merchant, M.S.; Reid, J.M.; Kudgus, R.A.; Liu, X.; Minard, C.G.; Voss, S.; Berg, S.L.; Weigel, B.J.; et al. Nivolumab in Children and Young Adults with Relapsed or Refractory Solid Tumours or Lymphoma (ADVL1412): A Multicentre, Open-Label, Single-Arm, Phase 1–2 Trial. Lancet Oncol. 2020, 21, 541–550. [Google Scholar] [CrossRef]
- Bollard, C.M.; Aguilar, L.; Straathof, K.C.; Gahn, B.; Huls, M.H.; Rousseau, A.; Sixbey, J.; Gresik, M.V.; Carrum, G.; Hudson, M.; et al. Cytotoxic T Lymphocyte Therapy for Epstein-Barr Virus+ Hodgkin’s Disease. J. Exp. Med. 2004, 200, 1623–1633. [Google Scholar] [CrossRef]
- Bollard, C.M.; Gottschalk, S.; Torrano, V.; Diouf, O.; Ku, S.; Hazrat, Y.; Carrum, G.; Ramos, C.; Fayad, L.; Shpall, E.J.; et al. Sustained Complete Responses in Patients with Lymphoma Receiving Autologous Cytotoxic T Lymphocytes Targeting Epstein-Barr Virus Latent Membrane Proteins. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 798–808. [Google Scholar] [CrossRef]
- Bollard, C.M.; Tripic, T.; Cruz, C.R.; Dotti, G.; Gottschalk, S.; Torrano, V.; Dakhova, O.; Carrum, G.; Ramos, C.A.; Liu, H.; et al. Tumor-Specific T-Cells Engineered to Overcome Tumor Immune Evasion Induce Clinical Responses in Patients with Relapsed Hodgkin Lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 1128–1139. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, L.P.; Rouce, R.; Gottschalk, S.; Torrano, V.; Carrum, G.; Wu, M.-F.; Hoq, F.; Grilley, B.; Marcogliese, A.M.; Hanley, P.J.; et al. EBV/LMP-Specific T Cells Maintain Remissions of T- and B-Cell EBV Lymphomas after Allogeneic Bone Marrow Transplantation. Blood 2018, 132, 2351–2361. [Google Scholar] [CrossRef]
- Jiang, L.; Xie, C.; Lung, H.L.; Lo, K.W.; Law, G.-L.; Mak, N.-K.; Wong, K.-L. EBNA1-Targeted Inhibitors: Novel Approaches for the Treatment of Epstein-Barr Virus-Associated Cancers. Theranostics 2018, 8, 5307–5319. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-M.; Wu, Z.-Q.; Wang, Y.; Guo, Y.-L.; Dai, H.-R.; Wang, X.-H.; Li, X.; Zhang, Y.-J.; Zhang, W.-Y.; Chen, M.-X.; et al. Autologous T Cells Expressing CD30 Chimeric Antigen Receptors for Relapsed or Refractory Hodgkin Lymphoma: An Open-Label Phase I Trial. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.A.; Ballard, B.; Zhang, H.; Dakhova, O.; Gee, A.P.; Mei, Z.; Bilgi, M.; Wu, M.-F.; Liu, H.; Grilley, B.; et al. Clinical and Immunological Responses after CD30-Specific Chimeric Antigen Receptor-Redirected Lymphocytes. J. Clin. Investig. 2017, 127, 3462–3471. [Google Scholar] [CrossRef]
- Ramos, C.A.; Grover, N.S.; Beaven, A.W.; Lulla, P.D.; Wu, M.-F.; Ivanova, A.; Wang, T.; Shea, T.C.; Rooney, C.M.; Dittus, C.; et al. Anti-CD30 CAR-T Cell Therapy in Relapsed and Refractory Hodgkin Lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 3794–3804. [Google Scholar] [CrossRef]
- Jean-Pierre, V.; Lupo, J.; Buisson, M.; Morand, P.; Germi, R. Main Targets of Interest for the Development of a Prophylactic or Therapeutic Epstein-Barr Virus Vaccine. Front. Microbiol. 2021, 12, 701611. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).